
Abstract
In this work, we present the synthesis and evaluation of magnetic resonance (MR) properties of novel phosphorus/iron-containing probes for dual 31P and 1H MR imaging and spectroscopy (MRI and MRS). The presented probes are composed of biocompatible semitelechelic and multivalent phospho-polymers based on poly(2-methacryloyloxyethyl phosphorylcholine) (pMPC) coordinated with small paramagnetic Fe3+ ions or superparamagnetic maghemite (γ-Fe2O3) nanoparticles via deferoxamine group linked to the end or along the polymer chains. All probes provided very short 1H T1 and T2 relaxation times even at low iron concentrations. The presence of iron had a significant impact on the shortening of 31P relaxation, with the effect being more pronounced for probes based on γ-Fe2O3 and multivalent polymer. While the water-soluble probe having one Fe3+ ion per polymer chain was satisfactorily visualized by both 31P-MRS and 31P-MRI, the probe with multiple Fe3+ ions could only be detected by 31P-MRS, and the probes consisting of γ-Fe2O3 nanoparticles could not be imaged by either technique due to their ultra-short 31P relaxations. In this proof-of-principle study performed on phantoms at a clinically relevant magnetic fields, we demonstrated how the different forms and concentrations of iron affect both the 1H MR signal of the surrounding water molecules and the 31P MR signal of the phospho-polymer probe. Thus, this double contrast can be exploited to simultaneously visualize body anatomy and monitor probe biodistribution.
Similar content being viewed by others

Introduction
Magnetic resonance imaging and spectroscopy (MRI and MRS) are advanced non-invasive techniques used in clinical and experimental medicine to visualize anatomical structures, analyze the chemical composition of tissues and metabolites, and monitor changes in physiological and pathological processes within the body. Currently, hydrogen (1H) is the most frequently imaged element in clinical MR methods because it is highly abundant in the human body (~ 10% of body mass), especially in water and fat, and its nucleus has favorable intrinsic physical properties spin quantum number (s = ½) and large gyromagnetic moment (γ = 42.6 MHz T−1) that is higher than any other isotopes, which allows its sensitive detection at relatively low magnetic fields of clinical scanners (1.5–3 T). Moreover, changes in the physicochemical properties of water correlate well with changes in biochemical processes in living tissue1.
To increase the contrast and improve the visibility of specific internal body areas, a contrast agent may be administrated to the patient before the examination2. The most commonly used type of contrast agents in clinical 1H-MR settings are paramagnetic coordination compounds based on gadolinium, which enhance the MR signal by dominant shortening the T1 relaxation time of nearby water protons3. Although these agents significantly improve the diagnostic capabilities of MRI, their use has been associated with some adverse effects such as allergic reactions, nephrotoxicity and rarely nephrogenic systemic fibrosis4. For some specific purposes, such as imaging the liver, transplanted cells or a certain type of cancer, superparamagnetic iron oxide-based nanoparticles, also called SPIONs (usually based on magnetite—Fe3O4), are clinically used as safer alternatives5,6,7,8. However, despite their biogenic nature, there are concerns that these T1/T2 relaxants also exhibit toxic effects in vivo, primarily due to non-specific binding to cellular components and blood plasma proteins, unless their surface is effectively coated, e.g. with hydrophilic polymers9. High hopes are also placed on low molecular weight iron(III) complexes, which have the advantage of high thermodynamic stability, low long-term toxicity and potential for multimodal imaging10. On the other hand, iron is a redox-active metal that can induce the production of dangerous reactive oxygen species in the body, so it is necessary to ensure its bioinertness by using a suitable ligand or a protective polymer layer11,12. Therefore, there is a growing need to The development of bioinert and tissue-specific contrast agents for a safer and more efficient contrast agent for in vivo MR imaging is therefore an important challenge.
While 1H-MR provides a unique insight into the anatomical distribution and morphology of organs and tissues within the body, advanced MR techniques have recently been developed to visualize the nuclei of other elements (called X-nuclei) to gather valuable information about tissue function, metabolism and physiology13. Among various imageable X-nuclei, such as 11B, 19F, 23Na, 31P, etc., 31P has received a considerable attention in recent years because it is present in many biomolecules such as nucleic acids, phospholipids, bioenergetic molecules, etc., which play an important role in many vital processes. Although the concentration of phosphorus in the human body is ~ 10 times lower (~ 1% of body mass) than that of hydrogen, and its monoisotope 31P has ~ 2.5 times lower gyromagnetic moment (s = ½, γ = 17.2 MHz T−1), and therefore lower MR sensitivity than the proton 1H, clinical scanners equipped with sensitive radiofrequency coils together with advanced imaging sequences (e.g. chemical shift imaging) allow its reliable detection. In clinical practice, 31P MR techniques are mainly used to study cell membrane composition, phosphorylated metabolite levels, bioenergetic status of organs and tissues, and intracellular pH levels14,15,16. Moreover, 31P MR techniques are also able to follow the biological fate of various exogenous phosphorus-containing compounds such as drug delivery carriers or biosensors of pathological conditions, which, in synergy with 1H MR, can provide comprehensive information on their biodistribution and pharmacokinetics or on the occurrence of physiological abnormalities in the body17,18.
In order to effectively integrate 1H with 31P MRI and thus take full advantage of the information provided by both methods, it is desirable to use a contrast agent/probe that would improve the visibility of internal body structures in 1H MRI while ensuring its traceability by 31P MRI. Such a compound should optimally contain in its structure both a (super)paramagnetic metal amplifying the 1H MR signal of nearby water protons and phosphorus in its structure to amplify the 1H MR signal of nearby water protons as well as to providing high and distinguishable 31P MR signal. In addition, the presence of paramagnetic metal can also affect the relaxation of 31P nuclei, allowing a 31P MR signal of sufficient intensity to be obtained19,20. In terms of clinical application, the contrast agent/probe while it should be well tolerated by the organism, have a It is also advantageous if the contrast agent/probe has a long biological half-life, specifically accumulate in target tissues, and should be metabolized or eliminated from the body after performing its function. One material that could meet these requirements is hydrophilic biocompatible phospho-polymers or phosphate-based colloids coordinated with iron.
