Simple and Rapid HPLC Separation and Quantification of Flavonoid, Flavonolignans, and 2,3-Dehydroflavonolignans in Silymarin

 and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Material
2.2. Analytical Methods
2.2.1. Isocratic Method
2.2.2. Gradient Method
2.2.3. Gradient Method for 2,3-dehydroflavonolignans
2.2.4. Chiral Separation of Enantiomers of 2,3-dehydroflavonolignans
2.3. Validation of the Analytical Chromatographic Method
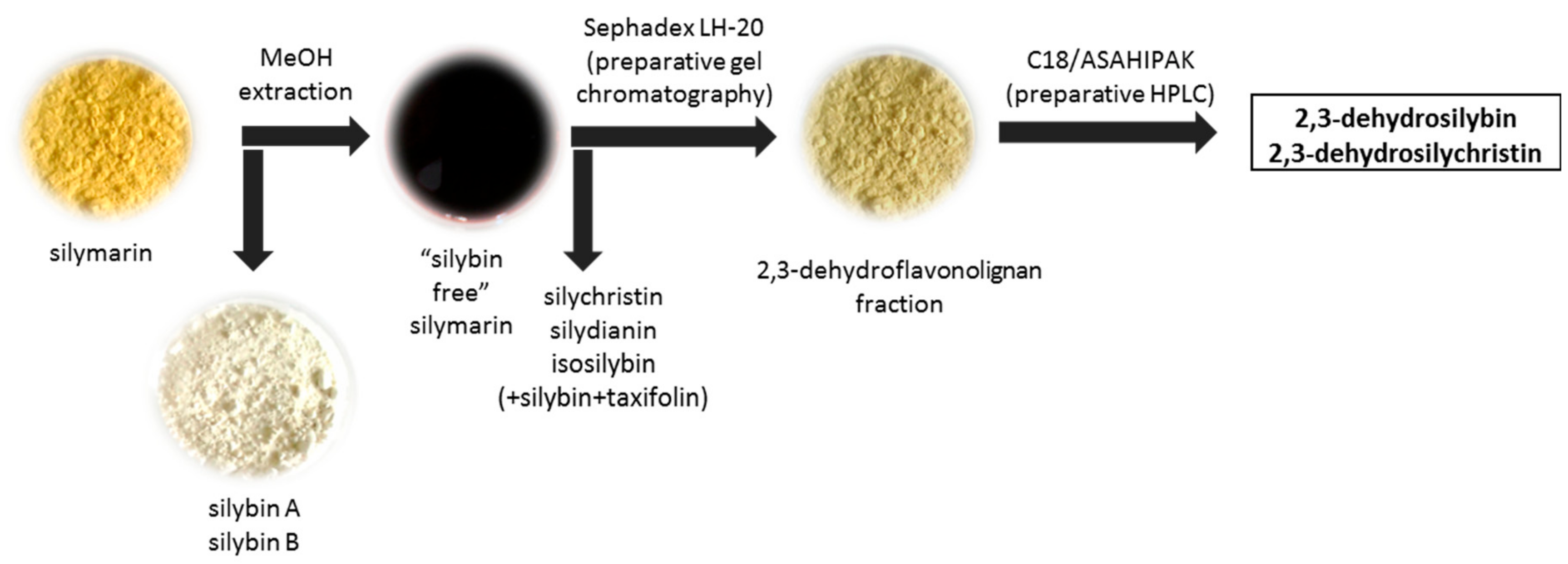
2.4. Semi-Preparative Chromatography
2.5. NMR Spectroscopy
3. Results and Discussion
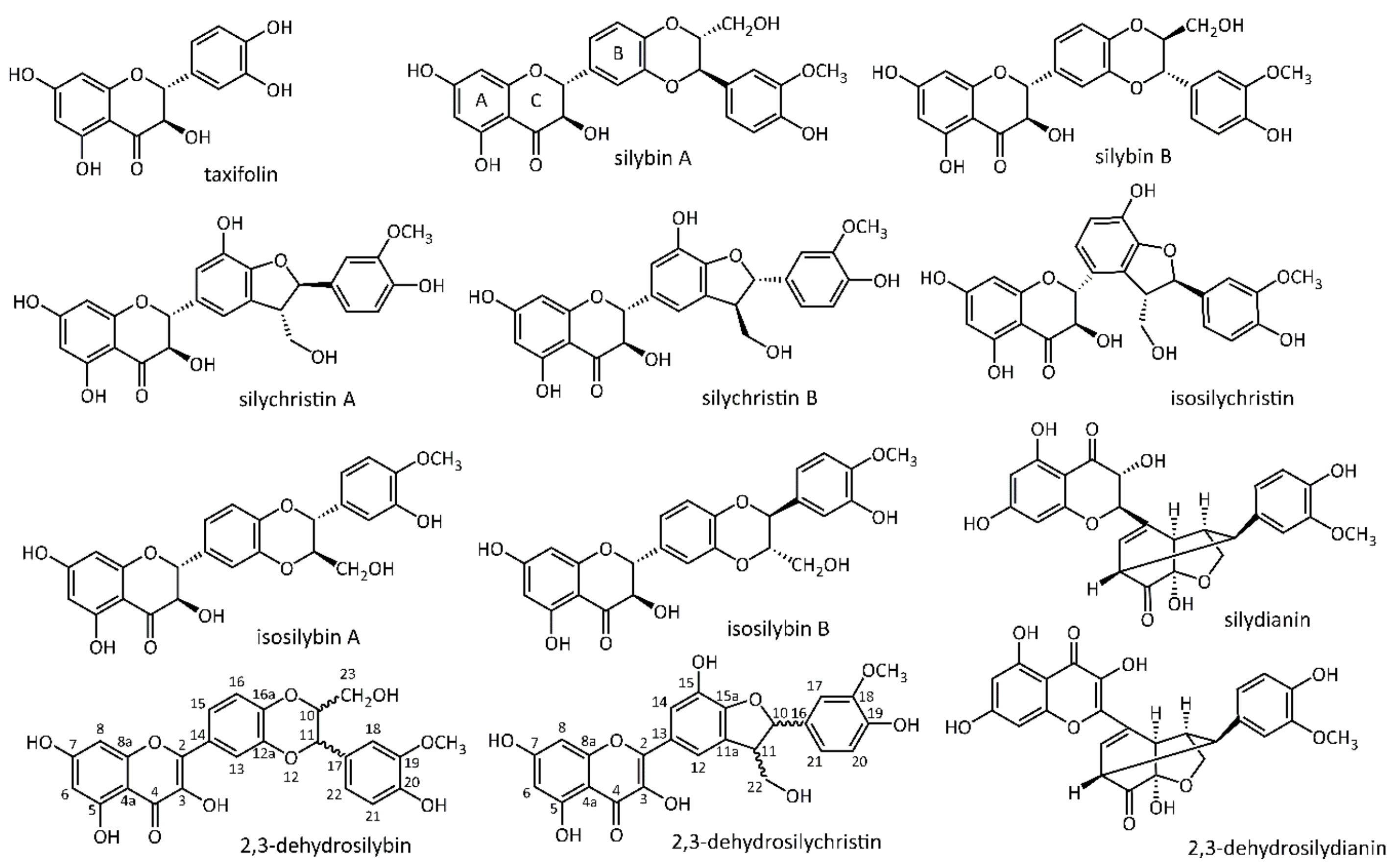
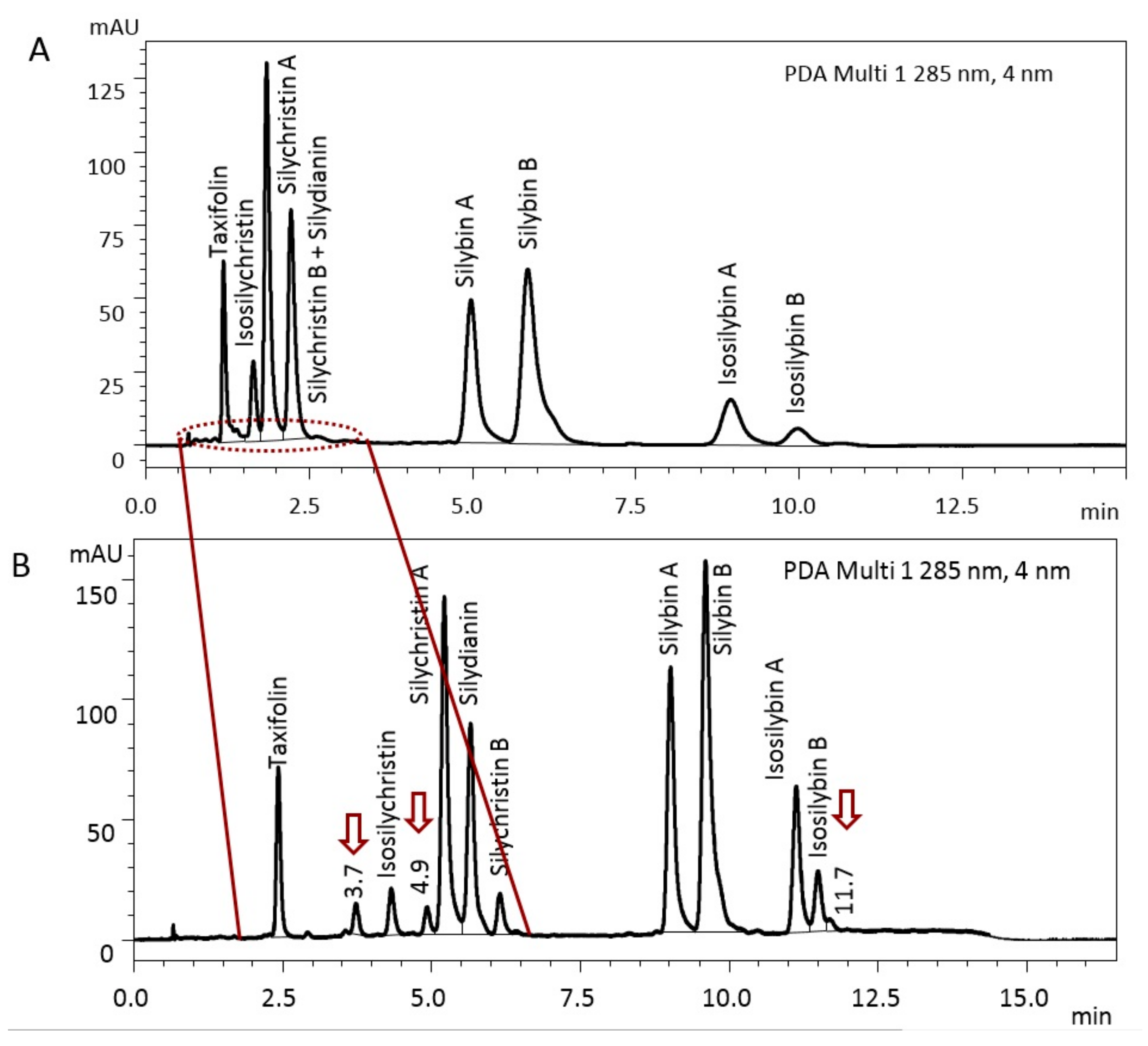
3.1. Optimization and Standardization of Analytical Techniques
3.2. Calibration and Validation of the Method
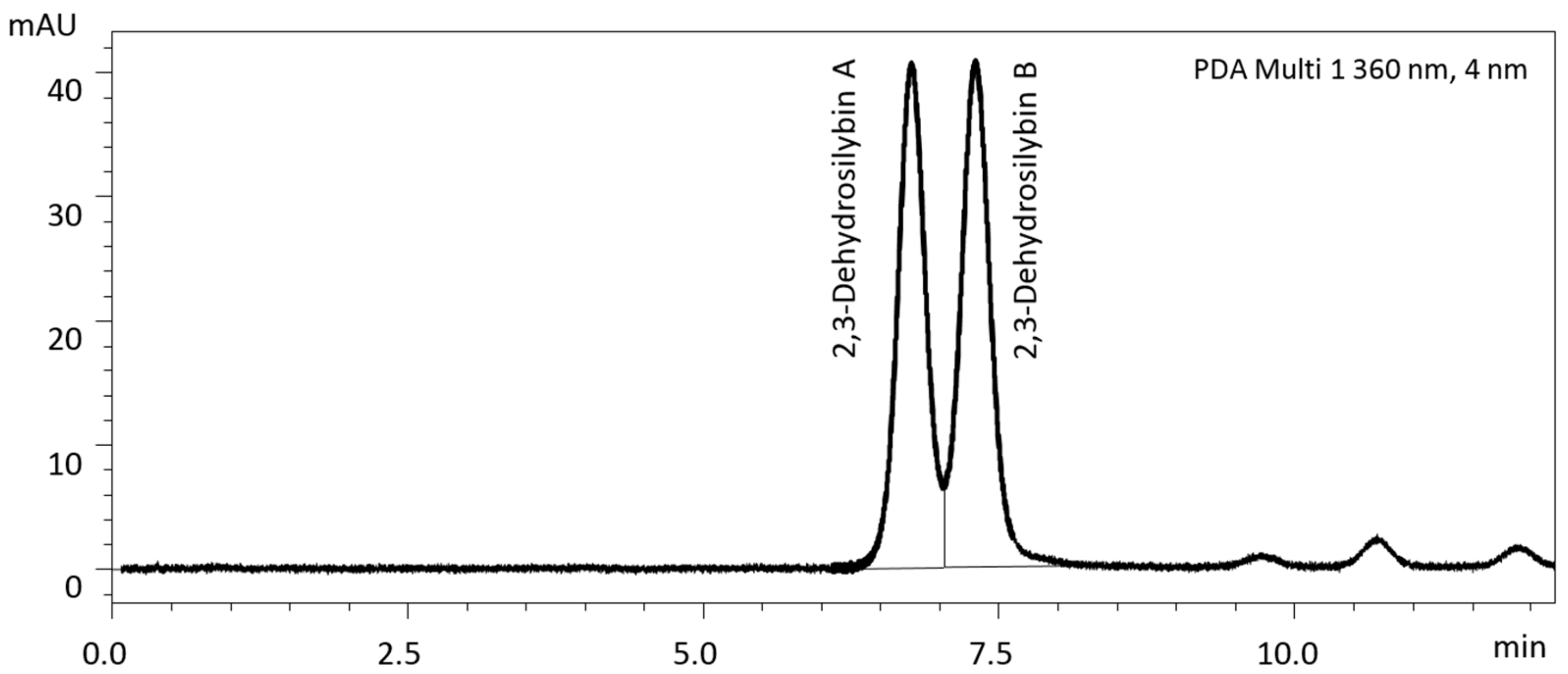
3.3. Separation of Enantiomers of 2,3-Dehydrosilybin and 2,3-Dehydrosilychristin
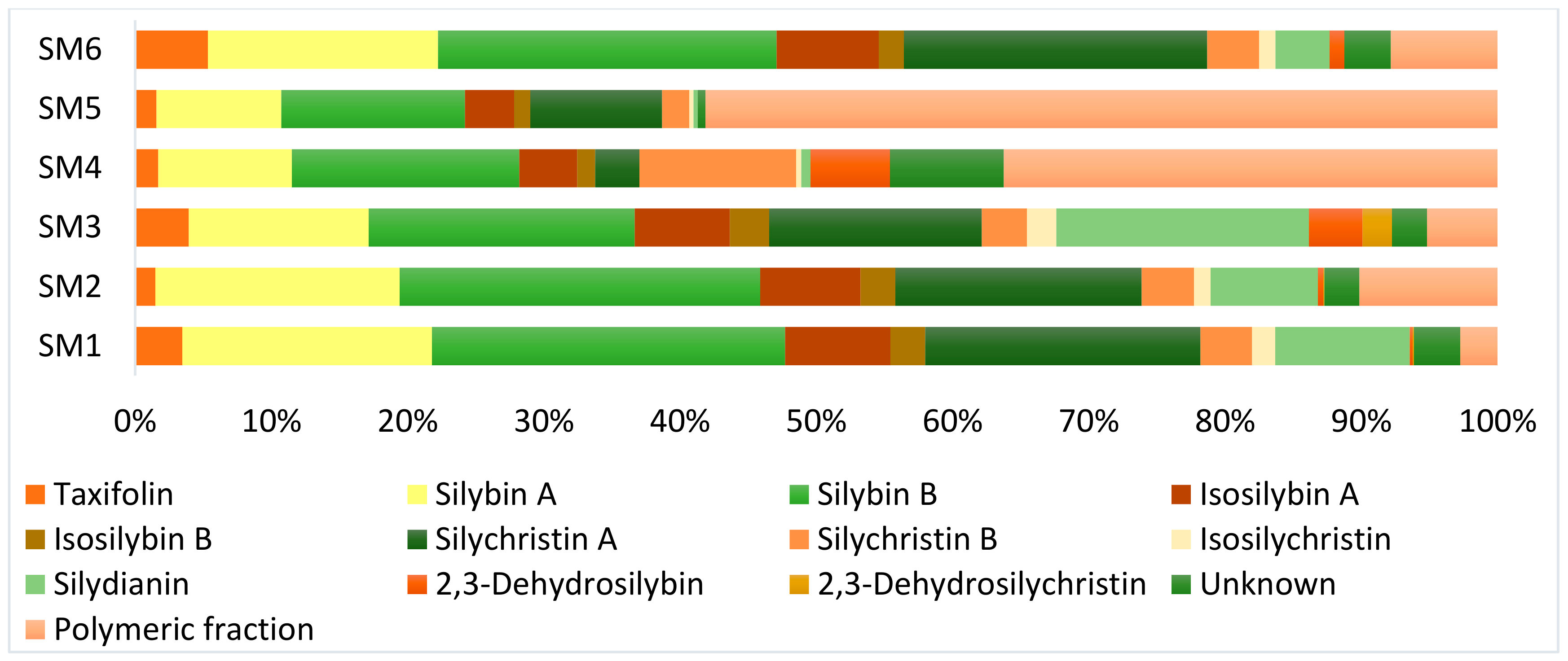
3.4. Flavonoid, Flavonolignans, and Dehydroflavonolignans Quantification
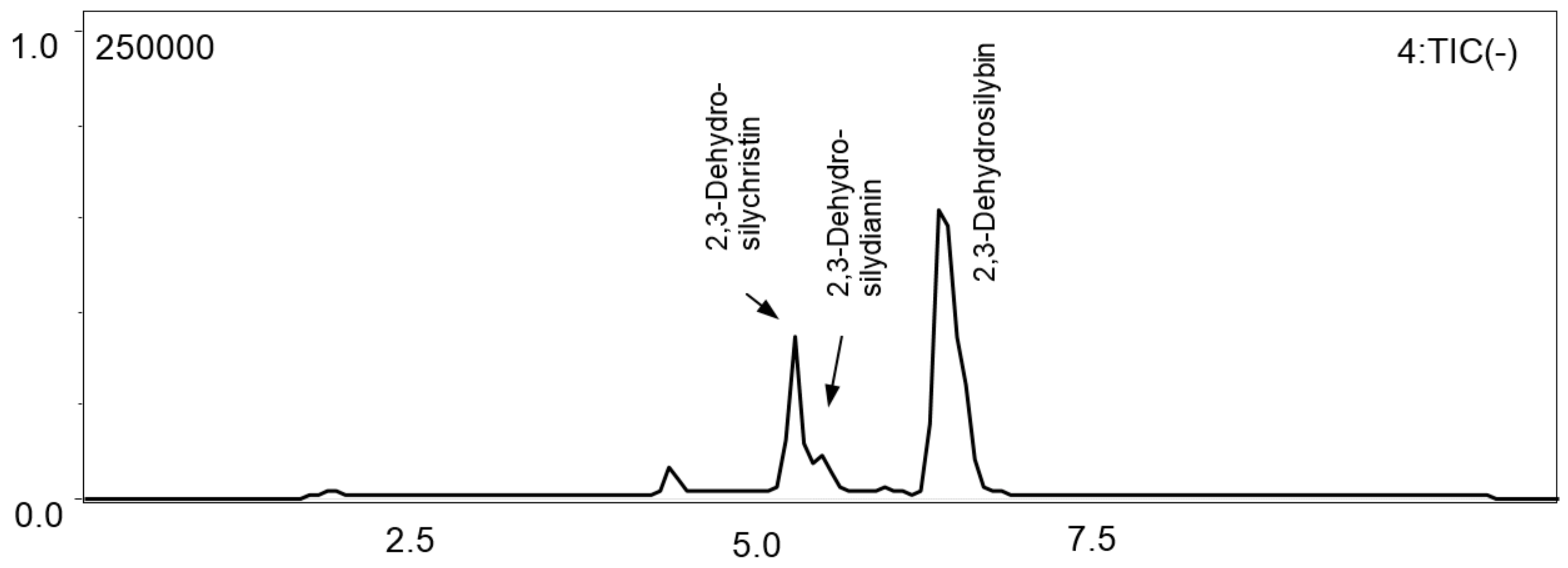
3.5. Polymeric Fraction
3.6. Isolation and Identification of 2,3-Dehydroflavonolignans Using Semi-Preparative Chromatography
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2,3-Dehydrosilybin | 2,3-Dehydrosilychristin | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Atom Nº | δC | m. | δH | m. | JHH (Hz) | δC | m. | δH | m. | JHH (Hz) |
| 2 | 145.49 | s | 146.67 | s | - | |||||
| 3 | n.a. | n.a. | - | |||||||
| 4 | 176.04 | s | 175.75 | s | - | |||||
| 4a | 102.87 | s | 102.88 | s | - | |||||
| 5 | 160.55 | s | 160.62 | s | - | |||||
| 6 | 98.22 | d | 6.189 | d | 1.8 | 98.12 | d | 6.187 | d | 2.0 |
| 7 | 164.25 | s | 163.93 | s | - | |||||
| 8 | 93.46 | d | 6.450 | d | 1.8 | 93.26 | d | 6.406 | d | 2.0 |
| 8a | 156.12 | s | 156.06 | s | - | |||||
