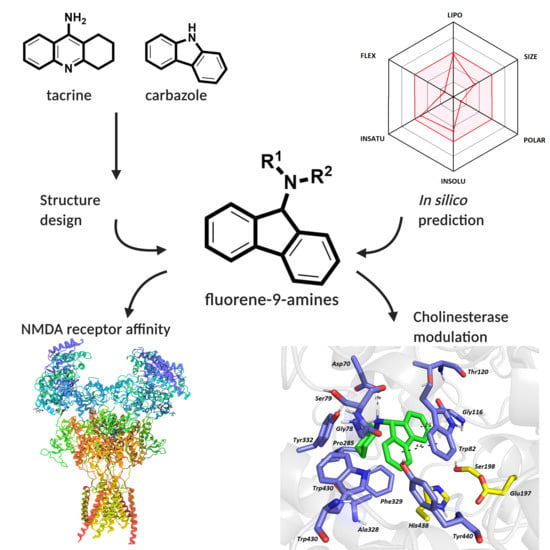
Pursuing the Complexity of Alzheimer’s Disease: Discovery of Fluoren-9-Amines as Selective Butyrylcholinesterase Inhibitors and N-Methyl-d-Aspartate Receptor Antagonists

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Chemistry
2.2. General Procedure for the Preparation of 9-Bromofluorene hydrochlorides (3a–o)
2.2.1. N-Propan-2-yl-9H-fluoren-9-amine hydrochloride (3a)
2.2.2. N-Methyl-9H-fluoren-9-amine hydrochloride (3b)
2.2.3. N-Cyclohexyl-9H-fluoren-9-amine hydrochloride (3c)
2.2.4. N-Cyclopropyl-9H-fluoren-9-amine hydrochloride (3d)
2.2.5. N-Cyclobutyl-9H-fluoren-9-amine hydrochloride (3e)
2.2.6. 1-(9H-fluoren-9-yl)piperidine hydrochloride (3f)
2.2.7. N-(2-methoxyethyl)-9H-fluoren-9-amine hydrochloride (3g)
2.2.8. N-Ethyl-9H-fluoren-9-amine hydrochloride (3h)
2.2.9. N,N-Diethyl-9H-fluoren-9-amine hydrochloride (3i)
2.2.10. 1-(9H-fluorene-9-yl)-4-methylpiperazine dihydrochloride (3j)
2.2.11. 1-(9H-fluorene-9-yl)-4-ethylpiperazine dihydrochloride (3k)
2.2.12. 4-(9H-fluorene-9-yl)morpholine hydrochloride (3l)
2.2.13. N-Butyl-9H-fluoren-9-amine hydrochloride (3m)
2.2.14. N-(2-methylpropyl)-9H-fluoren-9-amine hydrochloride (3n)
2.2.15. 1-(9H-fluoren-9-yl)pyrrolidine hydrochloride (3o)
2.3. In Vitro Anti-ChE Assay
2.4. Kinetic Study of hBChE Inhibition
2.5. Antagonist Activity Towards the NMDA receptor
2.5.1. HEK293 Cell Culture and Transfection
2.5.2. Electrophysiology
2.6. In Vitro Cell Viability Assessment
2.7. Determination of In Vitro BBB Permeation
- A: area of the well/cell monolayer,
- dC/dt: amount in the receiver compartment in given time,
- Vr: volume of the receiver compartment, and
- C0: the initial concentration of tested compounds.
2.8. Molecular Modeling Studies
2.9. In Silico Pharmacokinetics and Drug-Likeness Prediction
3. Results and Discussion
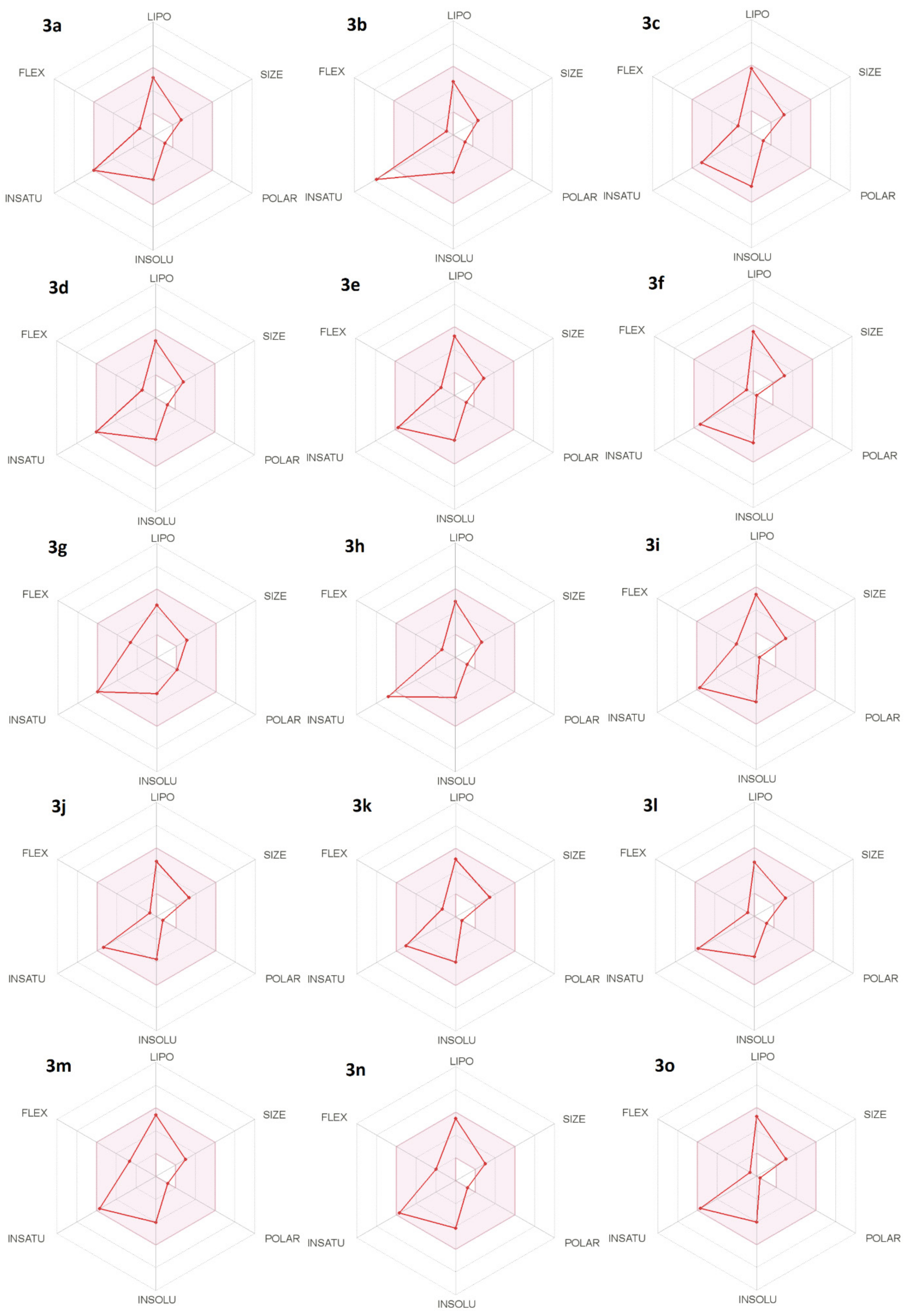
3.1. In Silico Prediction of the CNS and Oral Availability
3.2. Synthesis
3.3. Evaluation of Cholinesterase Inhibitory Activity
3.4. Kinetic Study of hBChE Inhibition
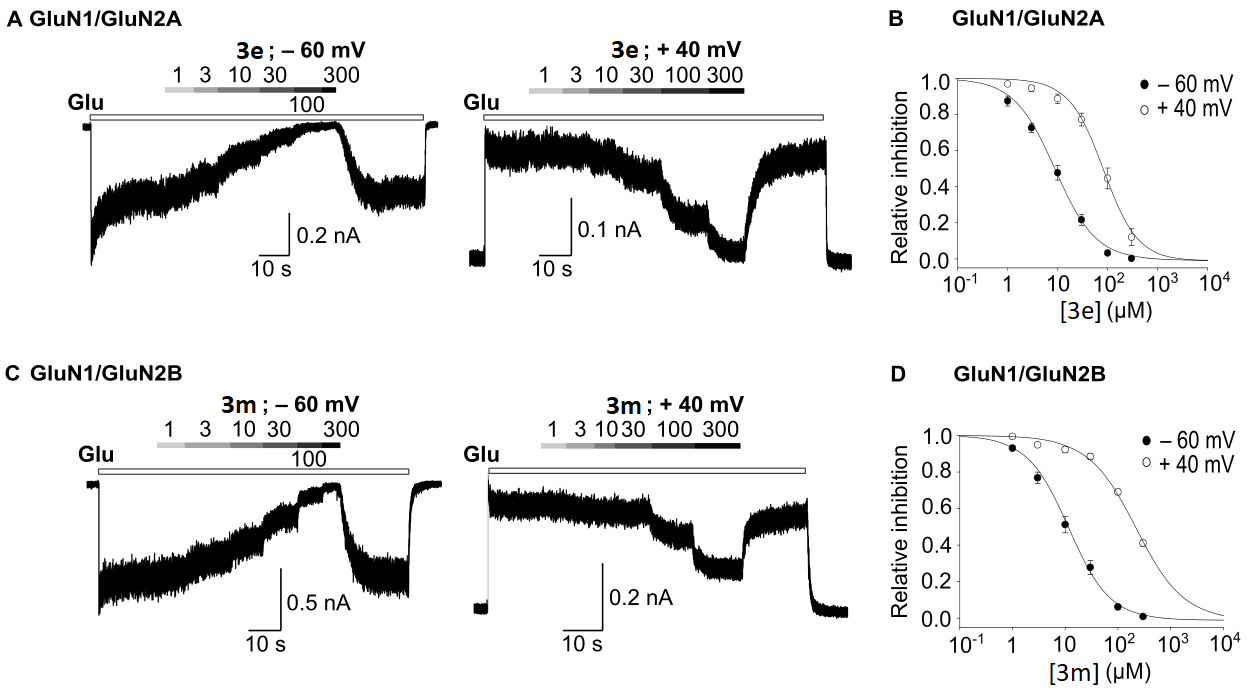
3.5. Evaluation of Antagonist Activity towards the NMDA Receptor
3.6. In Vitro Cell Viability Assessment
3.7. In Vitro BBB Permeation
3.8. In Silico Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferri, C.P.; Prince, M.; Brayne, C.; Brodaty, H.; Fratiglioni, L.; Ganguli, M.; Hall, K.; Hasegawa, K.; Hendrie, H.; Huang, Y.; et al. Global Prevalence of Dementia: A Delphi Consensus Study. Lancet 2005, 366, 2112–2117. [Google Scholar] [CrossRef]
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. 2017, 23, 818–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirova, A.-M.; Bays, R.B.; Lagalwar, S. Working Memory and Executive Function Decline across Normal Aging, Mild Cognitive Impairment, and Alzheimer’s Disease. Biomed. Res. Int. 2015, 2015, 748212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimers Dement 2011, 7, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Weller, J.; Budson, A. Current Understanding of Alzheimer’s Disease Diagnosis and Treatment. F1000Res 2018, 7. [Google Scholar] [CrossRef] [Green Version]