In this regard, the most frequently studied phospho-polymers are water-soluble poly(organophosphazene)s, poly(phosphoester)s, or poly(2-methacryloyloxyethyl phosphorylcholine) (pMPC), all experimentally applied in many biomedical fields, including drug delivery, regenerative medicine, antifouling coating technology or biosensing21,22,23. However, since The results of our recent comparative study of chemically and structurally different phospho-polymers showed that the best results in terms of MR properties are achieved by linear pMPC achieved the best results in terms of MR properties, we decided to focus just on this type of polymer17. We believe that the favorable MR properties of the pMPC polymer are due to both the high content of phosphorus in its structure and the polyzwitterionic character ensuring the very high hydrophilicity of its chains. In addition, the linear arrangement of its chains allows the anchoring of chelating groups both at the end and along the backbone, which can effectively control the concentration of coordinated Fe3+ ions or the coverage density of SPION nanoparticles and thereby influence their colloidal and biological stability. Other advantages of Finally, pMPC include that it can be prepared relatively easily and reproducibly by controlled polymerization techniques which not only that provide a well-defined material with tunable structural properties24. But also allow the incorporation of ligands for iron complexation.Therefore, we have focused on this type of polymer in this work.
For these purposes, two types of pMPC-based polymers were synthesized in this work: (i) semitelechelic with one iron-chelating deferoxamine (DFA) group at the end of the polymer chain and (ii) multivalent with multiple DFA groups distributed along the polymer chain. These polymers were complexed with small paramagnetic Fe3+ ions or colloidal superparamagnetic maghemite (γ-Fe2O3) nanoparticles. The influence of the density (single vs multiple) and form (soluble vs colloidal) of iron complexed to MPC-based polymers on 31P and 1H MR properties was evaluated under experimental conditions on a 4.7 T MR scanner with the aim of finding the optimal composition of complexes trackable in vitro/in vivo using dual 1H/31P MR spectroscopy and imaging. We believe that the development of new biosensors based on complexes of phospho-polymers with iron has great potential to expand the application possibilities of MR techniques usable in both anatomical and functional imaging.
Materials and methods
Chemicals
3-Aminopropanoic acid, 4,4′-azobis(4-cyanopentanoic acid) (ACVA), 2,2′-azobis(2-methylpropionitrile) (AIBN), N,N'-dicyclohexylcarbodiimide (DCC), 4-dimethylaminopyridine (DMAP), methacryloyl chloride, 2-methacryloyloxyethyl phosphorylcholine (MPC), thiazolidine-2-thione (TT) and triethylamine (TEA) were purchased from TCI Europe, Belgium. Ammonium hydroxide, 4-cyano-4-(phenylcarbonothioylthio)pentanoic acid (CPP), 2-cyano-2-propyl benzodithioate (CPB), deferoxamine methanesulphonate salt (DFA), iron(III) chloride hexahydrate, iron(II) chloride tetrahydrate and sodium bicarbonate were purchased from Sigma-Aldrich, Czech Republic. 2,2'-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) was from FUJIFILM Wako Chemicals Europe, Germany. All solvents were of HPLC grade (obtained from VWR International, Czech Republic) and dried over a layer of activated molecular sieves (4 Å) before use.
Synthesis of maghemite nanoparticles
Maghemite nanoparticles (γ-Fe2O3) were produced by aqueous co-precipitation of FeCl3∙6H2O and FeCl2∙4H2O salts in the presence of ammonium hydroxide under sonication followed by oxidation of the generated magnetite (Fe3O4) with sodium hypochlorite9. The z-average hydrodynamic diameter determined by Dynamic Light Scattering (DLS) (DhDLS) and the number-average diameter determined by Transmission Electron Microscopy (TEM) (DnTEM) of γ-Fe2O3 nanoparticles were 60.9 nm and 10.3 nm, respectively.
Synthesis of monomer
3-(3-Methacrylamidopropanoyl)thiazolidine-2-thione (Ma-βAla-TT) monomer was synthesized by the acylation of 3-aminopropanoic acid with methacryloyl chloride in an aqueous alkaline medium followed by the reaction of formed 3-methacrylamidopropanoic acid with TT in tetrahydrofuran in the presence of DCC and DMAP25.
Synthesis of RAFT agent and initiator
2-cyano-5-oxo-5-(2-sulphanylidene-1,3-thiazolidin-3-yl)pentan-2-yl benzenecarbodithioate (CTA-TT) RAFT agent was synthesized by the condensation of CPP acid with TT in dichloromethane in the presence of DCC and DMAP26.
2-[1-Cyano-1-methyl-4-oxo-4-(2-thioxo-thiazolidin-3-yl)-butylazo]-2-methyl-5-oxo-5-(2-thioxothiazolidin-3-yl)-pentanenitrile (ACVA-(TT)2) initiator was prepared by the reaction of ACVA with TT in tetrahydrofuran in the presence of DCC and DMAP26.
Synthesis of polymers
(1a) Semitelechelic CN-p(MPC)-DFA polymer was prepared in three synthetic steps as follows: First, a mixture of CTA-TT (15.8 mg, 41.7 µmol) and ACVA-(TT)2 (10 mg, 20,8 µmol) was dissolved in DMSO (0.4 mL) and added to a solution of MPC (500.0 mg, 1.7 mmol) in methanol (1.5 mL). The reaction mixture was thoroughly bubbled with argon and polymerized in a sealed glass ampoule at 70 °C for 16 h. After cooling to room temperature, the polymer was obtained by precipitation of the reaction mixture into acetone (40 mL) and purified by subsequent re-precipitation from methanol into acetone (40 mL). Centrifugation and vacuum drying of the precipitate yielded 400 mg (77%) of DTB-p(MPC)-TT polymer precursor as a pink powder. The number-average molecular weight (Mn) and the dispersity (ÐSEC) of the polymer were 18.1 kg mol−1 and 1.01, respectively.
Next, a mixture of DTB-p(MPC)-TT (150.0 mg, 7.6 µmol DTB groups) and AIBN (25.0 mg, 152 µmol) was dissolved in methanol (1.5 mL) and allowed to react at 80 °C for 2 h. The reaction mixture was cooled down to room temperature and the polymer was obtained by precipitation into acetone (40 mL). After purification on a column filled with Sephadex LH-20 in methanol, the polymer was isolated by precipitation into acetone and dried in vacuo to give 100 mg (67%) of CN-p(MPC)-TT polymer precursor as a yellow amorphous powder. The Mn and Ð of the polymer were 19.2 kg mol−1 and 1.02, respectively. The content of TT end groups of the polymer was 51.0 µmol g−1, corresponding to ~ 0.98 TT groups per polymer chain.
Finally, a mixture of DFA (14.2 mg, 25.3 µmol) and TEA (7.0 µL, 50.3 µmol) was dissolved in DMSO (0.330 mL) under heat, added to a solution of CN-p(MPC)-TT (100 mg, 5.1 µmol TT) in methanol (0.660 mL), and the mixture was allowed to react for 48 h at room temperature. After purification on a column filled with Sephadex LH-20 in methanol, the polymer was isolated by precipitation into acetone and dried in vacuo to give 49 mg (49%) of the resulting CN-p(MPC)-DFA polymer as a white powder. The Mn and ÐSEC of the polymer were 19.7 kg mol−1 and 1.02, respectively. The content of DFA end groups of the polymer was 49.7 µmol g−1, corresponding to ~ 0.98 DFA groups per polymer chain.