| 10 | 78.40 | d | 4.268 | ddd | 2.5, 4.6, 7.9 | 87.40 | d | 5.544 | d | 6.6 |
| 11 | 75.75 | d | 4.964 | d | 7.9 | 52.88 | d | 3.553 | m | - |
| 11a | 129.73 | s | - | |||||||
| 12 | 115.45 | d | 7.606 | br s | - | |||||
| 12a | 143.25 | s | ||||||||
| 13 | 116.01 | d | 7.786 | br s | - | n.a. | s | - | ||
| 14 | 123.80 | s | 115.70 | d | 7.630 | d | 1.7 | |||
| 15 | 121.01 | d | 7.758 | br m | - | 140.82 | s | - | ||
| 15a | 148.60 | s | - | |||||||
| 16 | 116.68 | d | 7.111 | d | 8.7 | 132.02 | s | - | ||
| 16a | 144.80 | s | ||||||||
| 17 | 127.14 | s | 110.44 | d | 6.960 | d | 2.0 | |||
| 18 | 111.69 | d | 7.041 | d | 2.0 | 147.51 | s | - | ||
| 19 | 147.56 | s | 146.46 | s | - | |||||
| 20 | 147.01 | s | 115.29 | d | 6.771 | d | 8.1 | |||
| 21 | 115.24 | d | 6.819 | d | 8.0 | 118.68 | d | 6.811 | dd | 2.0, 8.1 |
| 22 | 120.45 | d | 6.887 | dd | 2.0, 8.0 | 62.89 | t | 3.740 | dd | 5.5, 10.9 |
| 3.694 | dd | 6.7, 10.9 | ||||||||
| 23 | 60.00 | t | 3.571 | dd | 2.5, 12.3 | |||||
| 3.376 | dd | 4.6, 12.3 | ||||||||
| 3-OH | n.a. | n.a. | ||||||||
| 5-OH | n.a. | - | 12.462 | br s | - | |||||
| 7-OH | n.a. | n.a. | ||||||||
| 18-OMe | 55.60 | q | 3.756 | s | - | |||||
| 19-OMe | 55.63 | q | 3.790 | s | - | |||||
| 20-OH | n.a. | n.a. | ||||||||
| 22-OH | - | n.d. | ||||||||
| 23-OH | n.d. | |||||||||
| x-OH | - | 9.575 | br s | - | ||||||
| 9.021 | br s | - | ||||||||
Appendix B

References
- Martin, R.J.; Lauren, D.R.; Smith, W.A.; Jensen, D.J.; Deo, B.; Douglas, J.A. Factors influencing silymarin content and composition in variegated thistle (Silybum marianum). N. Z. J. Crop Hortic. Sci. 2006, 34, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Graf, T.N.; Cech, N.B.; Polyak, S.J.; Oberlies, N.H. A validated UHPLC-tandem mass spectrometry method for quantitative analysis of flavonolignans in milk