- Schachter, A.S.; Davis, K.L. Alzheimer’s Disease. Dialogues Clin. Neurosci. 2000, 2, 91–100. [Google Scholar] [CrossRef]
- 2020 Alzheimer’s Disease Facts and Figures. Alzheimer’s Dement. 2020, 16, 391–460. [CrossRef]
- Vaz, M.; Silvestre, S. Alzheimer’s Disease: Recent Treatment Strategies. Eur. J. Pharm. 2020, 887, 173554. [Google Scholar] [CrossRef]
- Abeysinghe, A.A.D.T.; Deshapriya, R.D.U.S.; Udawatte, C. Alzheimer’s Disease; a Review of the Pathophysiological Basis and Therapeutic Interventions. Life Sci. 2020, 256, 117996. [Google Scholar] [CrossRef]
- Goedert, M.; Crowther, R.A. Amyloid Plaques, Neurofibrillary Tangles and Their Relevance for the Study of Alzheimer’s Disease. Neurobiol. Aging 1989, 10, 405–406; discussion 412–414. [Google Scholar] [CrossRef]
- Lewis, D.A.; Higgins, G.A.; Young, W.G.; Goldgaber, D.; Gajdusek, D.C.; Wilson, M.C.; Morrison, J.H. Distribution of Precursor Amyloid-Beta-Protein Messenger RNA in Human Cerebral Cortex: Relationship to Neurofibrillary Tangles and Neuritic Plaques. Proc. Natl. Acad. Sci. USA 1988, 85, 1691–1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondragón-Rodríguez, S.; Basurto-Islas, G.; Santa-Maria, I.; Mena, R.; Binder, L.I.; Avila, J.; Smith, M.A.; Perry, G.; García-Sierra, F. Cleavage and Conformational Changes of Tau Protein Follow Phosphorylation during Alzheimer’s Disease. Int. J. Exp. Pathol. 2008, 89, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Mena, R.; Binder, L.I.; Smith, M.A.; Perry, G.; García-Sierra, F. Conformational Changes and Cleavage of Tau in Pick Bodies Parallel the Early Processing of Tau Found in Alzheimer Pathology. Neuropathol. Appl. Neurobiol. 2008, 34, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Mondragón-Rodríguez, S.; Perry, G.; Luna-Muñoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of Tau Protein at Sites Ser(396-404) Is One of the Earliest Events in Alzheimer’s Disease and Down Syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective Loss of Central Cholinergic Neurons in Alzheimer’s Disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Sarter, M.; Bruno, J.P. Cognitive Functions of Cortical Acetylcholine: Toward a Unifying Hypothesis. Brain Res. Brain Res. Rev. 1997, 23, 28–46. [Google Scholar] [CrossRef]
- Hasselmo, M.E.; Anderson, B.P.; Bower, J.M. Cholinergic Modulation of Cortical Associative Memory Function. J. Neurophysiol. 1992, 67, 1230–1246. [Google Scholar] [CrossRef]
- Wang, R.; Reddy, P.H. Role of Glutamate and NMDA Receptors in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1041–1048. [Google Scholar] [CrossRef] [Green Version]