(1b) Multivalent CN-p(MPC-co-Ma-βAla-DFA)-CN polymer was prepared in three synthetic steps as follows: First, a mixture of CPB (8.0 mg, 36.6 µmol) and V-70 (5.6 mg, 18.3 µmol) was dissolved in DMSO (0.3 mL) and added to a solution of MPC (400 mg, 1.4 mmol) and Ma-βAla-TT (39.0 mg, 0.2 mmol) in methanol (1.2 mL). The reaction mixture was thoroughly bubbled with argon and polymerized in a sealed glass ampoule at 40 °C for 48 h. After cooling to room temperature, the polymer was obtained by precipitation of the reaction mixture into acetone–diethyl ether (1:1, 40 mL) and purified by subsequent re-precipitation from methanol into the same precipitant. Centrifugation and vacuum drying of the precipitate yielded 350 mg (78%) of DTB-p(MPC-co-Ma-βAla-TT)-CN polymer precursor as an orange powder. The Mn and the ÐSEC of the polymer were 12.7 kg mol−1 and 1.10, respectively.
Next, a mixture of DTB-p(MPC-co-Ma-βAla-TT)-CN (350.0 mg, 26.9 µmol DTB groups) and AIBN (88.2 mg, 537.2 µmol) was dissolved in methanol (3.5 mL) and allowed to react at 80 °C for 2 h. The reaction mixture was cooled down to room temperature and the polymer was obtained by precipitation into acetone–diethyl ether (1:1, 40 mL). After purification on a column filled with Sephadex LH-20 in methanol, the polymer was isolated by precipitation into acetone–diethyl ether (1:1) and dried in vacuo to give 262 mg (75%) of CN-p(MPC-co-Ma-βAla-TT)-CN polymer precursor as a yellow amorphous powder. The Mn and ÐSEC of the polymer were 13.0 kg mol−1 and 1.10, respectively. The content of TT groups along the polymer chain was 7.7 mol%.
Finally, a mixture of DFA (45.2 mg, 69.8 μmol) and TEA (9.5 µL, 69.8 μmol) was dissolved in dimethylacetamide (3.4 mL) under heat, added to a solution of CN-p(MPC-co-Ma-βAla-TT)-CN (262.0 mg, 69.8 mmol TT) in methanol (6.8 mL), and the mixture was allowed to react for 48 h at room temperature. After purification on a column filled with Sephadex LH-20 in methanol, the polymer was isolated by precipitation into acetone–diethyl ether (1:1, 40 mL) and dried in vacuo to give 165 mg (63%) of the resulting CN-p(MPC-co-Ma-βAla-DFA)-CN polymer as a white powder. The Mn and ÐSEC of the polymer were 12.3 kg mol−1 and 1.10, respectively. The content of DFA groups along the polymer chain was 4.9 mol%.
Complexation of polymers with Fe3+ ions
FeCl3∙6H2O (5 eq.) was added to a solution of DFA groups-containing polymer 1a or 1b (1 eq.) in methanol (10% w/v) and allowed to react for 1 h at room temperature. After purification on a column filled with Sephadex LH-20 in methanol, the polymer/Fe3+ complexes were isolated by precipitation into acetone–diethyl ether (1:1) and dried in vacuo to form a brownish red powder product. The Mn and ÐSEC were 20.8 kg mol−1 and 1.10 for the complexes 1a/Fe3+, and 14.8 kg mol−1 and 1.03 for the complexes 1b/Fe3+.
Complexation of polymers with γ-Fe2O3 nanoparticles
A sterile stock solution of neat γ-Fe2O3 nanoparticles in water for injections was drop-wise added to an aqueous solution of DFA groups-containing polymers 1a or 1b (4:1 w/w) under sonication with a 1-mm2 tipped ultrasonic horn (Bandelin UW 3200, Germany). The final concentration of polymer/γ-Fe2O3 complexes was 0.5 mg mL−1 and the total volume of the sample was 2.0 mL. All reactions were performed in a sterile box, with the prepared polymer/γ-Fe2O3 complexes stored in septum-sealed vials to prevent bacterial contamination. The DhDLS and DnTEM were 102.3 nm and 11.8 nm for the complexes 1a/γ-Fe2O3, and 81.8 nm and 11.3 nm for the complexes 1b/γ-Fe2O3.
UV–VIS spectrophotometry
Spectrophotometric analysis of functionalized polymers was carried out in quartz glass cuvettes on a Specord Plus UV–VIS spectrophotometer (Analytik Jena, Germany). The molar content of DTB and TT groups in the polymers was determined at 302 and 305 nm in methanol using the molar absorption coefficient of 12 100 and 10 300 L mol−1 cm−1, respectively26. DFA group content was measured indirectly at 432 nm upon addition of FeCl3∙6H2O (2 eq. compared to the theoretical content of DFA groups) to the aqueous solution of polymers using a molar absorption coefficient of 2 585 L mol−1 cm−127. The resulting values of functional group contents were obtained by arithmetic average of 3 independent measurements.
Size-exclusion chromatography
The number- and weight-averages of molecular weights (Mn and Mw) and dispersities (ÐSEC, ÐSEC = Mw/Mn) of the polymers were determined by size-exclusion chromatography (SEC) on a HPLC system (Shimadzu, Japan) equipped with an internal UV–VIS diode array detector (SPD-M20A), an external differential refractometer (Optilab T-rEX), and a multi-angle light-scattering detector (DAWN HELEOS II, both Wyatt Technology, USA). TSKgel SuperAW3000 and SuperAW4000 columns (Tosoh Bioscience, USA) in series were used to analyze samples in the mobile phase of 80% methanol and 20% sodium acetate buffer (0.3 M, pH 6.5) at a flow rate of 0.6 mL·min−1. The dn/dc values of 0.125 mL g−1 were used to calculate the molecular weights of the MPC-based polymers18. The resulting molecular weight values were obtained by arithmetic average of 2 independent analyses.
Dynamic light scattering
The z-averages of the hydrodynamic diameters (DhDLS) and the polydispersity indexes (PDI) of neat γ-Fe2O3 nanoparticles and polymer/γ-Fe2O3 complexes were determined by the DLS technique at a scattering angle of 173° using a Nano-ZS instrument (Malvern Instruments, UK) equipped with a 4 mW, 633 nm laser. Samples of polymer/γ-Fe2O3 complexes with a weight concentration of 0.5 mg mL−1 were measured in water and in PBS buffer (0.15 M, pH 7.4) after tenfold dilution at 25 °C. For the evaluation of the DLS data, the DTS (Nano) program was used. The resulting DhDLS values were arithmetic means of at least 10 independent measurements.
