
Genetic Complementation of ATP Synthase Deficiency Due to Dysfunction of TMEM70 Assembly Factor in Rat

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
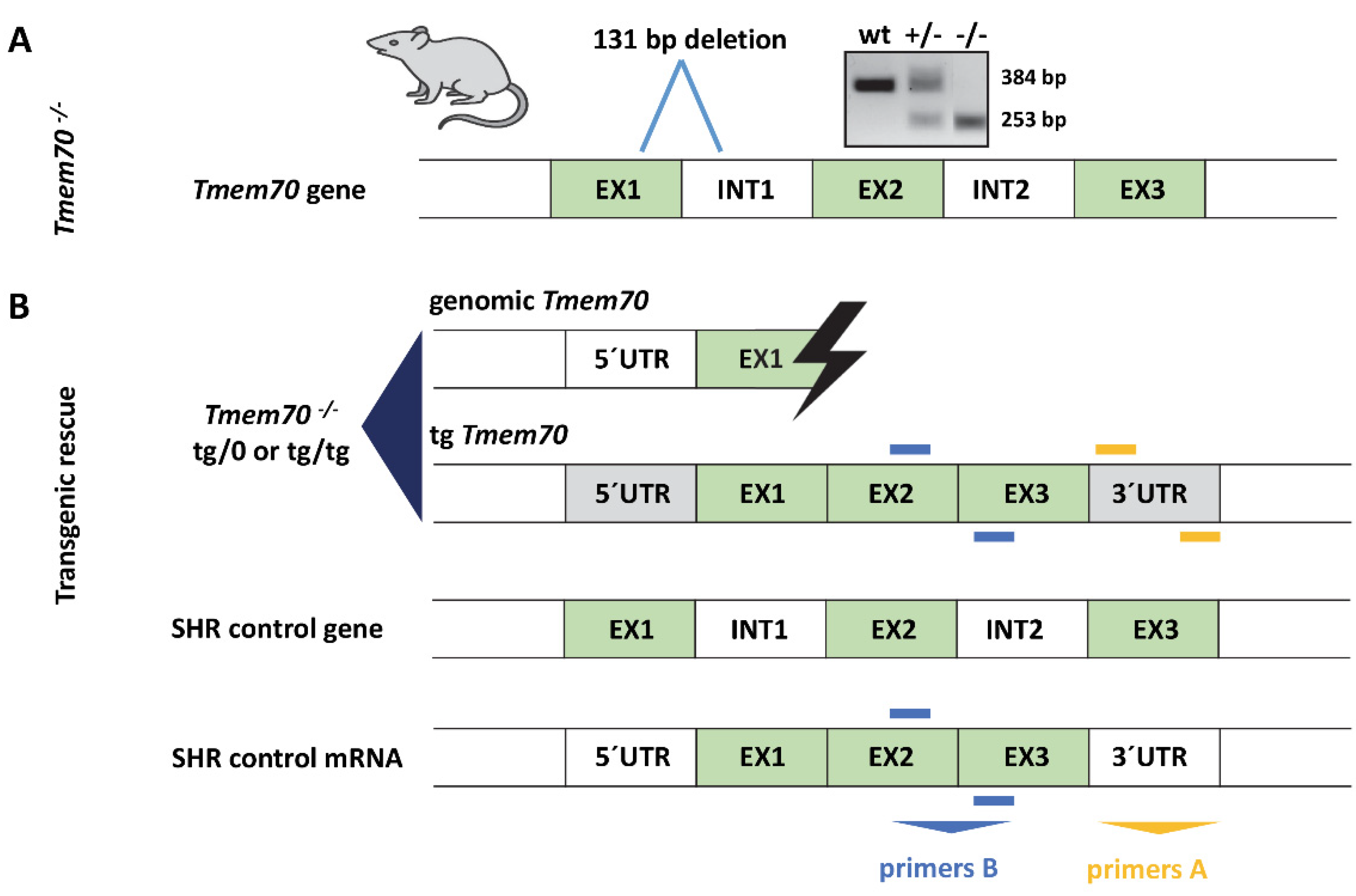
2.2. Generation of Tmem70 Knockout Rats
2.3. Generation of Tmem70 Transgene
2.4. Genotyping
2.5. Gene Expression Analysis
2.6. Tissue Homogenates
2.7. Electrophoretic Analyses
2.8. In-Gel Activity Staining
2.9. Western Blot Analysis
2.10. High Resolution Oxygraphy
2.11. Echocardiography
3. Results
3.1. Generation of Tmem70 Knockout SHR Expressing Tmem70 Wild-Type Transgene (SHR-Tmem70ko/ko,tg/tg)
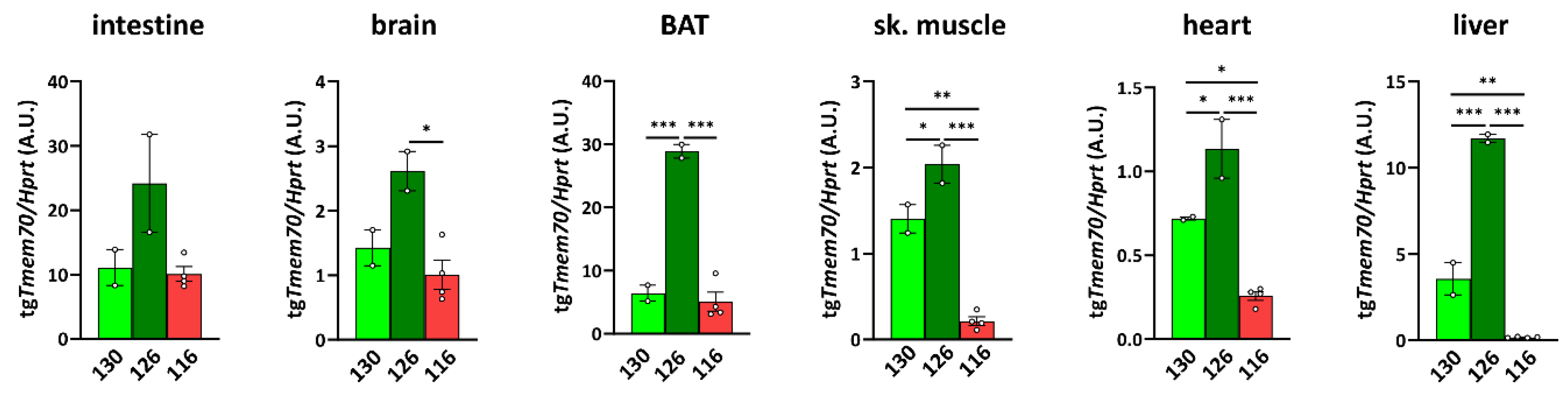
3.2. Tmem70 Transgene Expression in SHR Rats
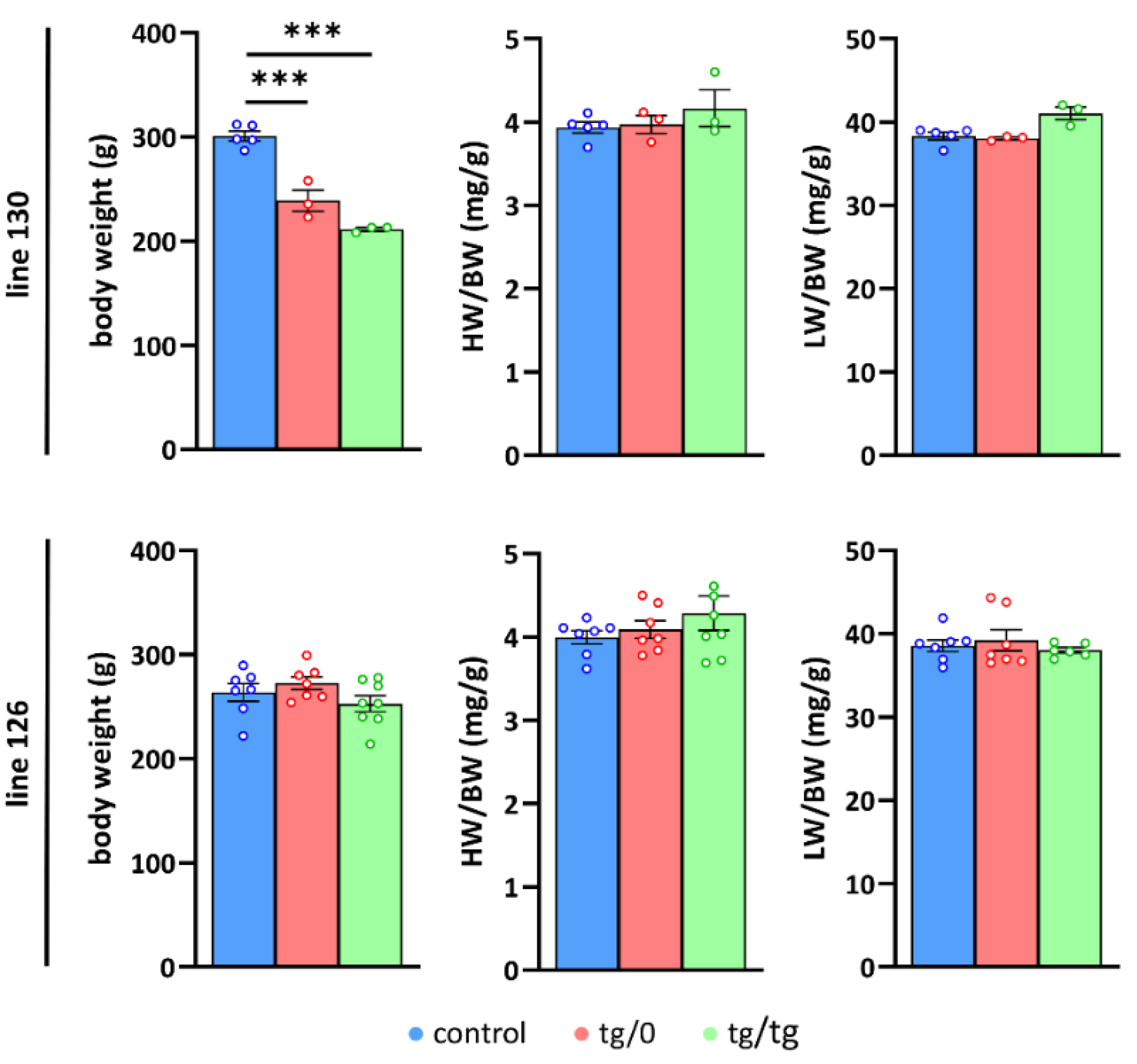
3.3. Impact of Tmem70 Transgene Expression on Growth Phenotype
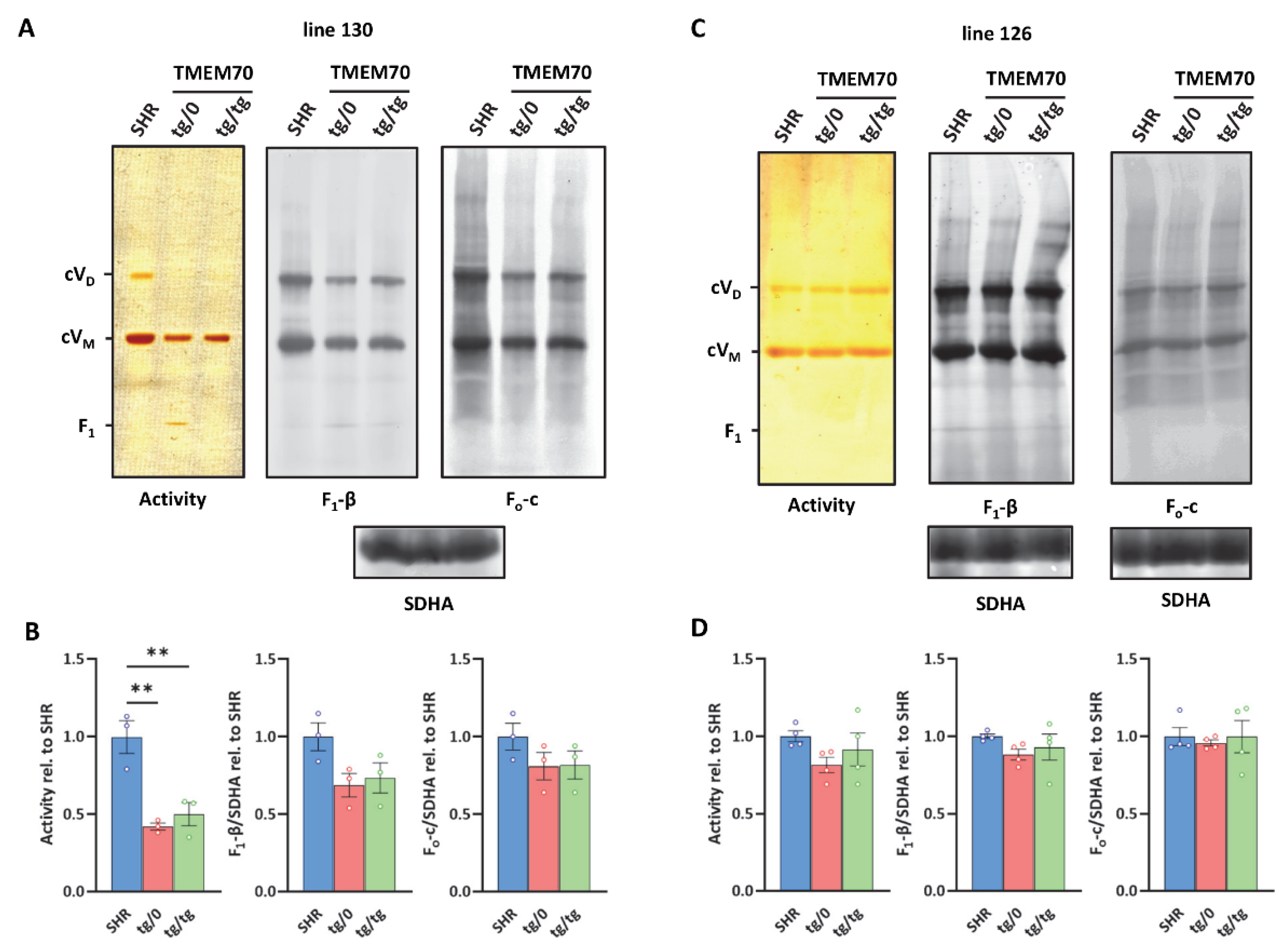
3.4. Complementation of ATP Synthase Deficiency
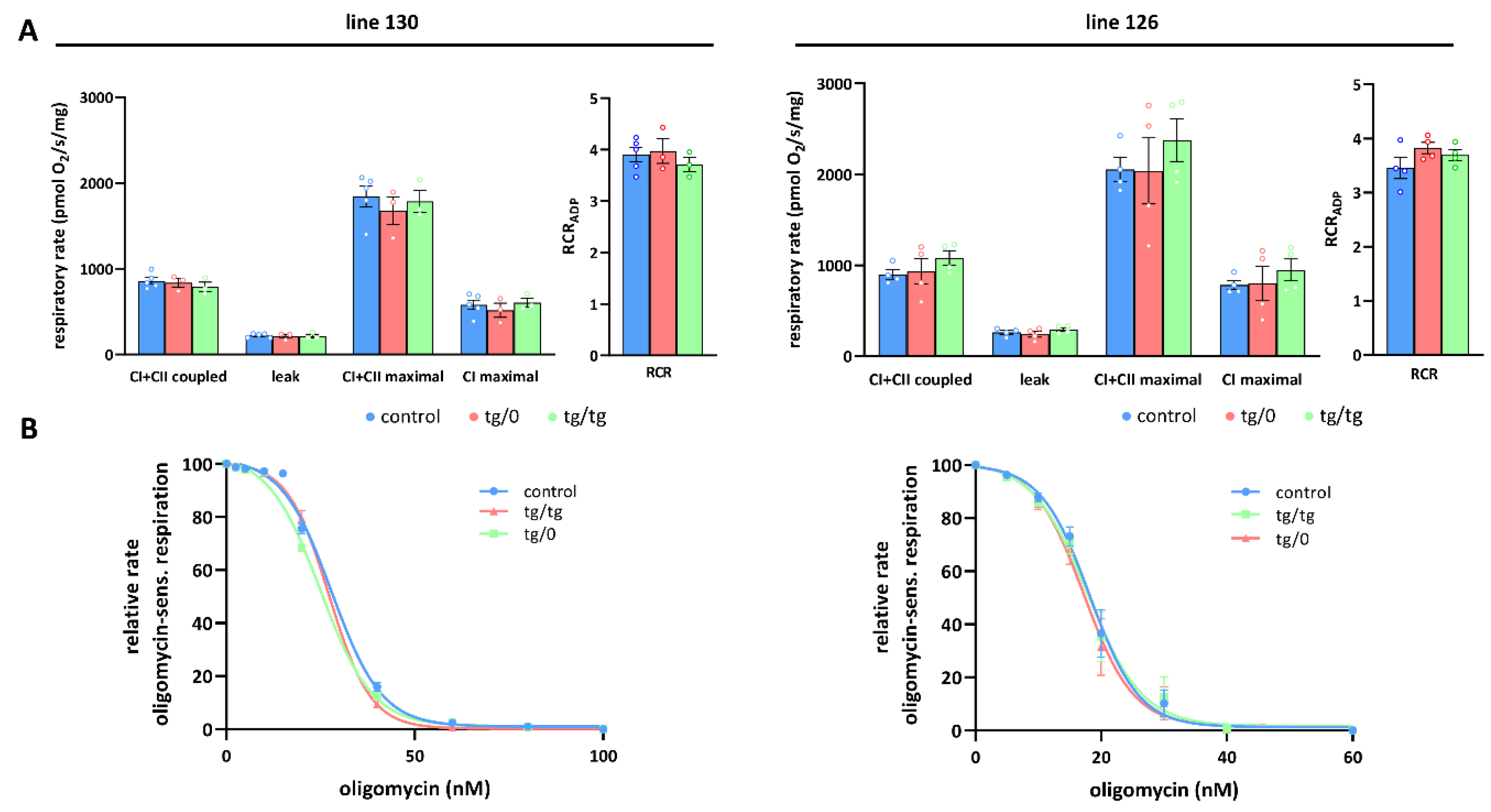
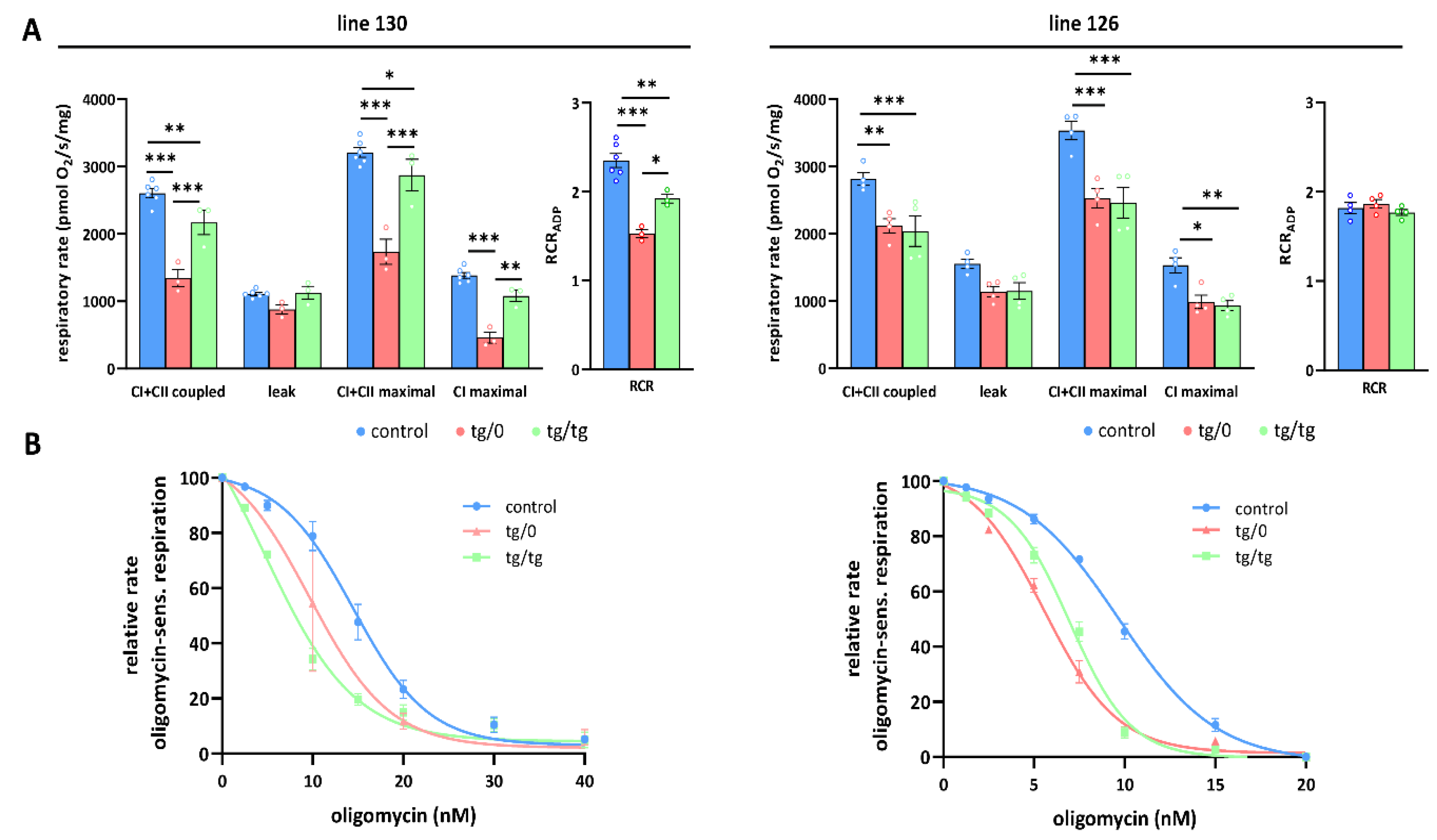
3.5. Mitochondrial Energetic Function
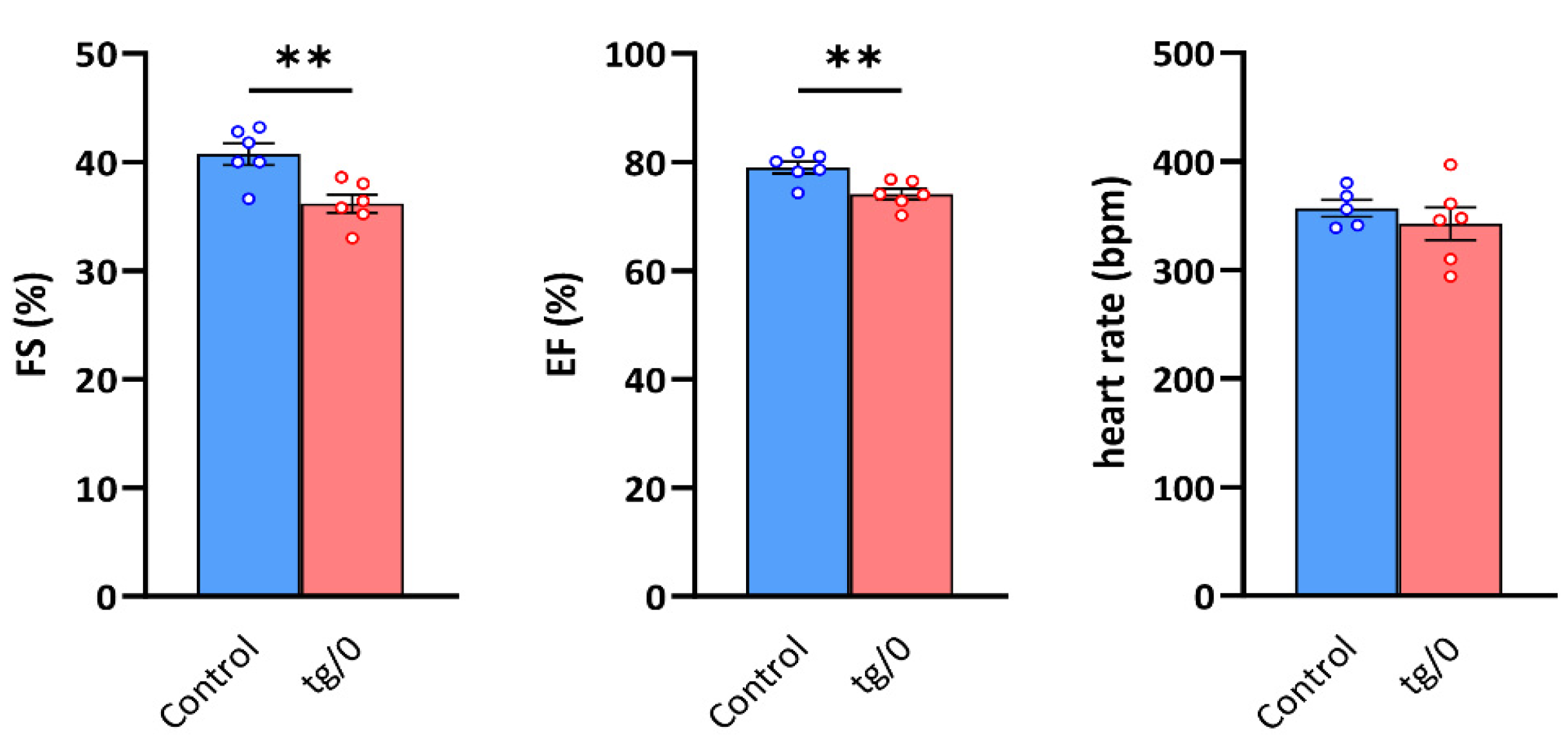
3.6. Cardiac Function
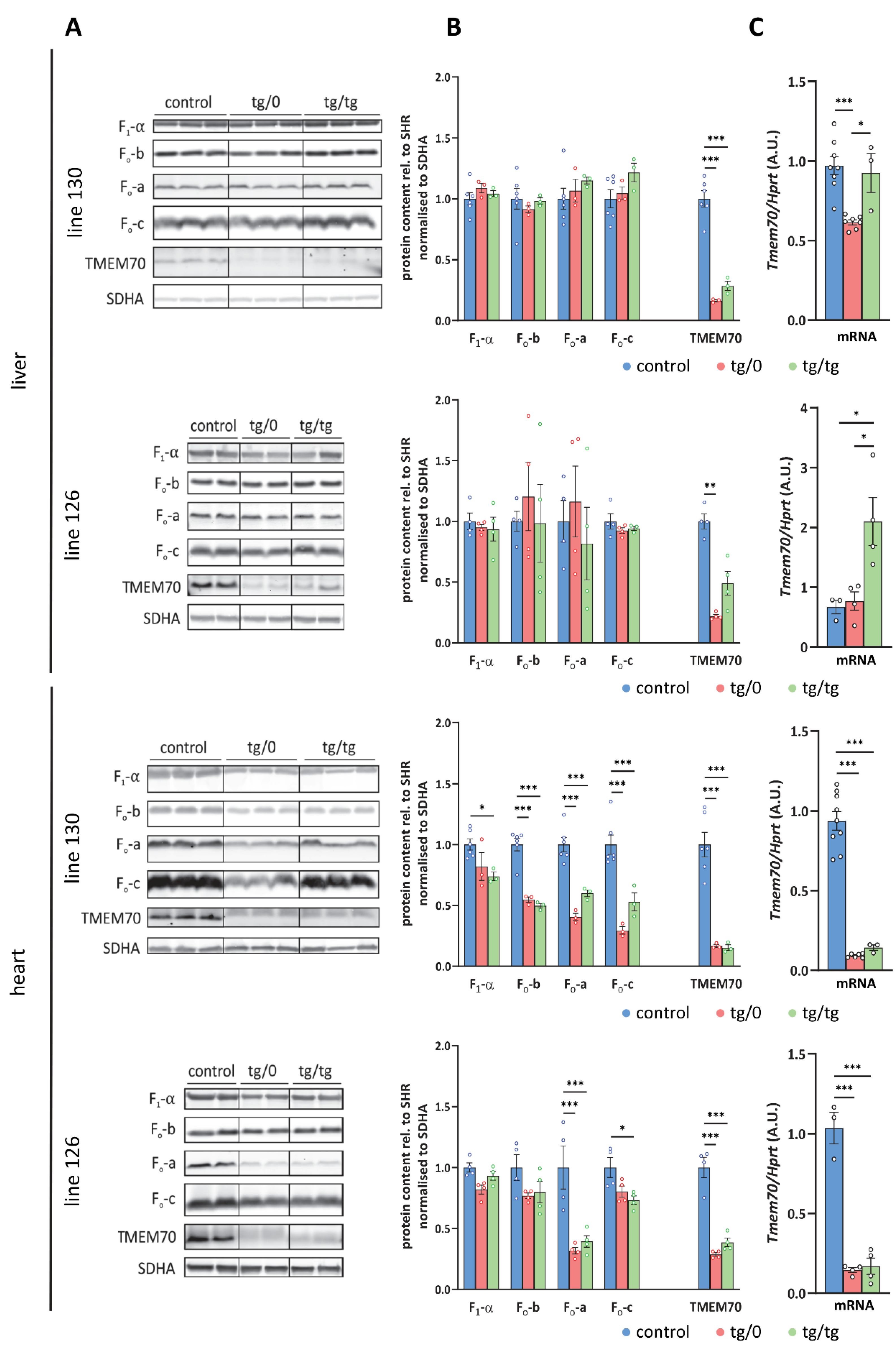
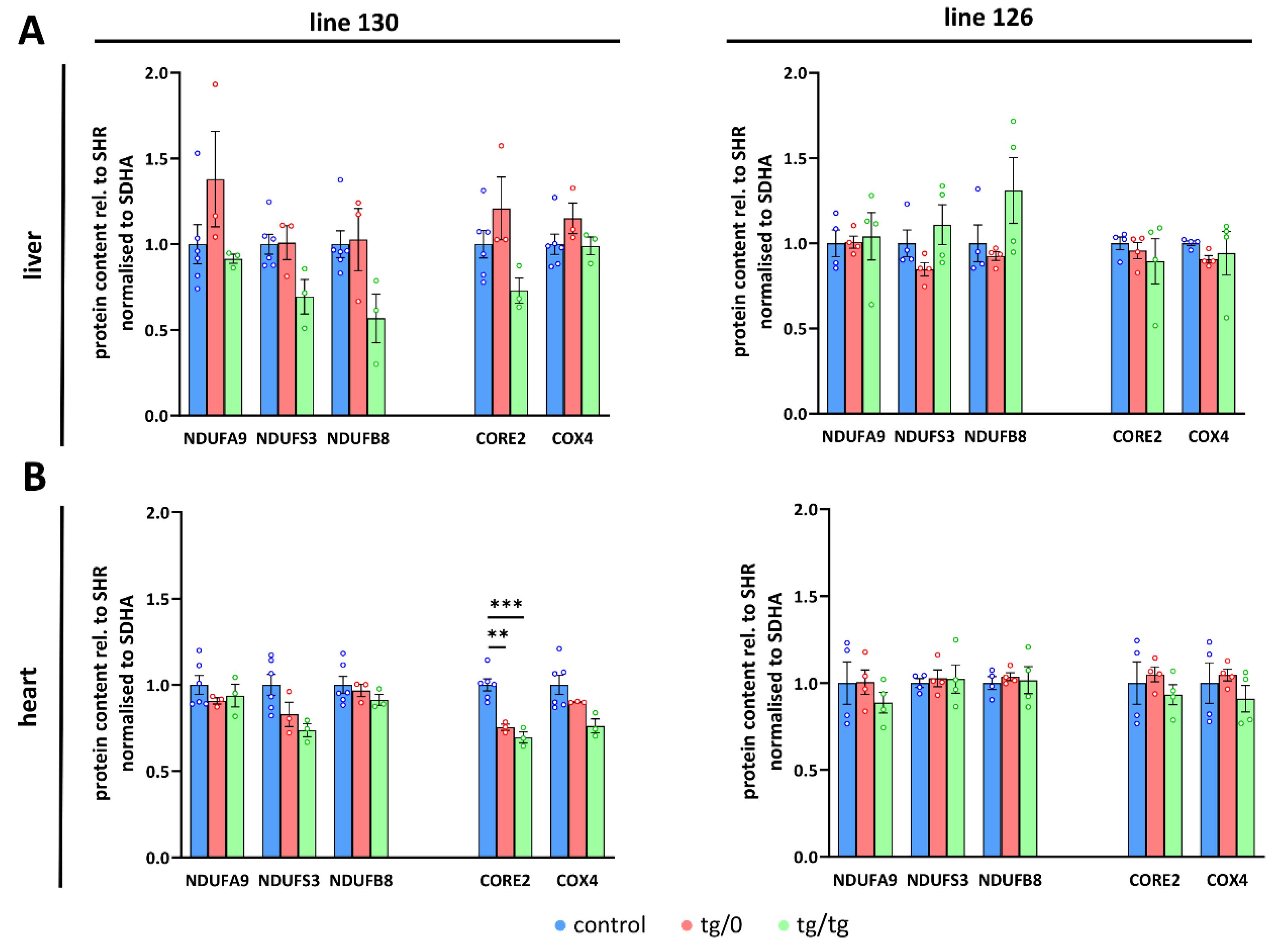
3.7. Restoration of ATP Synthase Biogenesis
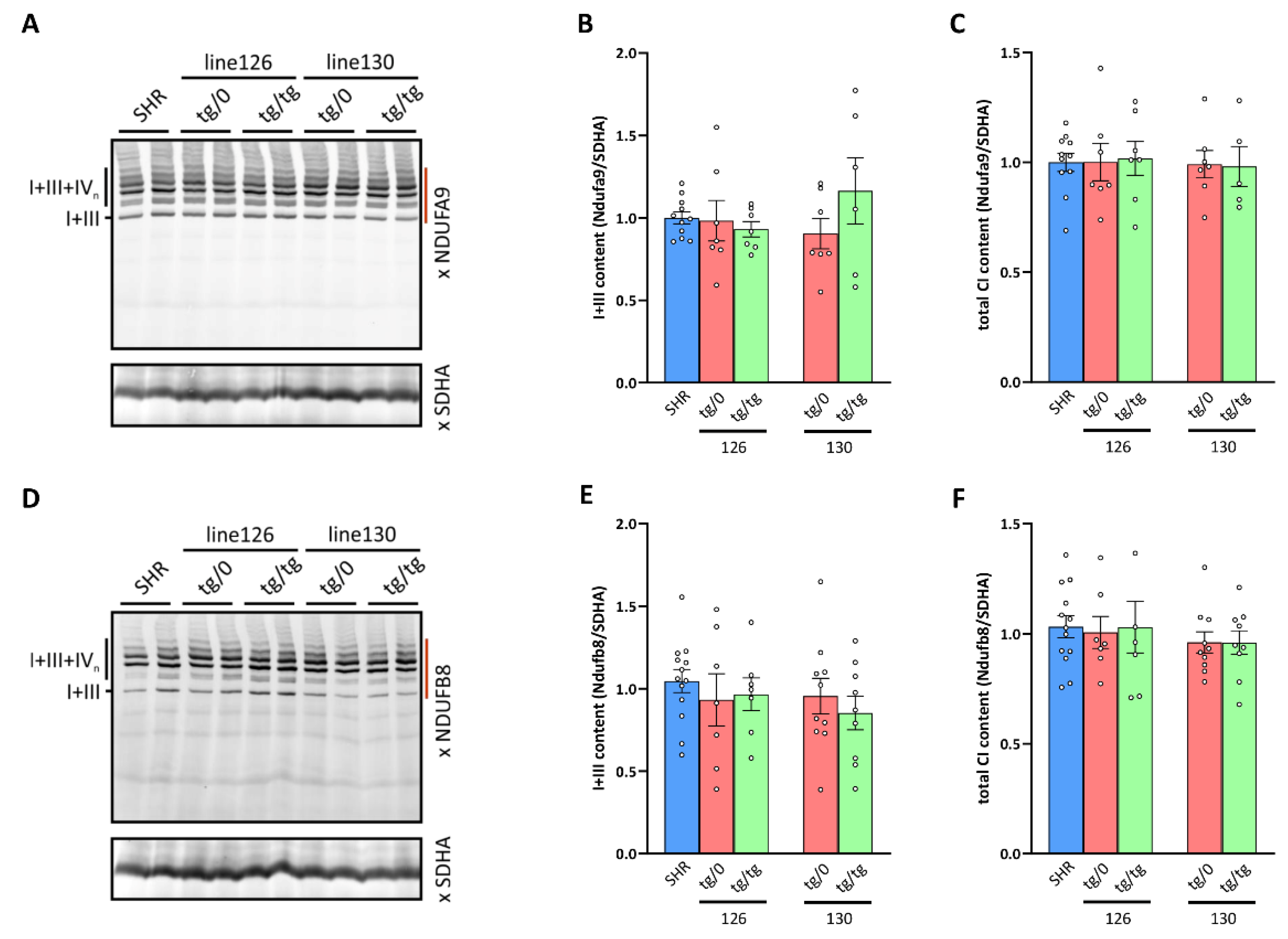
3.8. Respiratory Chain Complex I Assembly
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- DiMauro, S. A history of mitochondrial diseases. J. Inherit. Metab. Dis. 2011, 34, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Wagner, M.; Stenton, S.L.; Strom, T.M.; Wortmann, S.B.; Prokisch, H.; Meitinger, T.; Oexle, K.; Klopstock, T. Lifetime risk of autosomal recessive mitochondrial disorders calculated from genetic databases. EBioMedicine 2020, 54, 102730. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackerman, S.H.; Tzagoloff, A. Function, Structure, and Biogenesis of Mitochondrial ATP Synthase. Prog. Nucleic Acid Res. Mol. Biol. 2005, 80, 95–133. [Google Scholar] [CrossRef]
- Rak, M.; Gokova, S.; Tzagoloff, A. Modular assembly of yeast mitochondrial ATP synthase. EMBO J. 2011, 30, 920–930. [Google Scholar] [CrossRef] [Green Version]
- Rak, M.; Zeng, X.; Brière, J.-J.; Tzagoloff, A. Assembly of F0 in Saccharomyces cerevisiae. Biochim. Biophys. Acta 2009, 1793, 108–116. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Meyer, B.; Heide, H.; Steger, M.; Bleier, L.; Wumaier, Z.; Karas, M.; Schägger, H. Assembly and oligomerization of human ATP synthase lacking mitochondrial subunits a and A6L. Biochim. Biophys. Acta 2010, 1797, 1004–1011. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Carroll, J.; Ding, S.; Fearnley, I.M.; Montgomery, M.G.; Walker, J.E. Assembly of the peripheral stalk of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 29602–29608. [Google Scholar] [CrossRef]