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative Stress and the Amyloid Beta Peptide in Alzheimer’s Disease. Redox Biol. 2018, 14, 450–464. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Lauderback, C.M. Lipid Peroxidation and Protein Oxidation in Alzheimer’s Disease Brain: Potential Causes and Consequences Involving Amyloid Beta-Peptide-Associated Free Radical Oxidative Stress. Free Radic. Biol. Med. 2002, 32, 1050–1060. [Google Scholar] [CrossRef]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.d.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid Induction of Alzheimer A Beta Amyloid Formation by Zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Bolós, M.; Perea, J.R.; Avila, J. Alzheimer’s Disease as an Inflammatory Disease. Biomol. Concepts 2017, 8, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L.; Tong, G.; Ballard, C. Treatment Combinations for Alzheimer’s Disease: Current and Future Pharmacotherapy Options. J. Alzheimers Dis. 2019, 67, 779–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joe, E.; Ringman, J.M. Cognitive Symptoms of Alzheimer’s Disease: Clinical Management and Prevention. BMJ 2019, 367, l6217. [Google Scholar] [CrossRef] [Green Version]
- Zemek, F.; Drtinova, L.; Nepovimova, E.; Sepsova, V.; Korabecny, J.; Klimes, J.; Kuca, K. Outcomes of Alzheimer’s Disease Therapy with Acetylcholinesterase Inhibitors and Memantine. Expert Opin. Drug Saf. 2014, 13, 759–774. [Google Scholar] [CrossRef]
- Owen, R.T. Memantine and Donepezil: A Fixed Drug Combination for the Treatment of Moderate to Severe Alzheimer’s Dementia. Drugs Today 2016, 52, 239–248. [Google Scholar] [CrossRef]
- Calhoun, A.; King, C.; Khoury, R.; Grossberg, G.T. An Evaluation of Memantine ER + Donepezil for the Treatment of Alzheimer’s Disease. Expert Opin. Pharm. 2018, 19, 1711–1717. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands to Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-Target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- Cummings, J.L.; Morstorf, T.; Zhong, K. Alzheimer’s Disease Drug-Development Pipeline: Few Candidates, Frequent Failures. Alzheimer’s Res. Ther. 2014, 6, 37. [Google Scholar] [CrossRef] [Green Version]
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking Pathophysiological Processes in Alzheimer’s Disease: An Updated Hypothetical Model of Dynamic Biomarkers. Lancet Neurol. 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Panza, F.; Lozupone, M.; Watling, M.; Imbimbo, B.P. Do BACE Inhibitor Failures in Alzheimer Patients Challenge the Amyloid Hypothesis of the Disease? Expert Rev. Neurother. 2019, 19, 599–602. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Therapeutics of Neurotransmitters in Alzheimer’s Disease. J. Alzheimers Dis. 2017, 57, 1049–1069. [Google Scholar] [CrossRef] [Green Version]
- Fitzpatrick-Lewis, D.; Warren, R.; Ali, M.U.; Sherifali, D.; Raina, P. Treatment for Mild Cognitive Impairment: A Systematic Review and Meta-Analysis. CMAJ Open 2015, 3, E419–E427. [Google Scholar] [CrossRef] [Green Version]
- Watkins, P.B.; Zimmerman, H.J.; Knapp, M.J.; Gracon, S.I.; Lewis, K.W. Hepatotoxic Effects of Tacrine Administration in Patients With Alzheimer’s Disease. JAMA 1994, 271, 992–998. [Google Scholar] [CrossRef]