Transmission electron microscopy
The microphotographs of neat γ-Fe2O3 nanoparticles and polymer/γ-Fe2O3 complexes were measured using a transmission electron microscope (FEI-TEM, Tecnai, G2 Spirit, Oregon, USA). Ultrapure-water-diluted dispersions were directly dropped on a copper grid with a carbon film and dried at room temperature. Morphological parameters, such as the number- and weight-average of particle diameters (DnTEM and DwTEM) and dispersity (ÐTEM, ÐTEM = DwTEM/DnTEM), were calculated from the obtained TEM microphotographs using the following equations: \({\text{D}}_{\text{n}}\text{=}\boldsymbol{ }\sum {\text{n}}_{\text{i}}{{\text{D}}}_{\text{i}}\text{/}\sum {\text{n}}_{\text{i}}\), \({\text{D}}_{\text{w}}= \text{ } \sum {\text{n}}_{\text{i}}{{\text{D}}}_{\text{i}}^{4}\text{/}\sum {\text{n}}_{\text{i}}{{\text{D}}}_{\text{i}}^{3}\), where ni is the number of particles and Di is the particle diameter. Particle size analysis was performed using ImageJ software, measuring at least 300 objects in each sample. Particle diameters were measured in manual mode from multiple microphotographs of different regions in the sampling grid.
Magnetic resonance spectroscopy and imaging
MR characterization of the polymer/iron complexes was performed in aqueous solutions using a 4.7 T scanner (Bruker BioSpin, Ettlingen, Germany) equipped with a homemade dual 1H/31P radiofrequency (RF) surface coil as previously described18. The measurements of the samples were performed at a normalized molar concentration of phosphorus (cnP) of 100 mmol L−1 corresponding to a weight concentration (cwpol) of 29.5 mg mL−1 for polymer 1a and 32.0 mg mL−1 for polymer 1b. The molar concentrations of iron (cnFe) in the polymer/iron complexes were 0.03 mmol L−1 for the 1a/Fe3+ complex, 0.084 mmol L−1 for the 1b/Fe3+ complex and 6.26 mmol L−1 for both neat γ-Fe2O3 nanoparticles and 1a/γ-Fe2O3 and 1b/γ-Fe2O3 complexes. 1H MR images were obtained using standard 2D rapid acquisition and a relaxation enhancement (RARE) multi-spin echo MR sequence based on the following parameters: spatial resolution = 312 × 312 µm2, slice thickness = 2.5 mm, scan time (ST) = 1 min 16 s, T2w: repetition time (TR) = 3300 ms, echo time (TE) = 36 ms, turbo factor = 4 and T1w: TR = 294.8 ms, TE = 16.7 ms, turbo factor = 2. 31P MR images were obtained using a chemical shift imaging (CSI) sequence (TR = 500 ms, ST = 15 min, field of view FOV = 36 mm; resolution 2.25 × 2.25 × 5.8 mm3). 31P MRI SNR was calculated using SNR = 0.655 · S · σ−1, where S is signal intensity in the region of interest (ROI), σ is the standard deviation of background noise, and constant 0.655 reflects the Rician distribution of background noise in a magnitude MR image.
Relaxometry
Relaxometric experiments of aqueous solutions of polymer/iron complexes were performed on a 1.5 T Minispec 60 MHz relaxometer (Bruker Biospin, Germany) at 37 °C. The measurements of the samples were performed at a normalized molar concentration of phosphorus (cnP) of 100 mmol L−1 corresponding to a weight concentration (cwpol) of 29.5 mg mL−1 for polymer 1a and 32.0 mg mL−1 for polymer 1b. The molar concentrations of iron (cnFe) in the polymer/iron complexes were 0.03 mmol L−1 for the 1a/Fe3+ complex, 0.084 mmol L−1 for the 1b/Fe3+ complex and 6.26 mmol L−1 for both neat γ-Fe2O3 nanoparticles and 1a/γ-Fe2O3 and 1b/γ-Fe2O3 complexes. 1H T1 relaxation time was measured using an inversion recovery sequence (TR = 0.01–10 000 ms, recycle delay = 2 s, number of scans = 4, TE = 0.05 ms, points for fitting = 20). 1H T2 relaxation time was measured using the Carr-Purcell-Meiboom-Gill sequence (TR = 10 000 ms, recycle delay = 2 s, number of scans = 8, TE = 0.05 ms, points for fitting = 30 000). Relaxivities r1 and r2 were calculated using the least-squares curve fitting of R1 = 1/T1 and R2 = 1/T2 relaxation rates [s−1] versus iron concentration [mmol L−1]. 31P T1 relaxation time was measured using 10 spectroscopic single-pulse sequences with varying repetition times (TR = 200–4000 ms) and the 31P T2 relaxation time was measured using 10 spectroscopic Carr-Purcell-Meiboom-Gill (CPMG) sequences with varying echo times (TR = 5000 ms, TE = 20–1600 ms). Data were quantified by plotting amplitudes and fitting the appropriate curve (S ≈ S0 · (1—e−t/T1) for T1; S ≈ S0 · e−t/T2 for T2), where S is signal intensity (S0 signal intensity at equilibrium) and t is time: TR for T1 and TE for T2, respectively.
Results and discussion
Synthesis of phospho-polymer/Fe3+ ions complexes
Among many different types of hydrophilic phosphorus-containing polymers, we decided to use a polymer based on 2-methacryloyloxyethyl phosphorylcholine (MPC). This poly-zwitterion is characterized not only by high solubility in aqueous solutions, resistance to non-specific protein adsorption and cell adhesion, and ability to effectively penetrate cell membranes, but also by excellent 31P-MR properties, making it an optimal material for the purposes of this study17,24. In addition, MPC can be copolymerized by controlled polymerization techniques with functionalized monomers in the presence of functionalized chain transfer agents (CTA) allowing incorporation of reactive groups along or at the ends of the pMPC chains for subsequent post-polymerization modification with metal-chelating ligands23.
