
Computational Investigations on the Natural Small Molecule as an Inhibitor of Programmed Death Ligand 1 for Cancer Immunotherapy

 , and
, and
Abstract
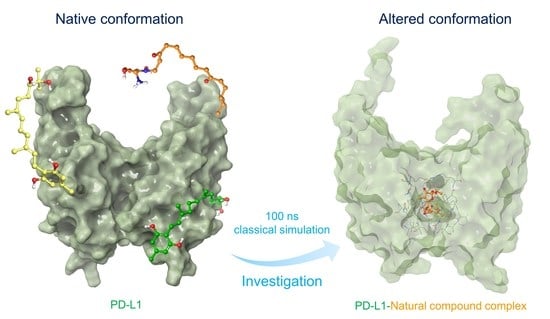
:
1. Introduction
2. Methods
2.1. Receptor and Ligand Library Collection
2.2. Multi-Step Virtual Screening and Pose Filtration
2.3. Molecular Dynamic Simulation
2.4. End-Point Binding Free Energy Calculation
3. Result and Discussion
3.1. Virtual Screening and ADMET Analysis
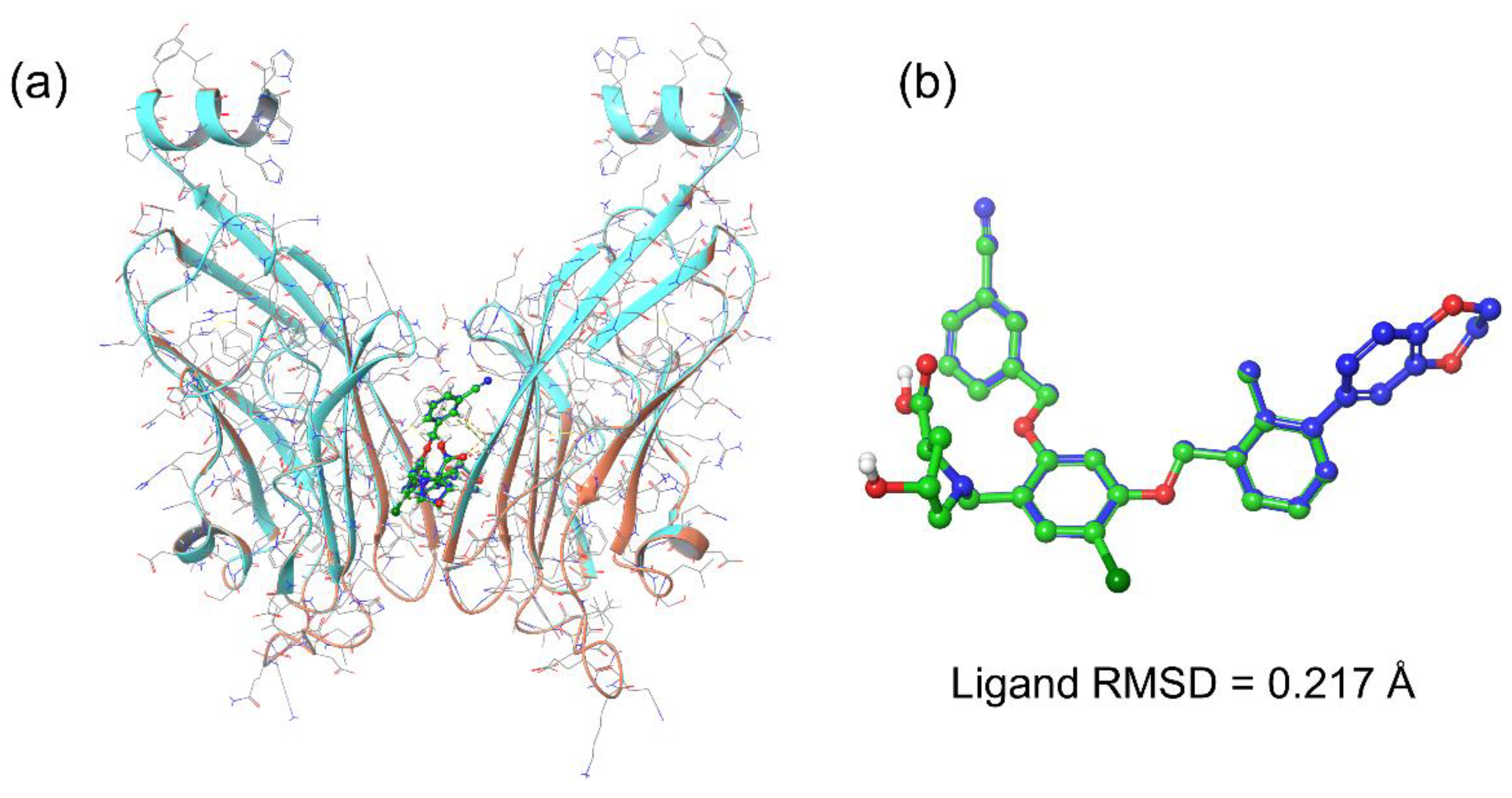
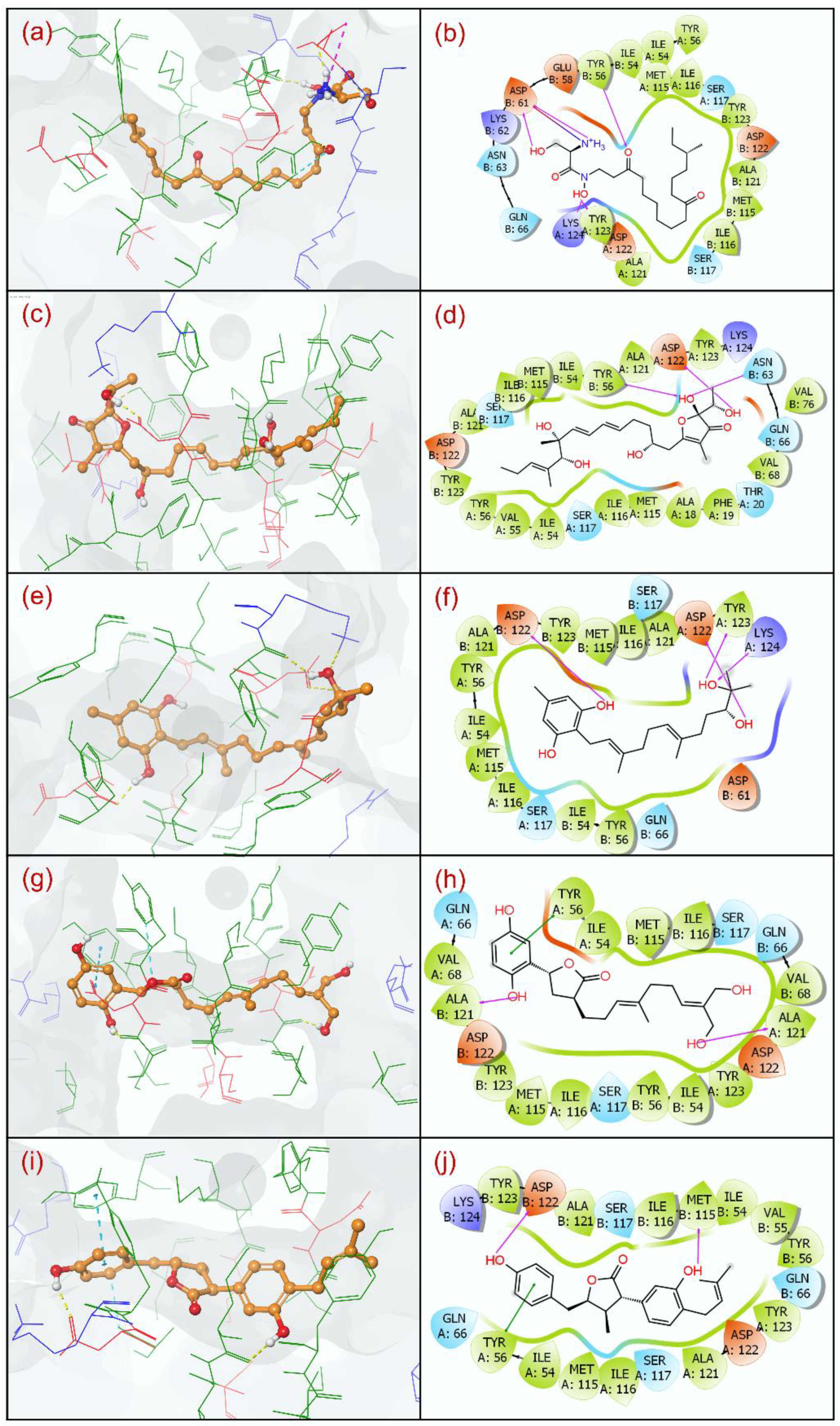
3.2. Docking Pose Validation and Interaction Analysis
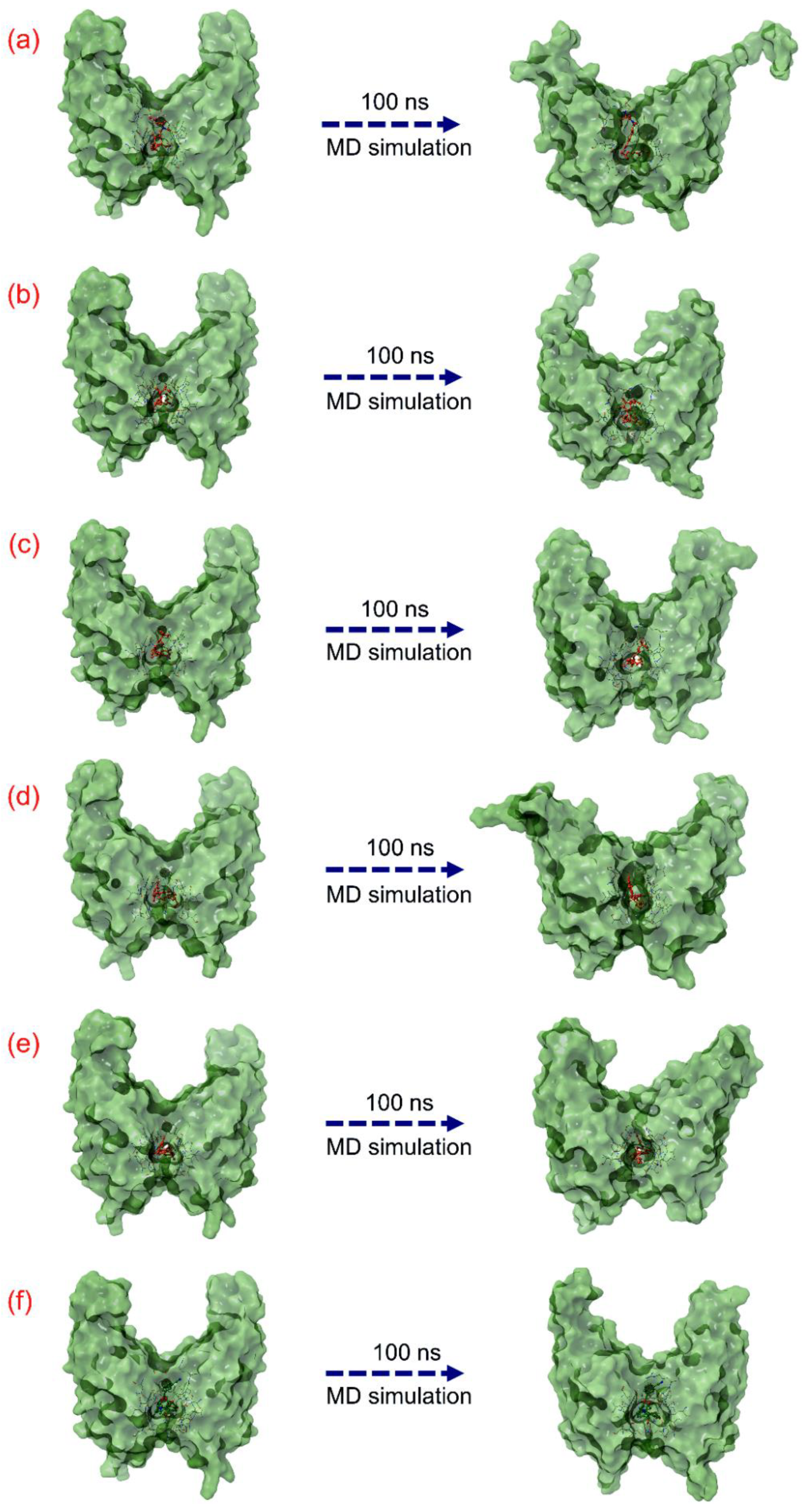
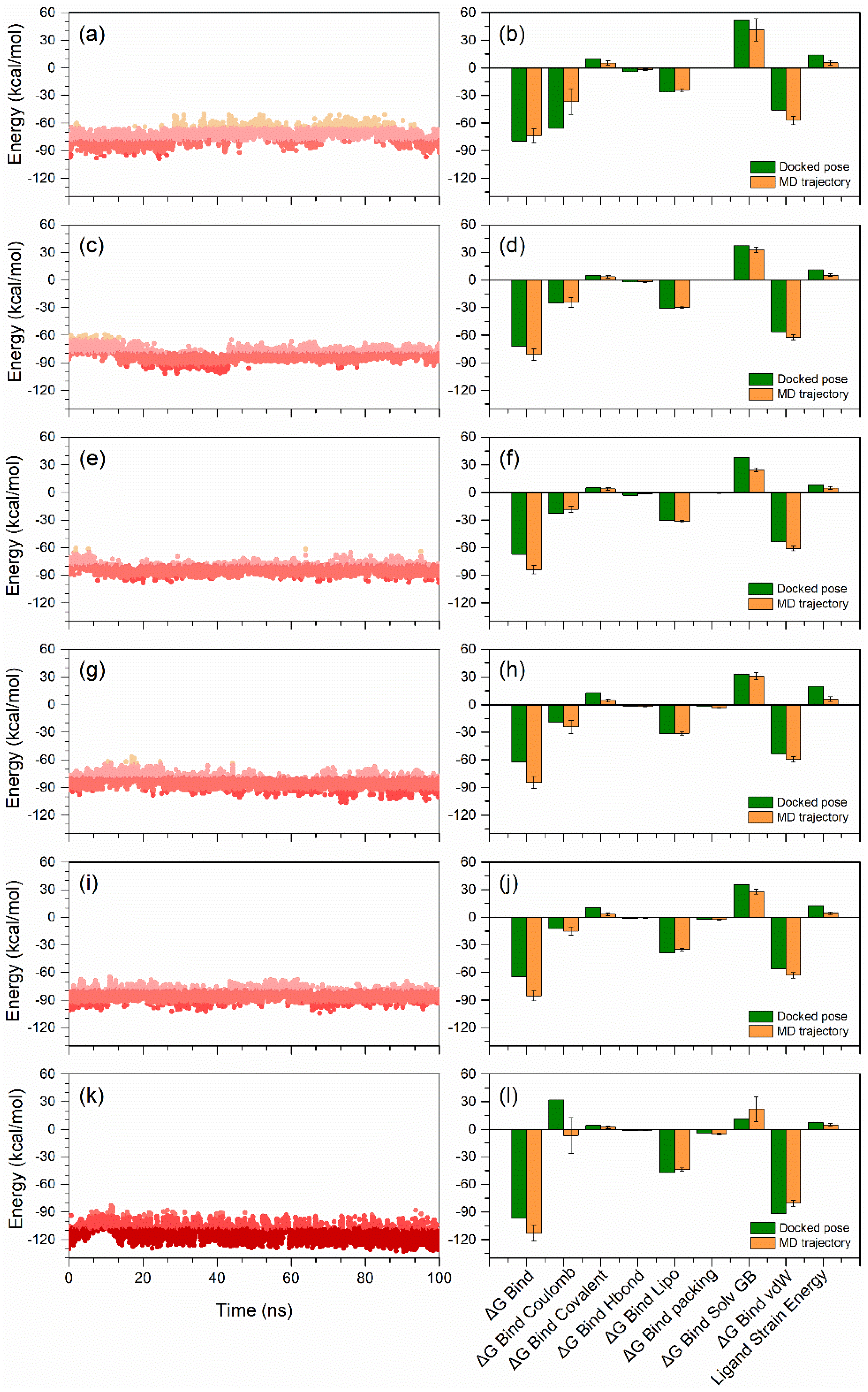
3.3. Molecular Dynamic Simulation Analysis
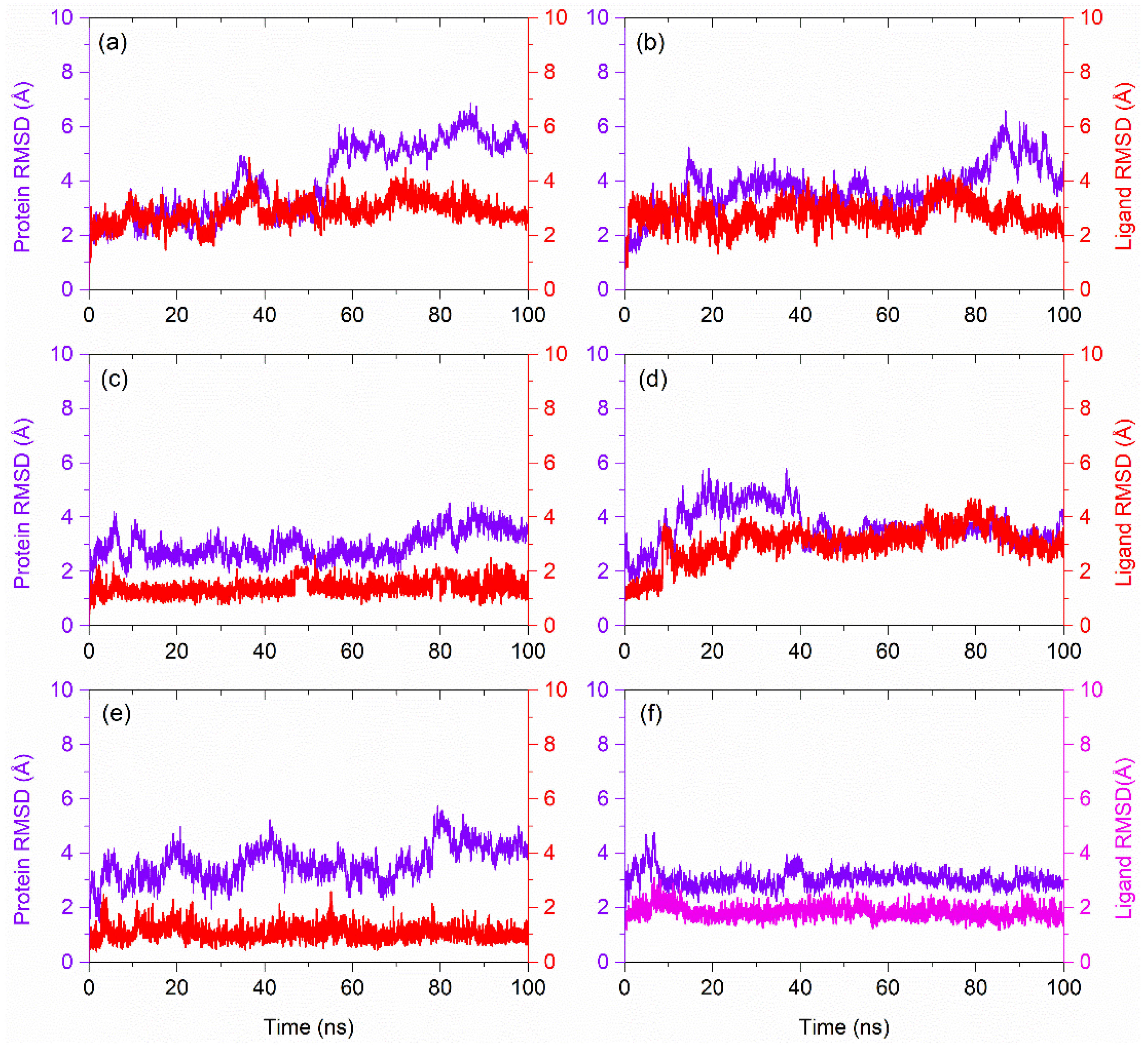
3.4. RMSD and RMSF Analysis
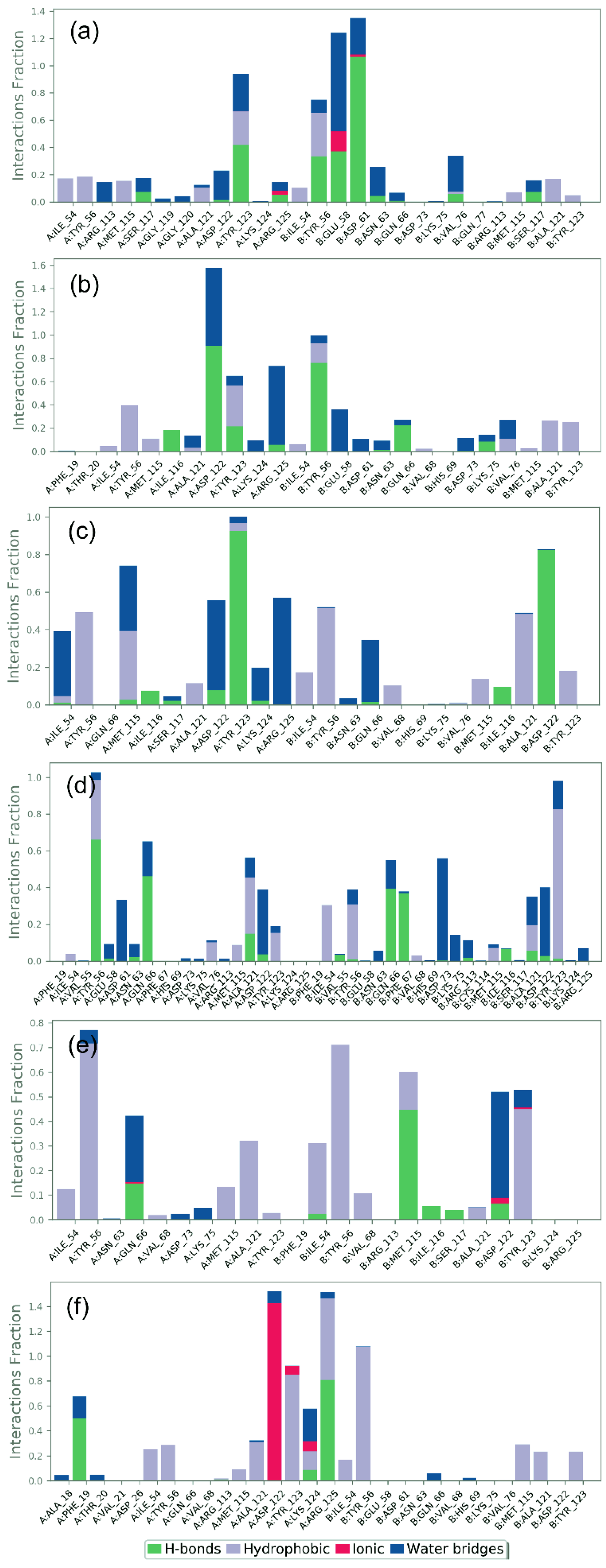
3.5. Protein–Ligand Interaction Mapping
3.6. End-Point Binding Free Energy Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, M.; Wu, H.X.; Xu, R.H. Advancing to the era of cancer immunotherapy. Cancer Commun. 2021, 41, 803–829. [Google Scholar] [CrossRef] [PubMed]
- Wyld, L.; Audisio, R.A.; Poston, G.J. The evolution of cancer surgery and future perspectives. Nat. Rev. Clin. Oncol. 2015, 12, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Aldeghaither, D.S.; Zahavi, D.J.; Murray, J.C.; Fertig, E.J.; Graham, G.T.; Zhang, Y.W.; O’Connell, A.; Ma, J.; Jablonski, S.A.; Weiner, L.M. A Mechanism of Resistance to Antibody-Targeted Immune Attack. Cancer Immunol. Res. 2019, 7, 230–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasan, N.; Baselga, J.; Hyman, D.M. A view on drug resistance in cancer. Nature 2019, 575, 299–309. [Google Scholar] [CrossRef] [Green Version]
- Ward, R.A.; Fawell, S.; Floc’h, N.; Flemington, V.; McKerrecher, D.; Smith, P.D. Challenges and Opportunities in Cancer Drug Resistance. Chem. Rev. 2021, 121, 3297–3351. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Mohanty, A.; Achuthan, S.; Kotnala, S.; Jolly, M.K.; Kulkarni, P.; Salgia, R. Group Behavior and Emergence of Cancer Drug Resistance. Trends Cancer 2021, 7, 323–334. [Google Scholar] [CrossRef]