thistle (Silybum marianum) extracts. J. Pharm. Biomed. Anal. 2016, 126, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Kuki, Á.; Nagy, L.; Deák, G.; Nagy, M.; Zsuga, M.; Kéki, S. Identification of silymarin constituents: An improved HPLC–MS method. Chromatographia 2012, 75, 175–180. [Google Scholar] [CrossRef]
- Wallace, S.; Carrier, D.J.; Beitle, R.R.; Clausen, E.C.; Griffis, C.L. HPLC-UV and LC-MS-MS characterization of silymarin in milk thistle seeds and corresponding products. J. Nutraceutic. Funct. Med. Food 2003, 4, 37–48. [Google Scholar] [CrossRef]
- Althagafy, H.S.; Meza-Aviña, M.E.; Oberlies, N.H.; Croatt, M.P. Mechanistic study of the biomimetic synthesis of flavonolignan diastereoisomers in milk thistle. J. Org. Chem. 2013, 78, 7594–7600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gažák, R.; Svobodová, A.; Psotová, J.; Sedmera, P.; Přikrylová, V.; Walterová, D.; Křen, V. Oxidised derivatives of silybin and their antiradical and antioxidant activity. Bioorg. Med. Chem. 2004, 12, 5677–5687. [Google Scholar] [CrossRef]
- Chambers, C.S.; Holečková, V.; Petrásková, L.; Biedermann, D.; Valentová, K.; Buchta, M.; Křen, V. The silymarin composition… and why does it matter? Food Res. Int. 2017, 100, 339–353. [Google Scholar] [CrossRef]
- Biedermann, D.; Vavříková, E.; Cvak, L.; Křen, V. Chemistry of silybin. Nat. Prod. Rep. 2014, 31, 1138–1157. [Google Scholar] [CrossRef]
- Plíšková, M.; Vondráček, J.; Křen, V.; Gažák, R.; Sedmera, P.; Walterová, D.; Psotová, J.; Šimánek, V.; Machala, M. Effects of silymarin flavonolignans and synthetic silybin derivatives on estrogen and aryl hydrocarbon receptor activation. Toxicology 2005, 215, 80–89. [Google Scholar] [CrossRef]