In this study, we used reversible addition−fragmentation chain-transfer (RAFT) polymerization of MPC in the presence of thiazolidine-2-thione (TT) group-functionalized CTA to produce a semitelechelic homopolymer with one terminal amino-reactive TT group; and RAFT copolymerization of MPC with a TT group-containing monomer (Ma-βAla-TT) to obtain a multivalent copolymer with multiple TT groups statistically distributed along the polymer backbone. The polymers were characterized by low dispersity (Ð ≤ 1.1) and molecular weight in the range of ~ 12–20 kg mol−1, which ensures both increased accumulation in tissues with more porous vasculature (e.g. solid tumors) via the enhanced permeability and retention effect and excretion from the body via the reticuloendothelial pathway after fulfilling their function28. The coordination of the polymers with iron was mediated by a compound called deferoxamine (DFA). DFA is a small natural siderophore secreted by Streptomyces pilosus, whose primary function is to transport complex Fe3+ ions from the extracellular region across cell membranes29. Due to its strong affinity for iron (the formation constant (logβ) of the dominant species of the Fe3+/DFA complex in 0.1 mol L−1 KCl solution at 25 °C is 41.430), DFA is clinically used to treat acute iron poisoning or haemochromatosis by binding free iron in the bloodstream and increasing its urinary excretion31. DFA was attached to the polymers by reacting its terminal amino group with the TT groups of the polymers to form an amide bond. As a source of iron, we used low-molecular-weight paramagnetic Fe3+ ions, which are considered one of the less toxic alternatives to clinically used Gd3+ ions12. The resulting complexes of MPC polymers with Fe3+ ions were obtained by incubating the DFA group-containing polymers in an aqueous FeCl3 solution. Neither the attachment of DFA groups nor the complexation with Fe3+ ions affected the solubility and molecular weights distribution of the polymers. The characteristics of the polymer/Fe3+ complexes are summarized in Table 1; their chemical structures are depicted in Figure 1.

Schematic representation and chemical structures of complexes of semitelechelic polymer 1a (left) and multivalent polymer 1b (right) with (A) Fe3+ ions and with (B) γ-Fe2O3 nanoparticles.
Synthesis of phospho-polymer/γ-Fe2O3 nanoparticle complexes
Most clinically approved iron-based contrast agents for MRI, including Feridex®, Resovist®, Lumirem®, etc., consist of various forms of superparamagnetic iron oxide nanoparticles that are characterized by high relaxivity, long-term circulation, or preferential passive accumulation in tissues with leaky vasculature, such as tumors32. However, the preparation of colloidally stable nanoparticles with surface properties allowing their further modification (e.g. by a phospho-polymer) is challenging. This is because nanoparticles based on bare iron oxides tend to agglomerate, and nanoparticles prepared in the presence of surfactants (e.g. oleic acid, oleylamine, dioctylamine, etc.) or hydrophilic polymers (e.g. dextran, polyethylene glycol, polylysine, etc.) to increase their stability in aqueous solutions have a protective layer around their surfaces that limits their subsequent functionalization9,33,34.
To avoid these difficulties, we used a special co-precipitation technique of Fe3+ and Fe2+ salts to produce maghemite (γ-Fe2O3) nanoparticles dielectrically stabilized with sodium citrate, which are characterized not only by reproducible preparation, high colloidal stability and easy access of functionalized (macro)molecules to their surface, but also by lower activity towards reactive oxygen species (ROS) and higher saturation magnetization values compared to commonly used magnetite (Fe3O4) nanoparticles35. DLS and TEM analyzes showed that the prepared nanoparticles were well-defined spheroids with DhDLS = 60.9 nm (PDI = 0.106) and DnTEM = 10.3 nm (ÐTEM = 1.113) (see Figure 2). In addition, elemental analysis (data not shown) proved that the γ-Fe2O3 nanoparticles contained a negligible amount of the citrate incorporated in their structure, which is an important precondition for subsequent surface modification.

Size characterization of γ-Fe2O3 nanoparticles complexed with semitelechelic polymer 1a and multivalent polymer 1b: (A) DLS analysis of aqueous particle solution plotted as hydrodynamic diameter (Dh) distribution by mean intensity; (B) TEM micrographs of dried nanoparticles.
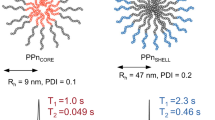
As in the case of polymer/Fe3+ ion complexes, the attachment of MPC-based polymers to the γ-Fe2O3 nanoparticle core was mediated through DFA moieties present either at the end (1a) or along (1b) the polymer chains. Based on the results presented in our previous study27, a γ-Fe2O3 to polymer ratio of 4:1 (w/w) was used to ensure efficient coating of the nanoparticle surface, with no free polymer detected in the solution, as documented by TEM (see Figure 2). More effective coating in terms of nanoparticles size occurred when the semitelechelic polymer 1a was used, where an increase in DhDLS of the nanoparticles by more than 40 nm was observed, which was approximately twice as much as in the case of the multivalent polymer 1b. Importantly, the hydrodynamic diameters of the nanoparticles are in range of previously clinically approved contrast agents based on superparamagnetic iron oxide (e.g. 62 nm and 150 nm)36. Also, the scattered light intensity (ILSDLS), proportional to the number (molecular weight) of bound polymer chains, increased by ~72% after coating the nanoparticles with polymer 1a, while it only increased by ~12% in the case of polymer 1b, indicating that polymer 1a covered the surface of the nanoparticles more densely (see Table 2 and Figure 2). We assume that this is due to the conformation of the polymer chains in solution; while in the case of a semitelechelic polymer its chains stick out from the surface of the nanoparticles to which they are densely attached via a single point, the chains of a copolymer with multiple binding sites are tightly attached to the nanoparticle surface via cooperative interactions. A schematic representation of the polymer/γ-Fe2O3 nanoparticle complexes is shown in Figure 1.
In order to most reliably describe the changes in size and morphology of γ-Fe2O3 nanoparticles coordinated to polymers without the influence of time and environmental, we monitored their properties immediately after complexation in pure water. However, for the purposes of the considered application, it was also interesting to see whether the nanoparticles would exhibit the same characteristics over time and under conditions simulating the physiological environment. Therefore, we performed DLS analysis of neat γ-Fe2O3 nanoparticles and polymer/γ-Fe2O3 nanoparticle complexes approximately 3 years after their preparation in both pure water and in PBS (Table 1). All types of nanoparticles prepared in pure water showed excellent colloidal stability even after three years of storage, documented by small changes in hydrodynamic sizes and low polydispersity indexes. In the case of the complex of semitelechelic polymer with γ-Fe2O3 nanoparticles (1a/γ-Fe2O3), however, there was an almost 2.5-fold decrease in the intensity of scattered light (ILSDLS) compared to the freshly prepared complex, which could be explain by the fact that a part of the polymer chains, linked to the nanoparticles through a single point, was detached from the surface and released into the solution over time. This theory is supported by only about a 10% decrease in ILSDLS for complexes with multivalent polymer (1b/γ-Fe2O3), which are attached to the nanoparticle surface via multiple points through a cooperative interaction and their cleavage during storage is therefore less likely. This corresponds to an increase in the size of these complexes over time, where the bonding between the DFA groups of the polymer and the particle surface is probably (at least) partially broken, causing a conformational change of the polymer chains, which then protrude from the particle surface similarly to the chains of the semitelechelic polymer. Tenfold dilution of aqueous solutions of nanoparticles with phosphate buffer (0.15 M PBS, pH 7.4) led to aggregation and subsequent sedimentation of neat nanoparticles, but their complexes with polymers still formed homogeneous dispersions with size < 100 nm and low PDI, indicating their high colloidal stability in conditions mimicking physiological environment.