- Allen, C.; Her, S.; Jaffray, D.A. Radiotherapy for Cancer: Present and Future. Adv. Drug Deliv. Rev. 2017, 109, 1–2. [Google Scholar] [CrossRef]
- Tay, R.E.; Richardson, E.K.; Toh, H.C. Revisiting the role of CD4(+) T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther. 2021, 28, 5–17. [Google Scholar] [CrossRef]
- Khalil, D.N.; Smith, E.L.; Brentjens, R.J.; Wolchok, J.D. The future of cancer treatment: Immunomodulation, CARs and combination immunotherapy. Nat. Rev. Clin. Oncol. 2016, 13, 273–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribas, A.; Wolchok, J.D. Cancer immunotherapy using checkpoint blockade. Science 2018, 359, 1350–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Allison, J.P. Dissecting the mechanisms of immune checkpoint therapy. Nat. Rev. Immunol. 2020, 20, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Park, W.; Heo, Y.J.; Han, D.K. New opportunities for nanoparticles in cancer immunotherapy. Biomater. Res. 2018, 22, 24. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z. Chapter 11-Using CAR-NK cells to overcome the host resistance to antibody immunotherapy and immune checkpoint blockade therapy. In Successes and Challenges of NK Immunotherapy; Bonavida, B., Jewett, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 193–212. [Google Scholar]
- Chowdhury, P.S.; Chamoto, K.; Honjo, T. Combination therapy strategies for improving PD-1 blockade efficacy: A new era in cancer immunotherapy. J. Intern. Med. 2018, 283, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Salmaninejad, A.; Valilou, S.F.; Shabgah, A.G.; Aslani, S.; Alimardani, M.; Pasdar, A.; Sahebkar, A. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J. Cell. Physiol. 2019, 234, 16824–16837. [Google Scholar] [CrossRef]