- He, J.; Ford, H.C.; Carroll, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc. Natl. Acad. Sci. USA 2017, 114, 3409–3414. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Ford, H.C.; Carroll, J.; Douglas, C.; Gonzales, E.; Ding, S.; Fearnley, I.M.; Walker, J.E. Assembly of the membrane domain of ATP synthase in human mitochondria. Proc. Natl. Acad. Sci. USA 2018, 115, 2988–2993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzagoloff, A.; Barrientos, A.; Neupert, W.; Herrmann, J.M. Atp10p Assists Assembly of Atp6p into the F0 Unit of the Yeast Mitochondrial ATPase. J. Biol. Chem. 2004, 279, 19775–19780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Barros, M.H.; Shulman, T.; Tzagoloff, A. ATP25, a New Nuclear Gene ofSaccharomyces cerevisiaeRequired for Expression and Assembly of the Atp9p Subunit of Mitochondrial ATPase. Mol. Biol. Cell 2008, 19, 1366–1377. [Google Scholar] [CrossRef] [Green Version]
- Zeng, X.; Hourset, A.; Tzagoloff, A. The Saccharomyces cerevisiae ATP22 Gene Codes for the Mitochondrial ATPase Subunit 6-Specific Translation Factor. Genetics 2007, 175, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, X.; Neupert, W.; Tzagoloff, A. The Metalloprotease Encoded byATP23Has a Dual Function in Processing and Assembly of Subunit 6 of Mitochondrial ATPase. Mol. Biol. Cell 2007, 18, 617–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytovchenko, O.; Naumenko, N.; Oeljeklaus, S.; Schmidt, B.; Von Der Malsburg, K.; Deckers, M.; Warscheid, B.; Van Der Laan, M.; Rehling, P. The INA complex facilitates assembly of the peripheral stalk of the mitochondrial F1F0-ATP synthase. EMBO J. 2014, 33, 1624–1638. [Google Scholar] [CrossRef]
- Li, Y.; Jourdain, A.A.; Calvo, S.E.; Liu, J.S.; Mootha, V.K. CLIC, a tool for expanding biological pathways based on co-expression across thousands of datasets. PLoS Comput. Biol. 2017, 13, e1005653. [Google Scholar] [CrossRef] [Green Version]
- Čížková, A.; Stranecky, V.; Mayr, J.A.; Tesarova, M.; Havlíčková, V.; Paul, J.; Ivánek, R.; Kuss, A.W.; Hansíková, H.; Kaplanová, V.; et al. TMEM70 mutations cause isolated ATP synthase deficiency and neonatal mitochondrial encephalocardiomyopathy. Nat. Genet. 2008, 40, 1288–1290. [Google Scholar] [CrossRef]
- Hejzlarová, K.; Tesařová, M.; Vrbacká-Čížková, A.; Vrbacký, M.; Hartmannová, H.; Kaplanová, V.; Nosková, L.; Kratochvílová, H.; Buzková, J.; Havlíčková, V.; et al. Expression and processing of the TMEM70 protein. Biochim. Biophys. Acta 2011, 1807, 144–149. [Google Scholar] [CrossRef] [Green Version]
- Kratochvílová, H.; Hejzlarová, K.; Vrbacky, M.; Mráček, T.; Karbanová, V.; Tesarova, M.; Gombitová, A.; Cmarko, D.; Wittig, I.; Zeman, J.; et al. Mitochondrial membrane assembly of TMEM70 protein. Mitochondrion 2014, 15, 1–9. [Google Scholar] [CrossRef]