MR properties of phospho-polymer/iron complexes
When examining both types of polymer (1a/Fe3+and 1b/Fe3+) and colloidal (1a/γ-Fe2O3 and 1b/γ-Fe2O3) probes, a significant effect of iron manifested by a shortening of 1H relaxation times was observed. The resulting 1H relaxivity ratio (r2/r1) for probes coordinated with small paramagnetic Fe3+ ions (1a/Fe3+and 1b/Fe3+) was in both cases approximately 1.7, indicating a slightly pronounced T2 effect, while in the case of probes coordinated with γ-Fe2O3 nanoparticles (1a/γ-Fe2O3 and 1b/γ- Fe2O3), the ratio was 15.9 and 30.9, respectively, corresponding to a much stronger T2 effect compared to T1. In general, MR contrast agents that increase signal by decreasing T1 are referred to as positive (mostly paramagnetic materials), whereas if their action leads to a decrease in signal by decreasing T2, they are referred to as negative (mostly superparamagnetic materials)37. Therefore, the probes used in our study can be categorized as “negative” contrast agents38. However, it should be noted that the very strong influence of these contrast agents on T2 relaxation times led to difficulties in obtaining the signal needed for accurate quantification. The main obstacle was the minimum echo time (0.04) that could be set on the relaxometer used. The average T2 relaxation times of probes 1a/γ-Fe2O3 and 1b/γ-Fe2O3 at 10–100% concentrations were in the range of 0.3 ± 0.001–0.05 ± 0.006 ms and 0.3 ± 0.001–0.05 ± 0.001 ms, respectively, so at the limit of measurability. Another important indicator of the effectiveness of contrast agents are r1 and r2 relaxivities. These values are the main physicochemical parameters that are considered for the development of an efficient MRI contrast agent and are dependent on the size, saturation magnetization, magnetic fields and chemical structure of the molecule as well as on the accessibility of water molecules to the magnetic center. Very high r2/r1 ratios are characteristic for SPION-type superparamagnetic colloids and typically range from 6 to 15 (10-40 MHz), making lower imaging fields more suitable for exploiting the T1 effect39. The r2/r1 values of both colloidal probes used in our study were slightly higher than those of structurally similar Resovist®, a clinically approved iron oxide-based contrast agent for MRI (see Table 3). This demonstrates their high efficiency, which allows them to be used at lower concentrations, thus reducing their potential adverse effects in vivo.
In 31P MR relaxometry study, we were able to measure T2 relaxation times only for 1a/Fe3+ and 1b/Fe3+ complexes, while T1 measurements could not be performed due to the inability to achieve a sufficiently short Repetition Time (TR) for capturing the relaxation curve using the saturation recovery method. This resulted in saturation of MR signal even at the shortest possible TR. Based on these results, we can only conclude that the 31P T1 is significantly shorter than 200 ms. The same was true for phantoms containing polymer/γ-Fe2O3 complexes, whose relaxation times were also too short to be reliably assessed (see Table 3). To summarize the results from 31P MR relaxometry, probes based on phospho-polymers coordinated with iron have significantly shorter relaxation times compared to phospho-polymers alone, whose T1 is typically in the range of 1000–2100 ms and T2 in the range of 30–200 ms17,18.
The conclusions resulting from the relaxometric measurement were also supported by the 1H MR imaging data, where a complete signal loss was observed in phantoms containing aqueous solutions of both phospho-polymers coordinated with small paramagnetic Fe3+ ions (1a/Fe3+and 1b/Fe3+) on T2-weighted images (Figure 3). In 1H MR spectroscopy, a more sensitive method, signal detection was achieved in phantoms containing both polymeric 1a/Fe3+ and 1b/Fe3+ complexes, although a lower signal-to-noise ratio and a broader peak were observed for the 1b/Fe3+ probe with higher content of iron (Figure 4). In the case of phantoms containing polymer/γ-Fe2O3 nanoparticle complexes (1a/γ-Fe2O3 and 1b/γ- Fe2O3), the relaxation times were so short that both imaging and spectroscopic measurements were not feasible for technical reasons, as insufficient signal was obtained for proper measurement setup. This is consistent with the very short T1 and, particularly, the ultrashort T2 relaxation times (~ 0.1 ms) obtained for those complexes by 1H MR relaxometry.

1H MRI (on the left), 31P MR CSI (in the middle) and overlapped 1H MRI/31P MR CSI measurements (on the right) of vials with water on the sides (controls) and a vial with a solution of (A) 1a/Fe3+ and (B) 1b/Fe3+ complexes between them.

31P MRS measurements of aqueous solutions of (A) 1a/Fe3+ and (B) 1b/Fe3+ complexes at different scan lengths. The inserted tables show the numerical values of the measured data.
Although a complete loss of proton signal was observed for both polymeric complexes (1a/Fe3+ and 1b/Fe3+) in 1H MR experiments, the phosphorus signal in 31P MR measurement still remained detectable. However, the complex of semitelechelic polymer with one iron atom per chain (1a/Fe3+) provided a high signal well detectable by both spectroscopy and imaging, while the complex of multivalent polymer with several iron atoms along the chain (1b/Fe3+) was detectable only by the more sensitive spectroscopic technique (Figure 4). Expectably, the iron concentration in both γ-Fe2O3 nanoparticle complexes was so high that no signal could be obtained from either MRS or MRI measurements. It suggests that the most suitable structure and composition of all studied probes has a water-soluble complex of semitelechelic polymer (1a/Fe3+), which has a sufficient concentration of phosphorus and iron to enable efficient dual 1H and 31P MR imaging. At the same time, the concentration of iron in the probe is so low that the toxic effect of the complex is very unlikely. Moreover, the easy copolymerizability of pMPC with functionalized methacrylate/mathacrylamide-based monomers (e.g. 3-(3-methacrylamidopropanoyl)thiazolidine-2-thione, N-(3-aminopropyl)methacrylamide, N-(3,4-dihydroxyphenethyl)methacrylamide, etc.) allows the post-polymerization introduction of various bioactive compounds (e.g. cancerostatic drugs, immunostimulatory molecules, nucleic acids, etc.) into its structure, extending the diagnostic potential of this probe to therapeutic applications. In addition to the diagnostic use of the probe in pharmacokinetic studies or in the monitoring of transplanted cells, it can be used, for example, in the treatment of tumor diseases after incorporation of a suitable low-molecular-weight cancerostatics.