- Muenst, S.; Soysal, S.D.; Tzankov, A.; Hoeller, S. The PD-1/PD-L1 pathway: Biological background and clinical relevance of an emerging treatment target in immunotherapy. Expert Opin. Ther. Targets 2015, 19, 201–211. [Google Scholar] [CrossRef]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Herbst, R.S.; Soria, J.C.; Kowanetz, M.; Fine, G.D.; Hamid, O.; Gordon, M.S.; Sosman, J.A.; McDermott, D.F.; Powderly, J.D.; Gettinger, S.N.; et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature 2014, 515, 563–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.M.; Tannock, I.F. The distribution of the therapeutic monoclonal antibodies cetuximab and trastuzumab within solid tumors. BMC Cancer 2010, 10, 255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maute, R.L.; Gordon, S.R.; Mayer, A.T.; McCracken, M.N.; Natarajan, A.; Ring, N.G.; Kimura, R.; Tsai, J.M.; Manglik, A.; Kruse, A.C.; et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc. Natl. Acad. Sci. USA 2015, 112, E6506–E6514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Hamid, O.; Robert, C.; Daud, A.; Hodi, F.S.; Hwu, W.J.; Kefford, R.; Wolchok, J.D.; Hersey, P.; Joseph, R.W.; Weber, J.S.; et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N. Engl. J. Med. 2013, 369, 134–144. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Tian, H. Development of small-molecule immune checkpoint inhibitors of PD-1/PD-L1 as a new therapeutic strategy for tumour immunotherapy. J. Drug Target. 2019, 27, 244–256. [Google Scholar] [CrossRef]
- Zhan, M.M.; Hu, X.Q.; Liu, X.X.; Ruan, B.F.; Xu, J.; Liao, C. From monoclonal antibodies to small molecules: The development of inhibitors targeting the PD-1/PD-L1 pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta Pharmacol. Sin. 2021, 42, 1–9. [Google Scholar] [CrossRef]
- Awadasseid, A.; Wu, Y.; Zhang, W. Advance investigation on synthetic small-molecule inhibitors targeting PD-1/PD-L1 signaling pathway. Life Sci. 2021, 282, 119813. [Google Scholar] [CrossRef]
- Ri, M.H.; Ma, J.; Jin, X. Development of natural products for anti-PD-1/PD-L1 immunotherapy against cancer. J. Ethnopharmacol. 2021, 281, 114370. [Google Scholar] [CrossRef]