- Vrbacky, M.; Kovalčíková, J.; Chawengsaksophak, K.; Beck, I.M.; Mráček, T.; Nůsková, H.; Sedmera, D.; Papoušek, F.; Kolar, F.; Sobol, M.; et al. Knockout of Tmem70 alters biogenesis of ATP synthase and leads to embryonal lethality in mice. Hum. Mol. Genet. 2016, 25, 4674–4685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalčíková, J.; Vrbacký, M.; Pecina, P.; Tauchmannová, K.; Nůsková, H.; Kaplanová, V.; Brázdová, A.; Alán, L.; Eliáš, J.; Čunátová, K.; et al. TMEM70 facilitates biogenesis of mammalian ATP synthase by promoting subunit c incorporation into the rotor structure of the enzyme. FASEB J. 2019, 33, 14103–14117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, J.; He, J.; Ding, S.; Fearnley, I.M.; Walker, J.E. TMEM70 and TMEM242 help to assemble the rotor ring of human ATP synthase and interact with assembly factors for complex I. Proc. Natl. Acad. Sci. USA 2021, 118, e2100558118. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Caballero, L.; Elurbe, D.M.; Baertling, F.; Guerrero-Castillo, S.; van den Brand, M.; van Strien, J.; van Dam, T.J.P.; Rodenburg, R.; Brandt, U.; Huynen, M.A.; et al. TMEM70 functions in the assembly of complexes I and V. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148202. [Google Scholar] [CrossRef]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; Di Rago, J.-P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Hejzlarová, K.; Mráček, T.; Vrbacký, M.; Kaplanová, V.; Karbanová, V.; Nůsková, H.; Pecina, P.; Houštěk, J. Nuclear Genetic Defects of Mitochondrial ATP Synthase. Physiol. Res. 2014, 63, S57–S71. [Google Scholar] [CrossRef]
- Jonckheere, A.I.; Renkema, G.H.; Bras, M.; van den Heuvel, L.P.; Hoischen, A.; Gilissen, C.; Nabuurs, S.B.; Huynen, M.A.; De Vries, M.C.; Smeitink, J.A.; et al. A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy. Brain 2013, 136, 1544–1554. [Google Scholar] [CrossRef] [Green Version]
- Oláhová, M.; Yoon, W.H.; Thompson, K.; Jangam, S.; Fernandez, L.; Davidson, J.M.; Kyle, J.E.; Grove, M.E.; Fisk, D.G.; Kohler, J.N.; et al. Biallelic Mutations in ATP5F1D, which Encodes a Subunit of ATP Synthase, Cause a Metabolic Disorder. Am. J. Hum. Genet. 2018, 102, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Mayr, J.A.; Havlíčková, V.; Zimmermann, F.; Magler, I.; Kaplanová, V.; Ješina, P.; Pecinová, A.; Nůsková, H.; Koch, J.; Sperl, W.; et al. Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 epsilon subunit. Hum. Mol. Genet. 2010, 19, 3430–3439. [Google Scholar] [CrossRef] [Green Version]
- De Meirleir, L.; Seneca, S.; Lissens, W.; De Clercq, I.; Eyskens, F.; Gerlo, E.; Smet, J.; Van Coster, R. Respiratory chain complex V deficiency due to a mutation in the assembly gene ATP12. J. Med. Genet. 2004, 41, 120–124. [Google Scholar] [CrossRef] [Green Version]