- Horak, M.; Holubova, K.; Nepovimova, E.; Krusek, J.; Kaniakova, M.; Korabecny, J.; Vyklicky, L.; Kuca, K.; Stuchlik, A.; Ricny, J.; et al. The Pharmacology of Tacrine at N-Methyl-d-Aspartate Receptors. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 75, 54–62. [Google Scholar] [CrossRef]
- Kaniakova, M.; Kleteckova, L.; Lichnerova, K.; Holubova, K.; Skrenkova, K.; Korinek, M.; Krusek, J.; Smejkalova, T.; Korabecny, J.; Vales, K.; et al. 7-Methoxyderivative of Tacrine Is a “foot-in-the-Door” Open-Channel Blocker of GluN1/GluN2 and GluN1/GluN3 NMDA Receptors with Neuroprotective Activity in Vivo. Neuropharmacology 2018, 140, 217–232. [Google Scholar] [CrossRef]
- Vorobjev, V.S.; Sharonova, I.N. Tetrahydroaminoacridine Blocks and Prolongs NMDA Receptor-Mediated Responses in a Voltage-Dependent Manner. Eur. J. Pharmacol. 1994, 253, 1–8. [Google Scholar] [CrossRef]
- Nelson, M.E.; Albuquerque, E.X. 9-Aminoacridines Act at a Site Different from That for Mg2+ in Blockade of the N-Methyl-D-Aspartate Receptor Channel. Mol. Pharmacol. 1994, 46, 151–160. [Google Scholar] [PubMed]
- Soukup, O.; Jun, D.; Zdarova-Karasova, J.; Patocka, J.; Musilek, K.; Korabecny, J.; Krusek, J.; Kaniakova, M.; Sepsova, V.; Mandikova, J.; et al. A Resurrection of 7-MEOTA: A Comparison with Tacrine. Curr. Alzheimer Res. 2013, 10, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Choubdar, N.; Golshani, M.; Jalili-Baleh, L.; Nadri, H.; Küçükkilinç, T.T.; Ayazgök, B.; Moradi, A.; Moghadam, F.H.; Abdolahi, Z.; Ameri, A.; et al. New Classes of Carbazoles as Potential Multi-Functional Anti-Alzheimer’s Agents. Bioorg. Chem. 2019, 91, 103164. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Chen, M.; Li, M.; Luo, B.; Zhao, Y.; Huang, P.; Xue, F.; Rapposelli, S.; Pi, R.; Wen, S. Discovery of Novel N-Substituted Carbazoles as Neuroprotective Agents with Potent Anti-Oxidative Activity. Eur. J. Med. Chem. 2013, 68, 81–88. [Google Scholar] [CrossRef]
- Crismon, M.L. Tacrine: First Drug Approved for Alzheimer’s Disease. Ann. Pharm. 1994, 28, 744–751. [Google Scholar] [CrossRef]
- Ellman, G.L.; Courtney, K.D.; Andres, V.; Feather-Stone, R.M. A New and Rapid Colorimetric Determination of Acetylcholinesterase Activity. Biochem. Pharm. 1961, 7, 88–95. [Google Scholar] [CrossRef]
- Pohanka, M.; Jun, D.; Kuca, K. Improvement of Acetylcholinesterase-Based Assay for Organophosphates in Way of Identification by Reactivators. Talanta 2008, 77, 451–454. [Google Scholar] [CrossRef]
- Sepsova, V.; Karasova, J.Z.; Korabecny, J.; Dolezal, R.; Zemek, F.; Bennion, B.J.; Kuca, K. Oximes: Inhibitors of Human Recombinant Acetylcholinesterase. A Structure-Activity Relationship (SAR) Study. Int. J. Mol. Sci. 2013, 14, 16882–16900. [Google Scholar] [CrossRef] [Green Version]
- Skrenkova, K.; Hemelikova, K.; Kolcheva, M.; Kortus, S.; Kaniakova, M.; Krausova, B.; Horak, M. Structural Features in the Glycine-Binding Sites of the GluN1 and GluN3A Subunits Regulate the Surface Delivery of NMDA Receptors. Sci. Rep. 2019, 9, 12303. [Google Scholar] [CrossRef] [Green Version]