- Li, X.; Yao, Z.; Jiang, X.; Sun, J.; Ran, G.; Yang, X.; Zhao, Y.; Yan, Y.; Chen, Z.; Tian, L.; et al. Bioactive compounds from Cudrania tricuspidata: A natural anticancer source. Crit. Rev. Food Sci. Nutr. 2020, 60, 494–514. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Pandey, P.; Mishra, R.; Arif, M.; Kumar, A.; Jafri, A.; Mazumder, R. Elucidation of S-allylcysteine role in inducing apoptosis by inhibiting PD-L1 expression in human lung cancer cells. Anti Cancer Agents Med. Chem. 2021, 21, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Rugamba, A.; Kang, D.Y.; Sp, N.; Jo, E.S.; Lee, J.M.; Bae, S.W.; Jang, K.J. Silibinin Regulates Tumor Progression and Tumorsphere Formation by Suppressing PD-L1 Expression in Non-Small Cell Lung Cancer (NSCLC) Cells. Cells 2021, 10, 1632. [Google Scholar] [CrossRef]
- Zak, K.M.; Grudnik, P.; Guzik, K.; Zieba, B.J.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structural basis for small molecule targeting of the programmed death ligand 1 (PD-L1). Oncotarget 2016, 7, 30323–30335. [Google Scholar] [CrossRef] [Green Version]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Domling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef] [Green Version]
- Muszak, D.; Surmiak, E.; Plewka, J.; Magiera-Mularz, K.; Kocik-Krol, J.; Musielak, B.; Sala, D.; Kitel, R.; Stec, M.; Weglarczyk, K.; et al. Terphenyl-Based Small-Molecule Inhibitors of Programmed Cell Death-1/Programmed Death-Ligand 1 Protein-Protein Interaction. J. Med. Chem. 2021, 64, 11614–11636. [Google Scholar] [CrossRef]
- Van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A database of microbially-derived natural products. Nucleic Acids Res. 2021, 50, D1317–D1323. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4; Schrödinger, LLC: New York, NY, USA, 2020.
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins 2004, 55, 351–367. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-4: Prime; Schrödinger, LLC: New York, NY, USA, 2020.