- Diodato, D.; Invernizzi, F.; Lamantea, E.; Fagiolari, G.; Parini, R.; Menni, F.; Parenti, G.; Bollani, L.; Pasquini, E.; Donati, M.A.; et al. Common and Novel TMEM70 Mutations in a Cohort of Italian Patients with Mitochondrial Encephalocardiomyopathy. JIMD Rep. 2015, 15, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hirono, K.; Ichida, F.; Nishio, N.; Ogawa-Tominaga, M.; Fushimi, T.; Feichtinger, R.G.; Mayr, J.A.; Kohda, M.; Kishita, Y.; Okazaki, Y.; et al. Mitochondrial complex deficiency by novel compound heterozygous TMEM70 variants and correlation with developmental delay, undescended testicle, and left ventricular noncompaction in a Japanese patient: A case report. Clin. Case Rep. 2019, 7, 553–557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honzik, T.; Tesarova, M.; Mayr, J.A.; Hansíková, H.; Jesina, P.; Bodamer, O.; Koch, J.; Magner, M.; Freisinger, P.; Huemer, M.; et al. Mitochondrial encephalocardio-myopathy with early neonatal onset due to TMEM70 mutation. Arch. Dis. Child. 2010, 95, 296–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magner, M.; Dvorakova, V.; Tesarova, M.; Mazurova, S.; Hansikova, H.; Zahorec, M.; Brennerova, K.; Bzduch, V.; Spiegel, R.; Horovitz, Y.; et al. TMEM70 deficiency: Long-term outcome of 48 patients. J. Inherit. Metab. Dis. 2015, 38, 417–426. [Google Scholar] [CrossRef]
- Staretz-Chacham, O.; Wormser, O.; Manor, E.; Birk, O.S.; Ferreira, C.R. TMEM70 deficiency: Novel mutation and hypercitrullinemia during metabolic decompensation. Am. J. Med. Genet. A 2019, 179, 1293–1298. [Google Scholar] [CrossRef]
- Koňaříková, E.; Marković, A.; Korandová, Z.; Houštěk, J.; Mráček, T. Current progress in the therapeutic options for mitochondrial disorders. Physiol. Res. 2020, 69, 967–994. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Viscomi, C.; Zeviani, M. Strategies for fighting mitochondrial diseases. J. Intern. Med. 2020, 287, 665–684. [Google Scholar] [CrossRef]
- Di Meo, I.; Auricchio, A.; Lamperti, C.; Burlina, A.; Viscomi, C.; Zeviani, M. Effective AAV-mediated gene therapy in a mouse model of ethylmalonic encephalopathy. EMBO Mol. Med. 2012, 4, 1008–1014. [Google Scholar] [CrossRef]
- Torres-Torronteras, J.; Viscomi, C.; Cabrera-Pérez, R.; Cámara, Y.; Di Meo, I.; Barquinero, J.; Auricchio, A.; Pizzorno, G.; Hirano, M.; Zeviani, M.; et al. Gene Therapy Using a Liver-targeted AAV Vector Restores Nucleoside and Nucleotide Homeostasis in a Murine Model of MNGIE. Mol. Ther. 2014, 22, 901–907. [Google Scholar] [CrossRef] [Green Version]
- Yadak, R.; Cabrera-Pérez, R.; Torres-Torronteras, J.; Bugiani, M.; Haeck, J.C.; Huston, M.W.; Bogaerts, E.; Goffart, S.; Jacobs, E.H.; Stok, M.; et al. Preclinical Efficacy and Safety Evaluation of Hematopoietic Stem Cell Gene Therapy in a Mouse Model of MNGIE. Mol. Ther. Methods Clin. Dev. 2018, 8, 152–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertacchi, M.; Gruart, A.; Kaimakis, P.; Allet, C.; Serra, L.; Giacobini, P.; Delgado-García, J.M.; Bovolenta, P.; Studer, M. Mouse Nr2f1 haploinsufficiency unveils new pathological mechanisms of a human optic atrophy syndrome. EMBO Mol. Med. 2019, 11, e10291. [Google Scholar] [CrossRef] [PubMed]