- Davis-Searles, P.R.; Nakanishi, Y.; Kim, N.-C.; Graf, T.N.; Oberlies, N.H.; Wani, M.C.; Wall, M.E.; Agarwal, R.; Kroll, D.J. Milk thistle and prostate cancer: Differential effects of pure flavonolignans from Silybum marianum on antiproliferative end points in human prostate carcinoma cells. Cancer Res. 2005, 65, 4448–4457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biedermann, D.; Buchta, M.; Holečková, V.; Sedlák, D.; Valentová, K.; Cvačka, J.; Bednárová, L.; Křenková, A.; Kuzma, M.; Škuta, C.; et al. Silychristin: Skeletal alterations and biological activities. J. Nat. Prod. 2016, 79, 3086–3092. [Google Scholar] [CrossRef] [PubMed]
- Marhol, P.; Bednář, P.; Kolářová, P.; Večeřa, R.; Ulrichová, J.; Tesařová, E.; Vavříková, E.; Kuzma, M.; Křen, V. Pharmacokinetics of pure silybin diastereoisomers and identification of their metabolites in rat plasma. J. Funct. Food. 2015, 14, 570–580. [Google Scholar] [CrossRef]
- Filippopoulou, K.; Papaevgeniou, N.; Lefaki, M.; Paraskevopoulou, A.; Biedermann, D.; Křen, V.; Chondrogianni, N. 2,3-Dehydrosilybin A/B as a pro-longevity and anti-aggregation compound. Free Radic. Biol. Med. 2017, 103, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Rajnochová Svobodová, A.; Gabrielová, E.; Ulrichová, J.; Zálešák, B.; Biedermann, D.; Vostálová, J. A pilot study of the UVA-photoprotective potential of dehydrosilybin, isosilybin, silychristin, and silydianin on human dermal fibroblasts. Arch. Dermatol. Res. 2019, 311, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Kosina, P.; Paloncýová, M.; Rajnochová Svobodová, A.; Zálešák, B.; Biedermann, D.; Ulrichová, J.; Vostálová, J. Dermal delivery of selected polyphenols from Silybum marianum. Theoretical and experimental study. Molecules 2018, 24, 61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vostálová, J.; Tinková, E.; Biedermann, D.; Kosina, P.; Ulrichová, J.; Rajnochová Svobodová, A. Skin protective activity of silymarin and its flavonolignans. Molecules 2019, 24, 1022. [Google Scholar] [CrossRef] [Green Version]