- Kaniakova, M.; Lichnerova, K.; Vyklicky, L.; Horak, M. Single Amino Acid Residue in the M4 Domain of GluN1 Subunit Regulates the Surface Delivery of NMDA Receptors. J. Neurochem. 2012, 123, 385–395. [Google Scholar] [CrossRef]
- Vyklicky, V.; Krausova, B.; Cerny, J.; Ladislav, M.; Smejkalova, T.; Kysilov, B.; Korinek, M.; Danacikova, S.; Horak, M.; Chodounska, H.; et al. Surface Expression, Function, and Pharmacology of Disease-Associated Mutations in the Membrane Domain of the Human GluN2B Subunit. Front. Mol. Neurosci. 2018, 11, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gazova, Z.; Soukup, O.; Sepsova, V.; Siposova, K.; Drtinova, L.; Jost, P.; Spilovska, K.; Korabecny, J.; Nepovimova, E.; Fedunova, D.; et al. Multi-Target-Directed Therapeutic Potential of 7-Methoxytacrine-Adamantylamine Heterodimers in the Alzheimer’s Disease Treatment. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Hemelíková, K.; Kolcheva, M.; Skrenkova, K.; Kaniaková, M.; Horák, M. Lectins Modulate the Functional Properties of GluN1/GluN3-Containing NMDA Receptors. Neuropharmacology 2019. [Google Scholar] [CrossRef] [PubMed]
- Kaniakova, M.; Nepovimova, E.; Kleteckova, L.; Skrenkova, K.; Holubova, K.; Chrienova, Z.; Hepnarova, V.; Kucera, T.; Kobrlova, T.; Vales, K.; et al. Combination of Memantine and 6-Chlorotacrine as Novel Multi-Target Compound against Alzheimer’s Disease. Curr. Alzheimer Res. 2019, 16, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Soukup, O.; Benkova, M.; Dolezal, R.; Sleha, R.; Malinak, D.; Salajkova, S.; Markova, A.; Hympanova, M.; Prchal, L.; Ryskova, L.; et al. The Wide-Spectrum Antimicrobial Effect of Novel N-Alkyl Monoquaternary Ammonium Salts and Their Mixtures; the QSAR Study against Bacteria. Eur. J. Med. Chem. 2020, 206, 112584. [Google Scholar] [CrossRef]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.-Y. Crystal Structures of Human Cholinesterases in Complex with Huprine W and Tacrine: Elements of Specificity for Anti-Alzheimer’s Drugs Targeting Acetyl- and Butyryl-Cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [Green Version]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Spilovska, K.; Korabecny, J.; Sepsova, V.; Jun, D.; Hrabinova, M.; Jost, P.; Muckova, L.; Soukup, O.; Janockova, J.; Kucera, T.; et al. Novel Tacrine-Scutellarin Hybrids as Multipotent Anti-Alzheimer’s Agents: Design, Synthesis and Biological Evaluation. Molecules 2017, 22, 1006. [Google Scholar] [CrossRef] [Green Version]
- Mezeiova, E.; Korabecny, J.; Sepsova, V.; Hrabinova, M.; Jost, P.; Muckova, L.; Kucera, T.; Dolezal, R.; Misik, J.; Spilovska, K.; et al. Development of 2-Methoxyhuprine as Novel Lead for Alzheimer’s Disease Therapy. Molecules 2017, 22, 1265. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Wang, L.; Jin, Y.-H. An Effective PSO-Based Memetic Algorithm for Flow Shop Scheduling. IEEE Trans. Syst. ManCybern. Part B (Cybern.) 2007, 37, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood-Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. ILOGP: A Simple, Robust, and Efficient Description of n-Octanol/Water Partition Coefficient for Drug Design Using the GB/SA Approach. J. Chem. Inf. Model. 2014, 54, 3284–3301. [Google Scholar] [CrossRef] [PubMed]