- Sarzi, E.; Seveno, M.; Piro-Mégy, C.; Elzière, L.; Quilès, M.; Péquignot, M.; Müller, A.; Hamel, C.P.; Lenaers, G.; Delettre, C. OPA1 gene therapy prevents retinal ganglion cell loss in a Dominant Optic Atrophy mouse model. Sci. Rep. 2018, 8, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynaud-Dulaurier, R.; Benegiamo, G.; Marrocco, E.; Al-Tannir, R.; Surace, E.M.; Auwerx, J.; Decressac, M. Gene replacement therapy provides benefit in an adult mouse model of Leigh syndrome. Brain 2020, 143, 2468. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Cerutti, R.; Luna-Sanchez, M.; Zeviani, M.; Viscomi, C. A Single Intravenous Injection of AAV-PHP.B- hNDUFS4 Ameliorates the Phenotype of Ndufs4−/− Mice. Mol. Ther. Methods Clin. Dev. 2020, 17, 1071–1078. [Google Scholar] [CrossRef]
- Houštek, J.; Klement, P.; Floryk, D.; Antonicka, H.; Hermanská, J.; Kalous, M.; Hansíková, H.; Houšťková, H.; Chowdhury, S.K.R.; Rosipal, T.; et al. A novel deficiency of mitochondrial ATPase of nuclear origin. Hum. Mol. Genet. 1999, 8, 1967–1974. [Google Scholar] [CrossRef] [Green Version]
- Nůsková, H.; Mikesova, J.; Efimova, I.; Pecinova, A.; Pecina, P.; Drahota, Z.; Houstek, J.; Mracek, T. Biochemical thresholds for pathological presentation of ATP synthase deficiencies. Biochem. Biophys. Res. Commun. 2020, 521, 1036–1041. [Google Scholar] [CrossRef]
- Rossignol, R.; Letellier, T.; Malgat, M.; Rocher, C.; Mazat, J.P. Tissue variation in the control of oxidative phosphorylation: Implication for mitochondrial diseases. Biochem. J. 2000, 347 Pt 1, 45–53. [Google Scholar] [CrossRef]
- Rossignol, R.; Malgat, M.; Mazat, J.-P.; Letellier, T. Threshold effect and tissue specificity. Implication for mitochondrial cytopathies. J. Biol. Chem. 1999, 274, 33426–33432. [Google Scholar] [CrossRef] [Green Version]
- Jonckheere, A.I.; Huigsloot, M.; Lammens, M.; Jansen, J.; van den Heuvel, L.P.; Spiekerkoetter, U.; von Kleist-Retzow, J.-C.; Forkink, M.; Koopman, W.J.; Szklarczyk, R.; et al. Restoration of complex V deficiency caused by a novel deletion in the human TMEM70 gene normalizes mitochondrial morphology. Mitochondrion 2011, 11, 954–963. [Google Scholar] [CrossRef]
- Bader, M. Rat Models of Cardiovascular Diseases. Methods Mol. Biol. 2009, 597, 403–414. [Google Scholar] [CrossRef]
- Dillmann, W.H. The rat as a model for cardiovascular disease. Drug Discov. Today Dis. Model. 2008, 5, 173–178. [Google Scholar] [CrossRef]