- Tilley, C.; Deep, G.; Agarwal, C.; Wempe, M.F.; Biedermann, D.; Valentová, K.; Kren, V.; Agarwal, R. Silibinin and its 2,3-dehydro-derivative inhibit basal cell carcinoma growth via suppression of mitogenic signaling and transcription factors activation. Mol. Carcinog. 2016, 55, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Fenclová, M.; Nováková, A.; Viktorová, J.; Jonatová, P.; Džuman, Z.; Ruml, T.; Křen, V.; Hajšlová, J.; Vítek, L.; Stránská-Zachariášová, M. Poor chemical and microbiological quality of the commercial milk thistle-based dietary supplements may account for their reported unsatisfactory and non-reproducible clinical outcomes. Sci. Rep. 2019, 9, 11118. [Google Scholar] [CrossRef]
- Csupor, D.; Csorba, A.; Hohmann, J. Recent advances in the analysis of flavonolignans of Silybum marianum. J. Pharm. Biomed. Anal. 2016, 130, 301–317. [Google Scholar] [CrossRef]
- Karkanis, A.; Bilalis, D.; Efthimiadou, A. Cultivation of milk thistle (Silybum marianum L. Gaertn.), a medicinal weed. Ind. Crop. Prod. 2011, 34, 825–830. [Google Scholar] [CrossRef]
- Džubák, P.; Hajdúch, M.; Gažák, R.; Svobodová, A.; Psotová, J.; Walterová, D.; Sedmera, P.; Křen, V. New derivatives of silybin and 2,3-dehydrosilybin and their cytotoxic and P-glycoprotein modulatory activity. Bioorg. Med. Chem. 2006, 14, 3793–3810. [Google Scholar] [CrossRef]
- Biedermann, D.; Moravcová, V.; Valentová, K.; Kuzma, M.; Petrásková, L.; Císařová, I.; Křen, V. Oxidation of flavonolignan silydianin to unexpected lactone-acid derivative. Phytochem. Lett. 2019, 30, 14–20. [Google Scholar] [CrossRef]
- Křenek, K.; Marhol, P.; Peikerová, Ž.; Křen, V.; Biedermann, D. Preparatory separation of the silymarin flavonolignans by Sephadex LH-20 gel. Food Res. Int. 2014, 65, 115–120. [Google Scholar] [CrossRef]