- Naz, A.; Iqtadar, R.; Siddiqui, F.A.; Ul-Haq, Z. Degradation Kinetics of Fluvoxamine in Buffer Solutions: In Silico ADMET Profiling and Identification of Degradation Products by LC-MS/ESI. Arab. J. Chem. 2020, 13, 4134–4146. [Google Scholar] [CrossRef]
- Madden, S.; Spaldin, V.; Park, B.K. Clinical Pharmacokinetics of Tacrine. Clin. Pharm. 1995, 28, 449–457. [Google Scholar] [CrossRef]
- Liu, M.-Y.; Meng, S.-N.; Wu, H.-Z.; Wang, S.; Wei, M.-J. Pharmacokinetics of Single-Dose and Multiple-Dose Memantine in Healthy Chinese Volunteers Using an Analytic Method of Liquid Chromatography-Tandem Mass Spectrometry. Clin. Ther. 2008, 30, 641–653. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Martin, Y.C. A Bioavailability Score. J. Med. Chem. 2005, 48, 3164–3170. [Google Scholar] [CrossRef]
- Daina, A.; Zoete, V. A BOILED-Egg to Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules. ChemMedChem 2016, 11, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Bade, R.; Chan, H.-F.; Reynisson, J. Characteristics of Known Drug Space. Natural Products, Their Derivatives and Synthetic Drugs. Eur. J. Med. Chem. 2010, 45, 5646–5652. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple Selection Criteria for Drug-like Chemical Matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Pohanka, M.; Karasova, J.Z.; Kuca, K.; Pikula, J.; Holas, O.; Korabecny, J.; Cabal, J. Colorimetric Dipstick for Assay of Organophosphate Pesticides and Nerve Agents Represented by Paraoxon, Sarin and VX. Talanta 2010, 81, 621–624. [Google Scholar] [CrossRef]
- Nordberg, A.; Ballard, C.; Bullock, R.; Darreh-Shori, T.; Somogyi, M. A Review of Butyrylcholinesterase as a Therapeutic Target in the Treatment of Alzheimer’s Disease. Prim. Care Companion CNS Disord. 2013, 15. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.-S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective Butyrylcholinesterase Inhibition Elevates Brain Acetylcholine, Augments Learning and Lowers Alzheimer Beta-Amyloid Peptide in Rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef] [Green Version]
- Nepovimova, E.; Korabecny, J.; Dolezal, R.; Babkova, K.; Ondrejicek, A.; Jun, D.; Sepsova, V.; Horova, A.; Hrabinova, M.; Soukup, O.; et al. Tacrine-Trolox Hybrids: A Novel Class of Centrally Active, Nonhepatotoxic Multi-Target-Directed Ligands Exerting Anticholinesterase and Antioxidant Activities with Low In Vivo Toxicity. J. Med. Chem. 2015, 58, 8985–9003. [Google Scholar] [CrossRef]
- Sobolova, K.; Hrabinova, M.; Hepnarova, V.; Kucera, T.; Kobrlova, T.; Benkova, M.; Janockova, J.; Dolezal, R.; Prchal, L.; Benek, O.; et al. Discovery of Novel Berberine Derivatives with Balanced Cholinesterase and Prolyl Oligopeptidase Inhibition Profile. Eur. J. Med. Chem. 2020, 203, 112593. [Google Scholar] [CrossRef]