- Liška, F.; Landa, V.; Zídek, V.; Mlejnek, P.; Šilhavý, J.; Šimáková, M.; Strnad, H.; Trnovská, J.; Škop, V.; Kazdová, L.; et al. Downregulation of Plzf Gene Ameliorates Metabolic and Cardiac Traits in the Spontaneously Hypertensive Rat. Hypertension 2017, 69, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Olson, E.; Pravenec, M.; Landa, V.; Koh-Tan, H.H.C.; Dominiczak, A.F.; McBride, M.W.; Graham, D. Transgenic overexpression of glutathione S-transferase μ-type 1 reduces hypertension and oxidative stress in the stroke-prone spontaneously hypertensive rat. J. Hypertens. 2019, 37, 985–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pravenec, M.; Kajiya, T.; Zídek, V.; Landa, V.; Mlejnek, P.; Simakova, M.; Šilhavý, J.; Malínská, H.; Oliyarnyk, O.; Kazdová, L.; et al. Effects of Human C-Reactive Protein on Pathogenesis of Features of the Metabolic Syndrome. Hypertension 2011, 57, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pravenec, M.; Kazdová, L.; Landa, V.; Zídek, V.; Mlejnek, P.; Jansa, P.; Wang, J.; Qi, N.; Kurtz, T.W. Transgenic and Recombinant Resistin Impair Skeletal Muscle Glucose Metabolism in the Spontaneously Hypertensive Rat. J. Biol. Chem. 2003, 278, 45209–45215. [Google Scholar] [CrossRef] [Green Version]
- Pravenec, M.; Křen, V.; Landa, V.; Mlejnek, P.; Musilová, A.; Šilhavý, J.; Šimáková, M.; Zídek, V. Recent Progress in the Genetics of Spontaneously Hypertensive Rats. Physiol. Res. 2014, 63, S1–S8. [Google Scholar] [CrossRef]
- Mráček, T.; Mikešová, J.; Kaplanová, V.; Pecina, P.; Šilhavý, J.; Mlejnek, P.; Šimáková, M.; Liška, F.; Marková, I.; Malínská, H.; et al. Downregulation of Tmem70 gene induces cardiac oxidative stress, change in fuel utilization and systolic left ventricle dysfunction in spontaneously hypertensive rats. Physiol Genom. 2021. Submitted. [Google Scholar]
- Ivics, Z.; Mátés, L.; Yau, T.Y.; Landa, V.; Zidek, V.; Bashir, S.; Hoffmann, O.I.; Hiripi, L.; Garrels, W.; Kues, W.A.; et al. Germline transgenesis in rodents by pronuclear microinjection of Sleeping Beauty transposons. Nat. Protoc. 2014, 9, 773–793. [Google Scholar] [CrossRef]
- Pecinová, A.; Drahota, Z.; Nůsková, H.; Pecina, P.; Houštěk, J. Evaluation of basic mitochondrial functions using rat tissue homogenates. Mitochondrion 2011, 11, 722–728. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.-P.; Schägger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Carrozzo, R.; Santorelli, F.M.; Schägger, H. Functional assays in high-resolution clear native gels to quantify mitochondrial complexes in human biopsies and cell lines. Electrophoresis 2007, 28, 3811–3820. [Google Scholar] [CrossRef] [PubMed]
- Pecinová, A.; Alán, L.; Brázdová, A.; Vrbacký, M.; Pecina, P.; Drahota, Z.; Houštěk, J.; Mráček, T. Role of Mitochondrial Glycerol-3-Phosphate Dehydrogenase in Metabolic Adaptations of Prostate Cancer. Cells 2020, 9, 1764. [Google Scholar] [CrossRef] [PubMed]
- Moradi-Ameli, M.; Godinot, C. Characterization of monoclonal antibodies against mitochondrial F1-ATPase. Proc. Natl. Acad. Sci. USA 1983, 80, 6167–6171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pecina, P.; Čapková, M.; Chowdhury, S.K.; Drahota, Z.; Dubot, A.; Vojtíšková, A.; Hansíková, H.; Houšťková, H.; Zeman, J.; Godinot, C.; et al. Functional alteration of cytochrome c oxidase by SURF1 mutations in Leigh syndrome. Biochim. Biophys. Acta 2003, 1639, 53–63. [Google Scholar] [CrossRef] [Green Version]