- Pyszková, M.; Biler, M.; Biedermann, D.; Valentová, K.; Kuzma, M.; Vrba, J.; Ulrichová, J.; Sokolová, R.; Mojovic, M.; Popovic-Bijelic, A.; et al. Flavonolignan 2,3-dehydroderivatives: Preparation, antiradical and cytoprotective activity. Free Radic. Biol. Med. 2016, 90, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Novotná, M.; Gažák, R.; Biedermann, D.; Di Meo, F.; Marhol, P.; Kuzma, M.; Bednárová, L.; Fuksová, K.; Trouillas, P.; Křen, V. cis–trans Isomerization of silybins A and B. Beilstein J. Org. Chem. 2014, 10, 1047–1063. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Du, Z.; Yuan, Q. A novel rapid method for simultaneous determination of eight active compounds in silymarin using a reversed-phase UPLC-UV detector. J. Chromatogr. B 2009, 877, 4159–4163. [Google Scholar] [CrossRef]
- AbouZid, S.F.; Chen, S.N.; Pauli, G.F. Silymarin content in Silybum marianum populations growing in Egypt. Ind. Crop. Prod. 2016, 83, 729–737. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.C.; Graf, T.N.; Sparacino, C.M.; Wani, M.C.; Wall, M.E. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum). Org. Biomol. Chem. 2003, 1, 1684–1689. [Google Scholar] [CrossRef]
- Cai, X.; Li, D.; Qiao, J.; Lian, H.; Wang, S. Determination of silymarin flavonoids by HPLC and LC-MS and investigation of extraction rate of silymarin in Silybum marianum fruits by boiling water. Asian J. Chem. 2009, 21, 63–74. [Google Scholar]
- Keshavarz Afshar, R.; Chaichi, M.R.; Ansari Jovini, M.; Jahanzad, E.; Hashemi, M. Accumulation of silymarin in milk thistle seeds under drought stress. Planta 2015, 242, 539–543. [Google Scholar] [CrossRef]