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schrödinger Release 2020-4: Glide; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-4: LigPrep; Schrödinger, LLC: New York, NY, USA, 2020.
- Schrödinger Release 2020-4: QikProp; Schrödinger, LLC: New York, NY, USA, 2020.
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Culletta, G.; Gulotta, M.R.; Perricone, U.; Zappala, M.; Almerico, A.M.; Tutone, M. Exploring the SARS-CoV-2 Proteome in the Search of Potential Inhibitors via Structure-Based Pharmacophore Modeling/Docking Approach. Computation 2020, 8, 77. [Google Scholar] [CrossRef]
- Tutone, M.; Pibiri, I.; Lentini, L.; Pace, A.; Almerico, A.M. Deciphering the Nonsense Readthrough Mechanism of Action of Ataluren: An in Silico Compared Study. ACS Med. Chem. Lett. 2019, 10, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Morin, P.; Wang, W.; Kollman, P.A. Use of MM-PBSA in reproducing the binding free energies to HIV-1 RT of TIBO derivatives and predicting the binding mode to HIV-1 RT of efavirenz by docking and MM-PBSA. J. Am. Chem. Soc. 2001, 123, 5221–5230. [Google Scholar] [CrossRef]
- Lee, K.E.; Bharadwaj, S.; Yadava, U.; Kang, S.G. Computational and In Vitro Investigation of (-)-Epicatechin and Proanthocyanidin B2 as Inhibitors of Human Matrix Metalloproteinase 1. Biomolecules 2020, 10, 1379. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Dubey, A.; Yadava, U.; Mishra, S.K.; Kang, S.G.; Dwivedi, V.D. Exploration of natural compounds with anti-SARS-CoV-2 activity via inhibition of SARS-CoV-2 Mpro. Brief Bioinform. 2021, 22, 1361–1377. [Google Scholar] [CrossRef]
- Mena-Ulecia, K.; Tiznado, W.; Caballero, J. Study of the Differential Activity of Thrombin Inhibitors Using Docking, QSAR, Molecular Dynamics, and MM-GBSA. PLoS ONE 2015, 10, e0142774. [Google Scholar] [CrossRef] [Green Version]
- Schrödinger Release 2020-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2020.