- Neckar, J.; Šilhavý, J.; Zídek, V.; Landa, V.; Mlejnek, P.; Simakova, M.; Seidman, J.G.; Seidman, C.; Kazdová, L.; Klevstig, M.; et al. CD36 overexpression predisposes to arrhythmias but reduces infarct size in spontaneously hypertensive rats: Gene expression profile analysis. Physiol. Genom. 2012, 44, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Coan, P.M.; Hummel, O.; Diaz, A.G.; Barrier, M.; Alfazema, N.; Norsworthy, P.J.; Pravenec, M.; Petretto, E.; Hubner, N.; Aitman, T.J. Genetic, physiological and comparative genomic studies of hypertension and insulin resistance in the spontaneously hypertensive rat. Dis. Model. Mech. 2017, 10, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.D.; Mueller, M.; Adamowicz-Brice, M.; Collins, M.J.; Gellert, P.; Maratou, K.; Srivastava, P.K.; Rotival, M.; Butt, S.; Game, L.; et al. Genetic Analysis of the Cardiac Methylome at Single Nucleotide Resolution in a Model of Human Cardiovascular Disease. PLoS Genet. 2014, 10, e1004813. [Google Scholar] [CrossRef] [Green Version]
- Pravenec, M.; Kožich, V.; Krijt, J.; Sokolová, J.; Zídek, V.; Landa, V.; Simakova, M.; Mlejnek, P.; Šilhavý, J.; Oliyarnyk, O.; et al. Folate Deficiency Is Associated with Oxidative Stress, Increased Blood Pressure, and Insulin Resistance in Spontaneously Hypertensive Rats. Am. J. Hypertens. 2013, 26, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Pravenec, M.; Zidek, V.; Simakova, M.; Kren, V.; Krenova, D.; Horky, K.; Jachymova, M.; Mikova, B.; Kazdova, L.; Aitman, T.J.; et al. Genetics of Cd36 and the clustering of multiple cardiovascular risk factors in spontaneous hypertension. J. Clin. Investig. 1999, 103, 1651–1657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schäfer, S.; Adami, E.; Heinig, M.; Rodrigues, K.E.C.; Kreuchwig, F.; Silhavy, J.; van Heesch, S.; Simaite, D.; Rajewsky, N.; Cuppen, E.; et al. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat. Commun. 2015, 6, 7200. [Google Scholar] [CrossRef] [PubMed]
- Pravenec, M.; Zídek, V.; Landa, V.; Mlejnek, P.; Šilhavý, J.; Simakova, M.; Trnovská, J.; Skop, V.; Markova, I.; Malínská, H.; et al. Mutant Wars2 Gene in Spontaneously Hypertensive Rats Impairs Brown Adipose Tissue Function and Predisposes to Visceral Obesity. Physiol. Res. 2017, 66, 917–924. [Google Scholar] [CrossRef]
- Katter, K.; Geurts, A.M.; Hoffmann, O.; Mátés, L.; Landa, V.; Hiripi, L.; Moreno, C.; Lazar, J.; Bashir, S.; Zidek, V.; et al. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J. 2013, 27, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Ali Hosseini Rad, S.M.; Poudel, A.; Tan, G.M.Y.; McLellan, A.D. Promoter choice: Who should drive the CAR in T cells? PLoS ONE 2020, 15, e0232915. [Google Scholar] [CrossRef]