- Chalupova, K.; Korabecny, J.; Bartolini, M.; Monti, B.; Lamba, D.; Caliandro, R.; Pesaresi, A.; Brazzolotto, X.; Gastellier, A.-J.; Nachon, F.; et al. Novel Tacrine-Tryptophan Hybrids: Multi-Target Directed Ligands as Potential Treatment for Alzheimer’s Disease. Eur. J. Med. Chem. 2019, 168, 491–514. [Google Scholar] [CrossRef] [PubMed]
- Weksler, B.; Romero, I.A.; Couraud, P.-O. The HCMEC/D3 Cell Line as a Model of the Human Blood Brain Barrier. Fluids Barriers CNS 2013, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium Oligomannate Therapeutically Remodels Gut Microbiota and Suppresses Gut Bacterial Amino Acids-Shaped Neuroinflammation to Inhibit Alzheimer’s Disease Progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | GIA a | BBB b | BBB Score c | Lipinski d | Bio. Score e |
|---|---|---|---|---|---|
| 3a | High | Yes | 5.63 | Yes | 0.55 |
| 3b | High | Yes | 5.63 | Yes | 0.55 |
| 3c | High | Yes | 5.59 | Yes | 0.55 |
| 3d | High | Yes | 5.64 | Yes | 0.55 |
| 3e | High | Yes | 5.93 | Yes | 0.55 |
| 3f | High | Yes | 5.15 | Yes | 0.55 |
| 3g | High | Yes | 5.68 | Yes | 0.55 |
| 3h | High | Yes | 5.63 | Yes | 0.55 |
| 3i | High | Yes | 5.17 | Yes | 0.55 |
| 3j | High | Yes | 5.67 | Yes | 0.55 |
| 3k | High | Yes | 5.86 | Yes | 0.55 |
| 3l | High | Yes | 5.55 | Yes | 0.55 |
| 3m | High | Yes | 5.62 | Yes | 0.55 |
| 3n | High | Yes | 5.62 | Yes | 0.55 |
| 3o | High | Yes | 5.72 | Yes | 0.55 |
| THA | High | Yes | 5.38 | Yes | 0.55 |
| Memantine | High | Yes | 4.61 | Yes | 0.55 |
| Compound | IC50 ± SEM (µM) a | SI for BChE b | |
|---|---|---|---|
| hAChE | hBChE | ||
| 3a | n.a. | 2.95 ± 0,09 | - |
| 3b | n.a. | 8.91 ± 0.31 | - |
| 3c | 13.98 ± 0.35 | 0.47 ± 0.01 | 29.7 |
| 3d | n.a. | 6.42 ± 0.22 | - |
| 3e | n.a. | 2.03 ± 0.08 | - |
| 3f | n.a. | 2.13 ± 0.06 | - |
| 3g | n.a. | 2.56 ± 0.11 | - |
| 3h | n.a. | 6.58 ± 0.19 | - |
| 3i | n.a. | 5.38 ± 0.11 | - |
| 3j | n.a. | 23.5 ± 0.8 | - |
| 3k | n.a. | 12.8 ± 0.3 | - |
| 3l | n.a. | 54.7 ± 1.9 | - |
| 3m | 49.91 ± 3.05 | 1.02 ± 0.03 | 48.9 |
| 3n | n.a. | 0.90 ± 0,02 | - |
| 3o | n.a. | 1.80 ± 0.06 | - |
| THA c | 0.50 ± 0.10 c | 0.023 ± 0.003 c | 21.7 |
| Compound | GluN1/GluN2A | GluN1/GluN2B | ||
|---|---|---|---|---|
| RIa (%) ± SEM | n | RIa (%) ± SEM | n | |
| 3a | 33.29 ± 1.66 | 4 | 25.00 ± 1.82 | 4 |
| 3b | 36.16 ± 2.16 | 4 | 18.01 ± 0.78 | 4 |
| 3c | 41.02 ± 3.56 | 5 | 17.24 ± 2.39 | 4 |
| 3d | 26.56 ± 0.85 | 5 | 14.26 ± 0.35 | 5 |
| 3e | 52.30 ± 4.18 | 5 | 44.79 ± 3.43 | 5 |
| 3f | 33.01 ± 1.96 | 5 | 19.35 ± 2.65 | 5 |
| 3g | 18.68 ± 1.98 | 5 | 6.76 ± 1.00 | 4 |
| 3h | 26.90 ± 2.14 | 5 | 17.48 ± 2.46 | 5 |
| 3i | 25.66 ± 1.92 | 5 | 20.12 ± 2.82 | 5 |
| 3j | 29.15 ± 2.85 | 4 | 21.88 ± 2.10 | 4 |
| 3k | 32.08 ± 1.01 | 4 | 19.12 ± 1.48 | 6 |
| 3l | 14.89 ± 1.76 | 4 | 11.64 ± 0.52 | 5 |
| 3m | 36.23 ± 3.73 | 6 | 51.22 ± 3.24 | 5 |
| 3n | 37.87 ± 2.43 | 6 | 49.71 ± 1.72 | 5 |