- Bowers, K.J.; Chow, E.; Xu, H.; Dror, R.O.; Eastwood, M.P.; Gregersen, B.A.; Klepeis, J.L.; Kolossvary, I.; Moraes, M.A.; Sacerdoti, F.D. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the 2006 ACM/IEEE conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Schrödinger Release 2018-4: Maestro; Schrödinger, LLC: New York, NY, USA, 2018.
- Guo, J.X.; Hurley, M.M.; Wright, J.B.; Lushington, G.H. A docking score function for estimating ligand-protein interactions: Application to acetylcholinesterase inhibition. J. Med. Chem. 2004, 47, 5492–5500. [Google Scholar] [CrossRef] [Green Version]
- Guedes, I.A.; Pereira, F.S.S.; Dardenne, L.E. Empirical Scoring Functions for Structure-Based Virtual Screening: Applications, Critical Aspects, and Challenges. Front. Pharmacol. 2018, 9, 1089. [Google Scholar] [CrossRef]
- Li, H.J.; Sze, K.H.; Lu, G.; Ballester, P.J. Machine-learning scoring functions for structure-based drug lead optimization. Wires Comput. Mol. Sci. 2020, 10, e1465. [Google Scholar] [CrossRef] [Green Version]
- Rastelli, G.; Pinzi, L. Refinement and Rescoring of Virtual Screening Results. Front. Chem. 2019, 7, 498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinzelmann, G.; Gilson, M.K. Automation of absolute protein-ligand binding free energy calculations for docking refinement and compound evaluation. Sci. Rep. 2021, 11, 1116. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Genheden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015, 10, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Pu, C.; Yan, G.; Shi, J.; Li, R. Assessing the performance of docking scoring function, FEP, MM-GBSA, and QM/MM-GBSA approaches on a series of PLK1 inhibitors. Medchemcomm 2017, 8, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Rastelli, G.; Del Rio, A.; Degliesposti, G.; Sgobba, M. Fast and Accurate Predictions of Binding Free Energies Using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010, 31, 797–810. [Google Scholar] [CrossRef]
- Niinivehmas, S.P.; Virtanen, S.I.; Lehtonen, J.V.; Postila, P.A.; Pentikainen, O.T. Comparison of virtual high-throughput screening methods for the identification of phosphodiesterase-5 inhibitors. J. Chem. Inf. Model. 2011, 51, 1353–1363. [Google Scholar] [CrossRef]
- Roy, S.K.; Inouye, Y.; Nakamura, S.; Furukawa, J.; Okuda, S. Isolation, structural elucidation and biological properties of neoenactins B1, B2, M1 and M2, neoenactin congeners. J. Antibiot. 1987, 40, 266–274. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Cao, B.; Liu, C.; Guan, P.; Mu, Y.; Jiang, Y.; Han, L.; Huang, X. Actinofuranones DI from a lichen-associated actinomycetes, streptomyces gramineus, and their anti-inflammatory effects. Molecules 2018, 23, 2393. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, T.; Suzuki, T.; Ariefta, N.R.; Koseki, T.; Aboshi, T.; Murayama, T.; Widiyantoro, A.; Kurniatuhadi, R.; Malik, A.; Annas, S. Meroterpenoids produced by Pseudocosmospora sp. Bm-1-1 isolated from Acanthus ebracteatus Vahl. Phytochem. Lett. 2019, 31, 85–91. [Google Scholar] [CrossRef]
- Liao, G.-F.; Wu, Z.-H.; Liu, Y.; Yan, Y.-M.; Lu, R.-M.; Cheng, Y.-X. Ganocapenoids A–D: Four new aromatic meroterpenoids from Ganoderma capense. Bioorg. Med. Chem. Lett. 2019, 29, 143–147. [Google Scholar] [CrossRef]
- Awaad, A.S.; Nabilah, A.J.A.; Zain, M.E. New antifungal compounds from Aspergillus terreus isolated from desert soil. Phytother. Res. 2012, 26, 1872–1877. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Yadava, U.; Kang, S.G. Computational aided mechanistic understanding of Camellia sinensis bioactive compounds against co-chaperone p23 as potential anticancer agent. J. Cell. Biochem. 2019, 120, 19064–19075. [Google Scholar] [CrossRef] [PubMed]
- Filipe, H.A.L.; Loura, L.M.S. Molecular Dynamics Simulations: Advances and Applications. Molecules 2022, 27, 2105. [Google Scholar] [CrossRef] [PubMed]