Conclusions
In conclusion, our study focused on the synthesis and characterization of phospho-polymer/iron complexes as sensitive probes/contrast agents for dual 1H/31P MR imaging and spectroscopy. Specifically, we used RAFT polymerization technique to produce well-defined biocompatible (co)polymers based on 2-methacryloyloxyethyl phosphorylcholine (MPC) coordinated with small paramagnetic Fe3+ ions or superparamagnetic maghemite (γ-Fe2O3) nanoparticles via deferoxamine group linked to the ends or along the polymer chains. While water-soluble polymer probes with Fe3+ ions were used in this comparative study as a safer alternative to commonly used compounds containing Gd3+ ions, polymer-colloidal probes with iron in the form of maghemite served as a more effective and stable version of clinically approved nanoparticlulate iron oxide compounds. We have clearly demonstrated that the presence of iron—whether in the form of a soluble ion or a colloidal oxide—significantly affected the contrast of the imaged area in 1H MRI experiments. Particularly in the case of maghemite-based probes, the r2/r1 values were slightly higher than those of the structurally similar clinically approved T2 contrast agent Resovist®, indicating their high efficacy, which allows them to be used at lower concentrations and thus reduce their potential side effects. Coordinated iron also had a significant impact on the relaxation times of phosphorus, while we showed that the structure of the phospho-polymer and the form of the iron play an important role. We showed that the polymer with multiple Fe3+ ions along the chain as well as the polymers coordinated to maghemite nanoparticles had excessively high iron concentration, leading to quenching of both 1H and 31P MR signals. Conversely, the most suitable composition exhibited a polymer with only one Fe3+ ion per chain, which was reliably detected by both 1H and 31P MR imaging and spectroscopy. We believe that this dual 1H/31P MR probe could be highly valuable in various clinical and research applications where it could be used to monitor probe biodistribution while visualizing body anatomy. It could play a key role, for example, in the optimization of transplant procedures or in the monitoring of accumulation in solid tumors.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Jirák, D. & Vítek, F. Basics of Medical Physics (Karolinum Press, 2017).
Wahsner, J., Gale, E. M., Rodriguez-Rodriguez, A. & Caravan, P. Chemistry of MRI contrast agents: Current challenges and new frontiers. Chem. Rev. 119, 957–1057. https://doi.org/10.1021/acs.chemrev.8b00363 (2019).
Fatima, A. et al. Recent advances in gadolinium based contrast agents for bioimaging applications. Nanomaterials (Basel) https://doi.org/10.3390/nano11092449 (2021).
Rogosnitzky, M. & Branch, S. Gadolinium-based contrast agent toxicity: A review of known and proposed mechanisms. Biometals 29, 365–376. https://doi.org/10.1007/s10534-016-9931-7 (2016).
Dulinska-Litewka, J. et al. Superparamagnetic iron oxide nanoparticles-current and prospective medical applications. Materials (Basel) https://doi.org/10.3390/ma12040617 (2019).
Saudek, F. et al. Magnetic resonance imaging of pancreatic islets transplanted into the liver in humans. Transplantation 90, 1602–1606. https://doi.org/10.1097/TP.0b013e3181ffba5e (2010).
Patil, S., Jirák, D., Saudek, F., Hájek, M. & Scheffler, K. Positive contrast visualization of SPIO-labeled pancreatic islets using echo-dephased steady-state free precession. Eur. Radiol. 21, 214–220. https://doi.org/10.1007/s00330-010-1909-1 (2011).
Deligianni, X. et al. In vivo visualization of cells labeled with superparamagnetic iron oxides by a sub-millisecond gradient echo sequence. Magn. Reson. Mater. Phys. 27, 329–337. https://doi.org/10.1007/s10334-013-0422-3 (2014).
Babic, M. et al. Poly(L-lysine)-modified iron oxide nanoparticles for stem cell labeling. Bioconjug. Chem. 19, 740–750. https://doi.org/10.1021/bc700410z (2008).
Wang, R. et al. A class of water-soluble Fe(iii) coordination complexes as T1-weighted MRI contrast agents. J. Mater. Chem. B 9, 1787–1791. https://doi.org/10.1039/d0tb02716b (2021).
Palagi, L. et al. Fe(deferasirox): An Iron(III)-based magnetic resonance imaging T1 contrast agent endowed with remarkable molecular and functional characteristics. J. Am. Chem. Soc. 143, 14178–14188. https://doi.org/10.1021/jacs.1c04963 (2021).
Marasini, R., Rayamajhi, S., Moreno-Sanchez, A. & Aryal, S. Iron(iii) chelated paramagnetic polymeric nanoparticle formulation as a next-generation T1-weighted MRI contrast agent. RSC Adv. 11, 32216–32226. https://doi.org/10.1039/d1ra05544e (2021).
Hu, R. et al. X-nuclei imaging: Current state, technical challenges, and future directions. J. Magn. Reson. Imaging 51, 355–376. https://doi.org/10.1002/jmri.26780 (2020).
Liu, Y. C., Gu, Y. N. & Yu, X. Assessing tissue metabolism by phosphorous-31 magnetic resonance spectroscopy and imaging: A methodology review. Quant. Imaging Med. Surg. 7, 707–726. https://doi.org/10.21037/qims.2017.11.03 (2017).
Neeman, M., Rushkin, E., Kaye, A. M. & Degani, H. 31P-NMR studies of phosphate transfer rates in T47D human breast cancer cells. Biochim. Biophys. Acta 930, 179–192. https://doi.org/10.1016/0167-4889(87)90030-9 (1987).
Levine, S. R. et al. Human focal cerebral ischemia: Evaluation of brain pH and energy metabolism with P-31 NMR spectroscopy. Radiology 185, 537–544. https://doi.org/10.1148/radiology.185.2.1410369 (1992).
Kracikova, L. et al. Phosphorus-containing polymers as sensitive biocompatible probes for (31)P magnetic resonance. Molecules https://doi.org/10.3390/molecules28052334 (2023).
Kracikova, L. et al. Phosphorus-containing polymeric Zwitterion: A pioneering bioresponsive probe for (31) P-magnetic resonance imaging. Macromol. Biosci. 22, e2100523. https://doi.org/10.1002/mabi.202100523 (2022).