| 3o | 27.80 ± 1.74 | 5 | 32.39 ± 2.99 | 5 |
| GluN1/GluN2A a | GluN1/GluN2B a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| IC50 −60 mV | h −60 mV | IC50 40 mV | h 40 mV | n (−60/40 mV) | IC50 −60 mV | h −60 mV | IC50 40 mV | h 40 mV | n (−60/40 mV) | |
| 3e | 8.85 ± 1.21 | 1.07 ± 0.05 | 82.62 ± 14.17 | 1.28 ± 0.12 | 8/5 | 15.27 ± 2.38 | 1.17 ± 0.05 | 156.40 ± 14.94 | 1.10 ± 0.08 | 12/7 |
| 3m | 14.21 ± 1.60 | 1.19 ± 0.14 | 113.99 ± 26.63 | 1.23 ± 0.13 | 5/4 | 11.67 ± 1.72 | 1.12 ± 0.05 | 220.78 ± 12.24 | 0.97 ± 0.10 | 9/6 |
| THA b | 9.1 ± 0.5 | 1.7 ± 0.0 | 84.6 ± 1.6 | 1.6 ± 0.1 | 8/4 | 19.7 ± 1.8 | 1.8 ± 0.1 | 168.8 ± 9.3 | 1.4 ± 0.1 | 9/6 |
| Mem b | 1.34 ± 0.08 | 1.04 ± 0.06 | 27.04 ± 1.19 | 1.16 ± 0.05 | 6/7 | 0.78 ± 0.09 | 1.04 ± 0.03 | 10.57 ± 0.63 | 0.84 ± 0.04 | 5/6 |
| Compound. | IC50 ± SEM (µM) a | ClogP |
|---|---|---|
| 3a | 387 ± 32 | 3.76 |
| 3b | 427 ± 12 | 2.99 |
| 3c | 111 ± 10 | 4.79 |
| 3d | 353 ± 32 | 3.46 |
| 3e | 192 ± 3 | 3.90 |
| 3f | 872 ± 10 | 4.22 |
| 3g | 738 ± 1 | 2.94 |
| 3h | 417 ± 16 | 3.35 |
| 3i | 266 ± 3 | 4.09 |
| 3j | 259 ± 12 | 3.22 |
| 3k | 239 ± 35 | 3.58 |
| 3l | >800 b | 3.15 |
| 3m | 140 ± 15 | 4.31 |
| 3n | 236 ± 24 | 4.29 |
| 3o | 413 ± 15 | 3.78 |
| THA | 248 ± 11 c | 2.63 |
| Compound | Papp ± SEM (×10−6 cm s−1) | CNS (+/−) a | N b |
|---|---|---|---|
| 3e | 13.51 ± 1.54 | CNS − | 3 |
| 3n | 14.61 ± 1.88 | CNS − | 3 |
| 3c | 12.28 ± 2.33 | CNS − | 3 |
| 3m | 18.48 ± 2.37 | CNS + | 4 |
| THA | 22.29 ± 2.01 | CNS + | 4 |
| Donepezil | 24.19 ± 2.83 | CNS + | 4 |
| Rivastigmine | 28.63 ± 4.64 | CNS + | 5 |
| Propranolol | 23.06 ± 2.06 | CNS + | 5 |
| Antipyrine | 17.19 ± 1.82 | CNS + | 3 |
| Furosemide | 13.89 ± 0.92 | CNS − | 3 |
| Obidoxime | 14.35 ± 0.52 | CNS − | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Konecny, J.; Misiachna, A.; Hrabinova, M.; Pulkrabkova, L.; Benkova, M.; Prchal, L.; Kucera, T.; Kobrlova, T.; Finger, V.; Kolcheva, M.; et al. Pursuing the Complexity of Alzheimer’s Disease: Discovery of Fluoren-9-Amines as Selective Butyrylcholinesterase Inhibitors and N-Methyl-d-Aspartate Receptor Antagonists. Biomolecules 2021, 11, 3. https://doi.org/10.3390/biom11010003
Konecny J, Misiachna A, Hrabinova M, Pulkrabkova L, Benkova M, Prchal L, Kucera T, Kobrlova T, Finger V, Kolcheva M, et al. Pursuing the Complexity of Alzheimer’s Disease: Discovery of Fluoren-9-Amines as Selective Butyrylcholinesterase Inhibitors and N-Methyl-d-Aspartate Receptor Antagonists. Biomolecules. 2021; 11(1):3. https://doi.org/10.3390/biom11010003
Chicago/Turabian StyleKonecny, Jan, Anna Misiachna, Martina Hrabinova, Lenka Pulkrabkova, Marketa Benkova, Lukas Prchal, Tomas Kucera, Tereza Kobrlova, Vladimir Finger, Marharyta Kolcheva, and et al. 2021. "Pursuing the Complexity of Alzheimer’s Disease: Discovery of Fluoren-9-Amines as Selective Butyrylcholinesterase Inhibitors and N-Methyl-d-Aspartate Receptor Antagonists" Biomolecules 11, no. 1: 3. https://doi.org/10.3390/biom11010003