- Pravenec, M.; Landa, V.; Zidek, V.; Musilova, A.; Kren, V.; Kazdova, L.; Aitman, T.J.; Glazier, A.M.; Ibrahimi, A.; Abumrad, N.A.; et al. Transgenic rescue of defective Cd36 ameliorates insulin resistance in spontaneously hypertensive rats. Nat. Genet. 2001, 27, 156–158. [Google Scholar] [CrossRef]
- Slone, J.; Huang, T. The special considerations of gene therapy for mitochondrial diseases. NPJ Genom. Med. 2020, 5, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Pereira, C.V.; Peralta, S.; Arguello, T.; Bacman, S.R.; Diaz, F.; Moraes, C.T. Myopathy reversion in mice after restauration of mitochondrial complex I. EMBO Mol. Med. 2020, 12, e10674. [Google Scholar] [CrossRef]
- Zhang, Y.; Tian, Z.; Yuan, J.; Liu, C.; Liu, H.L.; Ma, S.Q.; Li, B. The Progress of Gene Therapy for Leber’s Optic Hereditary Neuropathy. Curr. Gene Ther. 2017, 17, 320–326. [Google Scholar] [CrossRef]
- Ghezzi, D.; Zeviani, M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem. 2018, 62, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karbanová, V.H.; Vrbacká, A.; Hejzlarová, K.; Nůsková, H.; Stránecký, V.; Potocká, A.; Kmoch, S.; Houštěk, J. Compensatory upregulation of respiratory chain complexes III and IV in isolated deficiency of ATP synthase due to TMEM70 mutation. Biochim. Biophys. Acta 2012, 1817, 1037–1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Control | Tg/0 | |
|---|---|---|
| AWTd (mm) | 1.89 ± 0.05 | 1.79 ± 0.04 |
| LVDd (mm) | 7.50 ± 0.16 | 7.38 ± 0.18 |
| PWTd (mm) | 1.86 ± 0.06 | 1.76 ± 0.04 |
| AWTs (mm) | 2.86 ± 0.05 | 2.62 ± 0.02 * |
| LVDs (mm) | 4.46 ± 0.15 | 4.71 ± 0.16 |
| PWTs (mm) | 2.93 ± 0.10 | 2.65 ± 0.04 * |
| HR (b.p.m.) | 339 ± 17 | 343 ± 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marković, A.; Tauchmannová, K.; Šimáková, M.; Mlejnek, P.; Kaplanová, V.; Pecina, P.; Pecinová, A.; Papoušek, F.; Liška, F.; Šilhavý, J.; et al. Genetic Complementation of ATP Synthase Deficiency Due to Dysfunction of TMEM70 Assembly Factor in Rat. Biomedicines 2022, 10, 276. https://doi.org/10.3390/biomedicines10020276
Marković A, Tauchmannová K, Šimáková M, Mlejnek P, Kaplanová V, Pecina P, Pecinová A, Papoušek F, Liška F, Šilhavý J, et al. Genetic Complementation of ATP Synthase Deficiency Due to Dysfunction of TMEM70 Assembly Factor in Rat. Biomedicines. 2022; 10(2):276. https://doi.org/10.3390/biomedicines10020276
Chicago/Turabian StyleMarković, Aleksandra, Kateřina Tauchmannová, Miroslava Šimáková, Petr Mlejnek, Vilma Kaplanová, Petr Pecina, Alena Pecinová, František Papoušek, František Liška, Jan Šilhavý, and et al. 2022. "Genetic Complementation of ATP Synthase Deficiency Due to Dysfunction of TMEM70 Assembly Factor in Rat" Biomedicines 10, no. 2: 276. https://doi.org/10.3390/biomedicines10020276