Ziolkowska, N., Vit, M., Laga, R. & Jirak, D. Iron-doped calcium phytate nanoparticles as a bio-responsive contrast agent in (1)H/(31)P magnetic resonance imaging. Sci. Rep. 12, 2118. https://doi.org/10.1038/s41598-022-06125-7 (2022).
Pechrova, Z., Lobaz, V., Konefał, M., Konefał, R. & Hruby, M. Colloidal probe based on iron(III)-doped calcium phytate nanoparticles for 31P NMR monitoring of bacterial siderophores. Colloid Interface Sci. Commun. 42, 100427. https://doi.org/10.1016/j.colcom.2021.100427 (2021).
Andrianov, A. K. Water-soluble polyphosphazenes for biomedical applications. J. Inorg. Organomet. Polym. Mater. 16, 397–406. https://doi.org/10.1007/s10904-006-9065-4 (2006).
Pelosi, C., Tinè, M. R. & Wurm, F. R. Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. https://doi.org/10.1016/j.eurpolymj.2020.110079 (2020).
Kojima, C. et al. Different antifouling effects of random and block copolymers comprising 2-methacryloyloxyethyl phosphorylcholine and dodecyl methacrylate. Eur. Polym. J. https://doi.org/10.1016/j.eurpolymj.2020.109932 (2020).
Goda, T., Ishihara, K. & Miyahara, Y. Critical update on 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer science. J. Appl. Polym. Sci. https://doi.org/10.1002/app.41766 (2015).
Subr, V. & Ulbrich, K. Synthesis and properties of new N-(2-hydroxypropyl)-methacrylamide copolymers containing thiazolidine-2-thione reactive groups. React. Funct. Polym. 66, 1525–1538. https://doi.org/10.1016/j.reactfunctpolym.2006.05.002 (2006).
Subr, V., Kostka, L., Strohalm, J., Etrych, T. & Ulbrich, K. Synthesis of Well-Defined Semitelechelic Poly[N-(2-hydroxypropyl)methacrylamide] Polymers with Functional Group at the α-End of the Polymer Chain by RAFT Polymerization. Macromolecules 46, 2100–2108. https://doi.org/10.1021/ma400042u (2013).
Kracíková, L. et al. Polymer-colloidal systems as MRI-detectable nanocarriers for peptide vaccine delivery. Eur. Polym. J. https://doi.org/10.1016/j.eurpolymj.2022.111704 (2022).
Maeda, H., Wu, J., Sawa, T., Matsumura, Y. & Hori, K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: A review. J. Control Release 65, 271–284. https://doi.org/10.1016/s0168-3659(99)00248-5 (2000).
Schupp, T., Waldmeier, U. & Divers, M. Biosynthesis of desferrioxamine-B in Streptomyces-Pilosus—Evidence for the involvement of lysine decarboxylase. Fems Microbiol. Lett. 42, 135–139 (1987).
Evers, A., Hancock, R. D., Martell, A. E. & Motekaitis, R. J. Metal ion recognition in ligands with negatively charged oxygen donor groups. Complexation of iron(III), gallium(III), indium(III), aluminum(III), and other highly charged metal ions. Inorg. Chem. 28, 2189–2195. https://doi.org/10.1021/ic00310a035 (1989).
Aaseth, J., Crisponi, G. & Andersen, O. Chelation Therapy in the Treatment of Metal Intoxication. Chelation Therapy in the Treatment of Metal Intoxication, 1–371. https://doi.org/10.1016/C2014-0-01302-0 (2016).
Vangijzegem, T. et al. Superparamagnetic iron oxide nanoparticles (SPION): From fundamentals to state-of-the-art innovative applications for cancer therapy. Pharmaceutics https://doi.org/10.3390/pharmaceutics15010236 (2023).
Babic, M. et al. Poly(N, N-dimethylacrylamide)-coated maghemite nanoparticles for stem cell labeling. Bioconjug. Chem. 20, 283–294. https://doi.org/10.1021/bc800373x (2009).
Laurent, S. et al. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physicochemical characterizations, and biological applications. Chem. Rev. 108, 2064–2110. https://doi.org/10.1021/cr068445e (2008).
Pollert, E. et al. Magnetic poly(glycidyl methacrylate) microspheres containing maghemite prepared by emulsion polymerization. J. Magn. Magn. Mater. 306, 241–247. https://doi.org/10.1016/j.jmmm.2006.03.069 (2006).
Reimer, P. & Balzer, T. Ferucarbotran (Resovist): A new clinically approved RES-specific contrast agent for contrast-enhanced MRI of the liver: Properties, clinical development, and applications. Eur. Radiol. 13, 1266–1276. https://doi.org/10.1007/s00330-002-1721-7 (2003).
Alzola-Aldamizetxebarria, S., Fernandez-Mendez, L., Padro, D., Ruiz-Cabello, J. & Ramos-Cabrer, P. A comprehensive introduction to magnetic resonance imaging relaxometry and contrast agents. ACS Omega 7, 36905–36917. https://doi.org/10.1021/acsomega.2c03549 (2022).
Huh, Y. M. et al. In vivo magnetic resonance detection of cancer by using multifunctional magnetic nanocrystals. J. Am. Chem. Soc. 127, 12387–12391. https://doi.org/10.1021/ja052337c (2005).
Gossuin, Y., Gillis, P., Hocq, A., Vuong, Q. L. & Roch, A. Magnetic resonance relaxation properties of superparamagnetic particles. Wires Nanomed. Nanobiotechnol. 1, 299–310. https://doi.org/10.1002/wnan.36 (2009).
Acknowledgements
This work was supported by the Ministry of Health of the Czech Republic (project no. NU20-08-00095), by the Ministry of Health CR-DRO (Institute for Clinical and Experimental Medicine IKEM, project no. IN00023001), by the Grant Agency of Charles University in Prague (project no. 271723), and by the National Institute for Research of Metabolic and Cardiovascular Diseases (Programme EXCELES, project no. LX22NPO5104)—Funded by the European Union—Next Generation EU.
Author information
Authors and Affiliations
Contributions
R.L. and D.J. conceived the experiments; L.K., L.A., M.B., M.Š., D.Č. and N.J.-Z. conducted the experiments, M.B., D.Č., N.J.-Z., R.L. and D.J. analysed the results; L.K. and R.L written original draft; M.B., N.J.-Z., and D.J. reviewed and edited draft; R.L. and D.J. supervised the students; R.L. administered the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kracíková, L., Androvič, L., Červený, D. et al. Iron-based compounds coordinated with phospho-polymers as biocompatible probes for dual 31P/1H magnetic resonance imaging and spectroscopy. Sci Rep 14, 3847 (2024). https://doi.org/10.1038/s41598-024-54158-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-54158-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