- Poppe, L.; Petersen, M. Variation in the flavonolignan composition of fruits from different Silybum marianum chemotypes and suspension cultures derived therefrom. Phytochemistry 2016, 131, 68–75. [Google Scholar] [CrossRef]
- Bilia, A.R.; Salvini, D.; Mazzi, G.; Vincieri, F.F. Characterization of calendula flower, milk-thistle fruit, and passion flower tinctures by HPLC-DAD and HPLC-MS. Chromatographia 2000, 53, 210–215. [Google Scholar] [CrossRef]
- Gažák, R.; Sedmera, P.; Marzorati, M.; Riva, S.; Křen, V. Laccase-mediated dimerization of the flavonolignan silybin. J. Mol. Catal. B Enzym. 2008, 50, 87–92. [Google Scholar] [CrossRef]





| Standard | Purity [%] | Slope [m] * | R2 | LOD [mg/mL] | LOQ [mg/mL] | RSD [%] |
|---|---|---|---|---|---|---|
| Taxifolin | 95.4 | 2,627,107 | 0.998 | 0.0010 | 0.0032 | 1.1 |
| Isosilychristin | 96.7 | 1,722,853 | 0.999 | 0.0015 | 0.0049 | 1.3 |
| Silychristin A | 99.0 | 2,020,320 | 0.999 | 0.0012 | 0.0043 | 1.3 |
| Silychristin B | 88.0 | 1,218,423 | 0.999 | 0.0021 | 0.0070 | 1.1 |
| Silydianin | 97.0 | 1,438,790 | 0.999 | 0.0025 | 0.0084 | 1.2 |
| Silybin A | 98.0 | 2,132,608 | 0.997 | 0.0014 | 0.0046 | 0.8 |
| Silybin B | 100.0 | 2,464,791 | 0.999 | 0.0010 | 0.0033 | 0.8 |
| Isosilybin A | 96.0 | 2,152,112 | 0.996 | 0.0014 | 0.0045 | 0.7 |
| Isosilybin B | 98.0 | 2,282,544 | 0.994 | 0.0015 | 0.0049 | 0.6 |
| 2,3-Dehydrosilybin | 98.0 | 33,994,912 | 0.964 | 0.0001 | 0.0005 | 0.4 |
| 2,3-Dehydrosilychristin | 100.0 | 41,737,232 | 0.973 | 0.0001 | 0.0003 | 0.1 |
| Component | Rt | [M-H]− | Content [mg/g] a | |||||
|---|---|---|---|---|---|---|---|---|
| [min] | m/z | SM 1 b | SM 2 c | SM 3 d | SM 4 e | SM 5 f | SM 6 g | |
| Taxifolin | 2.4 | 303 | 27.8 | 7.8 | 27.9 | 4.0 | 2.5 | 31.4 |
| Isosilychristin | 4.3 | 481 | 13.6 | 6.4 | 15.2 | 0.9 | 0.5 | 7.0 |
| Silychristin A | 5.2 | 481 | 162.0 | 94.6 | 110.4 | 7.6 | 15.5 | 130.8 |