- Plattner, N.; Doerr, S.; De Fabritiis, G.; Noe, F. Complete protein-protein association kinetics in atomic detail revealed by molecular dynamics simulations and Markov modelling. Nat. Chem. 2017, 9, 1005–1011. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Swanson, J.M.; Henchman, R.H.; McCammon, J.A. Revisiting free energy calculations: A theoretical connection to MM/PBSA and direct calculation of the association free energy. Biophys. J. 2004, 86, 67–74. [Google Scholar] [CrossRef]
- Adekoya, O.A.; Willassen, N.-P.; Sylte, I. Molecular insight into pseudolysin inhibition using the MM-PBSA and LIE methods. J. Struct. Biol. 2006, 153, 129–144. [Google Scholar] [CrossRef]
- Genheden, S.; Ryde, U. Comparison of end-point continuum-solvation methods for the calculation of protein-ligand binding free energies. Proteins 2012, 80, 1326–1342. [Google Scholar] [CrossRef] [Green Version]
- Shi, D.F.; An, X.L.; Bai, Q.F.; Bing, Z.T.; Zhou, S.Y.; Liu, H.X.; Yao, X.J. Computational Insight Into the Small Molecule Intervening PD-L1 Dimerization and the Potential Structure-Activity Relationship. Front. Chem. 2019, 7, 764. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Jin, Y.; Wang, B.; Liu, B. Molecular Mechanism of Small-Molecule Inhibitors in Blocking the PD-1/PD-L1 Pathway through PD-L1 Dimerization. Int. J. Mol. Sci. 2021, 22, 4766. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
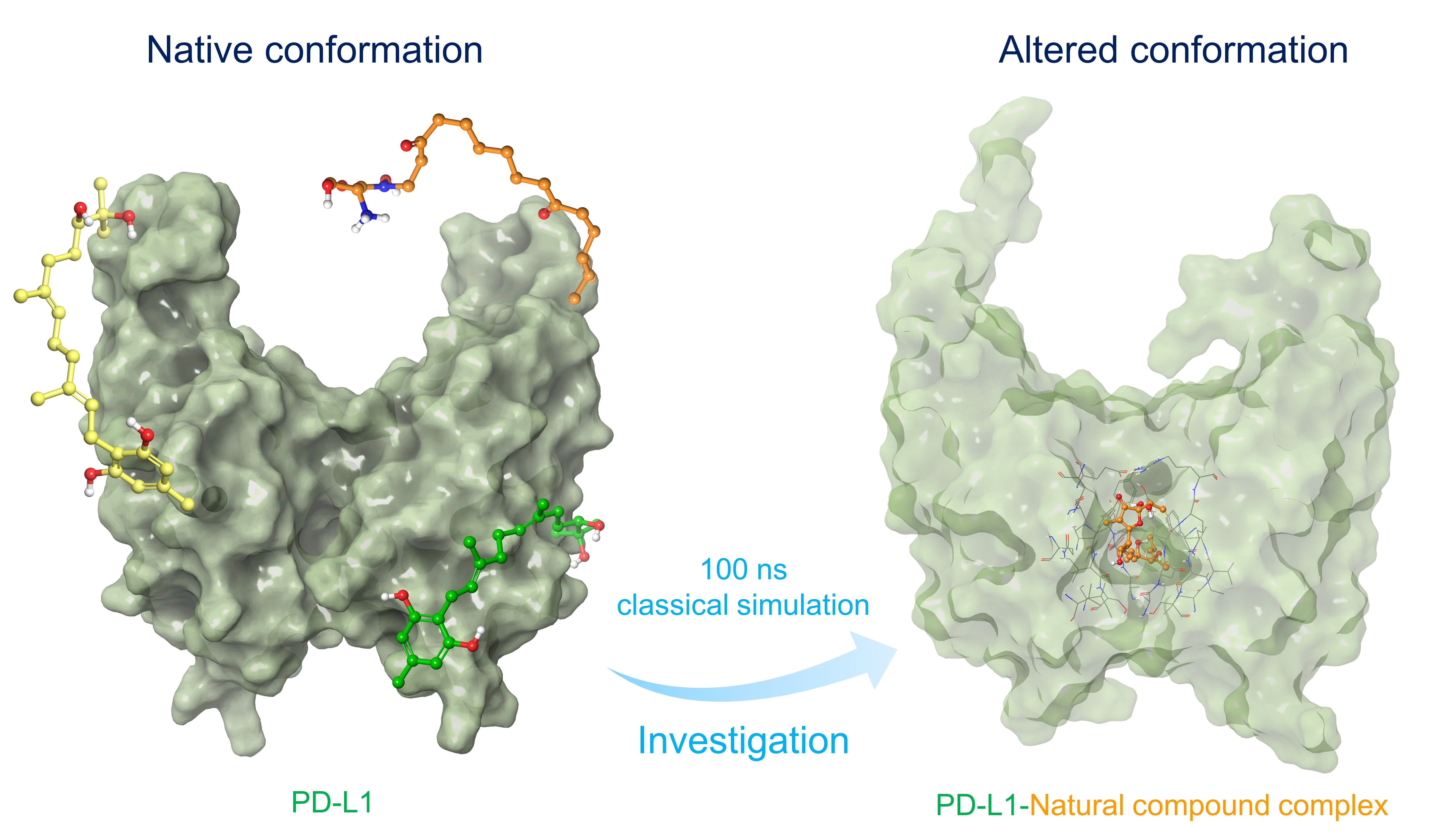
| S. No. | Title | Compound | Mol. Formula | Mol. wt. | Origin | Docking Score (kcal/mol) | ΔGBind (kcal/mol) |
|---|---|---|---|---|---|---|---|
| 1 | NPA020827 | Neoenactin B1 | C20H38N2O5 | 386.531 | Streptomyces olivoreticuli subsp. Neoenacticus | −10.36 | −79.63 |
| 2 | NPA027965 | Actinofuranone I | C23H36O7 | 424.533 | Streptomyces gramineus | −10.92 | −71.44 |
| 3 | NPA026024 | Cosmosporin A | C22H34O4 | 362.508 | Pseudocosmospora sp. Bm-1-1 | −10.28 | −67.43 |
| 4 | NPA026082 | Ganocapenoid A | C21H28O6 | 376.449 | Ganoderma capense | −10.54 | −66.92 |
| 5 | NPA013736 | 3-[3-hydroxy-4-(3-methylbut-2-enyl)phenyl]-5-(4-hydroxybenzyl)-4-methyldihydrofuran-2(3H)-one | C23H26O4 | 366.456 | Aspergillus terreus | −10.49 | −64.78 |
| 6 | NPA030364 | 4-carbglyceryl-3,3′-dihydroxy-5,5′-dimethyldiphenyl ether | C18H20O7 | 348.352 | Aspergillus versicolor SCSIO 41502 | −10.45 | −60.21 |
| 7 | NPA004673 | Not named | C19H16O3 | 292.334 | Burkholderia pseudomallei | −10.50 | −57.08 |
| 8 | NPA020009 | Sterin A | C16H20O6 | 308.33 | Stereum hirsutum | −11.39 | −55.39 |
| 9 | NPA027779 | Decarboxyunguidepside A | C19H20O5 | 328.364 | Aspergillus unguis | −10.39 | −54.51 |
| 10 | NPA025743 | Premacrophorintriol-I | C22H34O5 | 378.508 | Trichoderma sp. 1212-03 | −10.34 | −54.42 |
| 11 | NPA002619 | 4′′-Deoxy-5′-Desmethyl-Terphenyllin | C19H16O4 | 308.333 | Aspergillus sp. YXf3 | −10.56 | −54.41 |
| 12 | NPA018153 | Linieodolide A | C17H30O6 | 330.42 | Bacillus sp. 09ID194 | −10.30 | −53.94 |
| 13 | NPA017629 | 5′-O-desmethylterphenyllin | C19H16O5 | 324.332 | Aspergillus sp. YXf3 | −10.62 | −53.87 |
| 14 | NPA011065 | Nocarbenzoxazole E | C16H14N2O5 | 314.297 | Nocardiopsis lucentensis DSM 44048 | −10.73 | −53.37 |
| 15 | NPA022801 | Floricolin Q | C18H14O5 | 310.306 | Floricola striata | −10.81 | −52.11 |
| 16 | NPA015571 | Cylindrocarpol | C23H34O5 | 390.519 | Acremonium sp. | −11.86 | −49.25 |
| 17 | NPA014938 | Baciphelacin | C22H34N2O6 | 422.52 | Bacillus thiaminolyticus IFO 3967/B-1-7 | −10.89 | −40.41 |
| 18 | JQT inhibitor | BDBM363278 | C36H33ClN2O7 | 641.1 | Synthetic | −9.824 | −63.98 |
| S. No. | Complex | H-bond | Hydrophobic | Polar | π-π/*π-Cation | Salt Bridge | Positive | Negative |
|---|---|---|---|---|---|---|---|---|
| 1 | PD-L1-Neoenactin B1 | A:Tyr123, A:Lys124, B:Tyr56, B:Asp61(2) | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Ser117, B:Asn63, B:Gln66, B:Ser117 | - | B:Asp61 | A:Lys124, B:Lys62 | A:Asp122, B:Glu58, B:Asp61, B:Asp122 |
| 2 | PD-L1-Actinofuranone I | A:Asp122, B:Tyr56, B:Asn63 | A:Ala18, A:Phe19, A:Ile54, A:Val55, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Val68, B:Val76, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Thr20, A:Gln66, A:Ser117, B:Asn63, B:Ser117 | - | - | A:Lys124 | A:Asp122, B:Asp122 |