| Silydianin | 5.6 | 481 | 79.1 | 41.2 | 131.3 | 1.6 | 0.5 | 23.4 |
| Silychristin B | 6.1 | 481 | 30.4 | 20.1 | 23.5 | 27.0 | 3.2 | 22.4 |
| Silybin A | 9.0 | 481 | 146.8 | 93.8 | 93.4 | 23.0 | 14.7 | 99.2 |
| Silybin B | 9.6 | 481 | 207.9 | 138.4 | 138.3 | 39.2 | 21.6 | 146.0 |
| Isosilybin A | 11.1 | 481 | 61.9 | 38.6 | 49.4 | 10.0 | 5.8 | 44.0 |
| Isosilybin B | 11.4 | 481 | 20.5 | 13.3 | 20.5 | 3.1 | 1.9 | 10.8 |
| Unknown at 3.7 min | 3.7 | 481 | 10.9 | 6.8 | 7.1 | 8.8 | 0.7 | 16.2 |
| Unknown at 4.9 min | 4.9 | 481 | 9.7 | ND h | 6.9 | 3.3 | ND | ND |
| Unknown at 11.6 min | 11.6 | 481 | 6.6 | 6.7 | 4.3 | 7.5 | 0.2 | 3.8 |
| 2,3-Dehydrosilybin | 6.4 | 479 | 1.7 | 2.1 | 27.9 | 13.7 | ND | 6.4 |
| 2,3-Dehydrosilychristin | 5.2 | 479 | 0.8 | 0.4 | 15.2 | ND | ND | ND |
| Σ flavonoid, flavono-lignans and 2,3-dehy-droflavonolignans | 777.1 | 467.7 | 628.1 | 135.7 | 68.3 | 542.6 | ||
| Polymeric fraction (%) | 22.0 | 53.0 | 36.6 | 85.1 | 93.2 i | 46.0 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrásková, L.; Káňová, K.; Biedermann, D.; Křen, V.; Valentová, K. Simple and Rapid HPLC Separation and Quantification of Flavonoid, Flavonolignans, and 2,3-Dehydroflavonolignans in Silymarin. Foods 2020, 9, 116. https://doi.org/10.3390/foods9020116
Petrásková L, Káňová K, Biedermann D, Křen V, Valentová K. Simple and Rapid HPLC Separation and Quantification of Flavonoid, Flavonolignans, and 2,3-Dehydroflavonolignans in Silymarin. Foods. 2020; 9(2):116. https://doi.org/10.3390/foods9020116
Chicago/Turabian StylePetrásková, Lucie, Kristýna Káňová, David Biedermann, Vladimír Křen, and Kateřina Valentová. 2020. "Simple and Rapid HPLC Separation and Quantification of Flavonoid, Flavonolignans, and 2,3-Dehydroflavonolignans in Silymarin" Foods 9, no. 2: 116. https://doi.org/10.3390/foods9020116