| 3 | PD-L1-Cosmosporin A | A:Asp122, A:Tyr123, A:Lys124, B:Asp122 | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Ser117, B:Gln66 B:Ser117 | - | - | A:Lys124 | A:Asp122, B:Asp61 B:Asp122 |
| 4 | PD-L1-Ganocapenoid A | A:Ala121, B:Ala121 | A:Ile54, A:Tyr56, A:Val68, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Val68, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Gln66, A:Ser117, B:Gln66, B:Ser117 | A:Tyr56 | - | - | A:Asp122, B:Asp122 |
| 5 | PD-L1-3-[3-hydroxy-4-(3-methylbut-2-enyl)phenyl]-5-(4-hydroxybenzyl)-4-methyldihydrofuran-2(3H)-one | B:Ala122, B:Met115 | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Val55, B:Tyr56, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Gln66, A:Ser117, B:Gln66, B:Ser117 | A:Tyr56 | - | B:Lys124 | A:Asp122, B:Asp122 |
| 6 | PD-L1-JQT inhibitor | - | A:Ala18, A:Phe19, A:Ile54, A:Val55, A:Tyr56, A:Met115, A:Ile116, A:Ala121 A:Tyr123, B:Ile54, B:Tyr56, B:Val68, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Thr20, A:Gln66, A:Ser117, B:Asn63, B:Gln66, B:Ser117 | A:Lys124, B:Tyr56, *A:Lys124 | A:Lys124 | A:Lys124, A:Arg125 | A:Asp122, B:Asp61, B:Asp122 |
| S. No. | Complex | H-Bond | Hydrophobic | Polar | π-π/*π-Cation | Salt Bridge | Positive | Negative |
|---|---|---|---|---|---|---|---|---|
| 1 | PD-L1-Neoenactin B1 | B:Tyr56, B:Asp61 | A:Ile54, A:Val55, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Val76, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Ser117, B:Asn63, B:Gln66, B:Ser117 | - | B:Asp61 | B:Lys62 | A:Asp122, B:Asp61, B:Asp122 |
| 2 | PD-L1-Actinofuranone I | A:Asp122, B:Tyr56 | A:Ile54, A:Val55, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B: Ile54, B:Tyr56, B:Val68, B:Val76, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Ser117, B:Asn63, B:Ser117 | - | - | A:Lys124, A:Arg125 | A:Asp122, B:Glu58, B:Asp122 |
| 3 | PD-L1-Cosmosporin A | A:Tyr123, B:Asp122 | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B: Ile54, B:Val55, B:Tyr56, B:Val68, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Ser117, B:Gln66,B:Ser117 | - | - | A:Lys124, A:Arg125 | A:Asp122, B:Glu58, B:Asp122 |
| 4 | PD-L1-Ganocapenoid A | A:Tyr56, B:Gln66 | A:Ile54, A:Tyr56, A:Val76, A:Met115, A:Ala121, A:Tyr123, B:Ile54, B:Val55, B:Tyr56, B:Val68, B:Val76, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Asn63, A:Gln66, B:Gln66, B:Ser117 | - | - | B:Arg113 | A:Glu58, A:Asp122, B:Asp122 |
| 5 | PD-L1-3-[3-hydroxy-4-(3-methylbut-2-enyl)phenyl]-5-(4-hydroxybenzyl)-4-methyldihydrofuran-2(3H)-one | B:Met115 | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Val55, B:Tyr56, B:Val68, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Gln66, A:Ser117, B:Gln66, B:Ser117 | A:Tyr56 B:Tyr123 | - | - | A:Asp122, B:Asp122 |
| 6 | PD-L1-JQT inhibitor | - | A:Ile54, A:Tyr56, A:Met115, A:Ile116, A:Ala121, A:Tyr123, B:Ile54, B:Tyr56, B:Val68, B:Val76, B:Met115, B:Ile116, B:Ala121, B:Tyr123 | A:Thr20, A:Gln66, A:Ser117,B:Gln66, B:Ser117 | B:Tyr56, *A:Arg125 | A:Lys124 | A:Lys124, A:Arg125 | A:Asp122, B:Asp122 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, G.S.; Moustafa, M.; Sahoo, A.K.; Malý, P.; Bharadwaj, S. Computational Investigations on the Natural Small Molecule as an Inhibitor of Programmed Death Ligand 1 for Cancer Immunotherapy. Life 2022, 12, 659. https://doi.org/10.3390/life12050659
Kumar GS, Moustafa M, Sahoo AK, Malý P, Bharadwaj S. Computational Investigations on the Natural Small Molecule as an Inhibitor of Programmed Death Ligand 1 for Cancer Immunotherapy. Life. 2022; 12(5):659. https://doi.org/10.3390/life12050659
Chicago/Turabian StyleKumar, Geethu S, Mahmoud Moustafa, Amaresh Kumar Sahoo, Petr Malý, and Shiv Bharadwaj. 2022. "Computational Investigations on the Natural Small Molecule as an Inhibitor of Programmed Death Ligand 1 for Cancer Immunotherapy" Life 12, no. 5: 659. https://doi.org/10.3390/life12050659