Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma



1
Laboratory of Cancer Cell Biology, Institute of Molecular Genetics of the Czech Academy of Sciences, Videnska 1083, CZ14220 Prague, Czech Republic
2
Department of Developmental and Cell Biology, Faculty of Science, Charles University, Albertov 6, CZ12800 Prague, Czech Republic
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cells 2020, 9(9), 2068; https://doi.org/10.3390/cells9092068
Submission received: 10 July 2020
/
Revised: 4 September 2020
/
Accepted: 6 September 2020
/
Published: 10 September 2020
(This article belongs to the Special Issue Cell Cycle Control and Cancer)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Genome integrity is protected by the cell-cycle checkpoints that prevent cell proliferation in the presence of DNA damage and allow time for DNA repair. The transient checkpoint arrest together with cellular senescence represent an intrinsic barrier to tumorigenesis. Tumor suppressor p53 is an integral part of the checkpoints and its inactivating mutations promote cancer growth. Protein phosphatase magnesium-dependent 1 (PPM1D) is a negative regulator of p53. Although its loss impairs recovery from the G2 checkpoint and promotes induction of senescence, amplification of the PPM1D locus or gain-of-function truncating mutations of PPM1D occur in various cancers. Here we used a transgenic mouse model carrying a truncating mutation in exon 6 of PPM1D (Ppm1dT). As with human cell lines, we found that the truncated PPM1D was present at high levels in the mouse thymus. Truncated PPM1D did not affect differentiation of T-cells in the thymus but it impaired their response to ionizing radiation (IR). Thymocytes in Ppm1dT/+ mice did not arrest in the checkpoint and continued to proliferate despite the presence of DNA damage. In addition, we observed a decreased level of apoptosis in the thymi of Ppm1dT/+ mice. Moreover, the frequency of the IR-induced T-cell lymphomas increased in Ppm1dT/+Trp53+/− mice resulting in decreased survival. We conclude that truncated PPM1D partially suppresses the p53 pathway in the mouse thymus and potentiates tumor formation under the condition of a partial loss of p53 function.
1. Introduction
In healthy cells, genome stability is protected by cell-cycle checkpoints that prevent cell proliferation in the presence of DNA damage and allow time for DNA repair [1,2,3]. Induction of DNA double-strand breaks (DSBs, such as the ones generated by ionizing radiation (IR)) or the presence of structural chromosomal aberrancies emanating from mitotic defects activate ATM kinase that phosphorylates the tumor suppressor protein p53, which leads to its stabilization and transcriptional activation [4,5,6]. By triggering the expression of CDKN1Ap21, p53 induces cell-cycle arrest in the G1 checkpoint [7]. Replication stress (caused by various factors including oncogene activation) leads to activation of the Ataxia telangiectasia and Rad3-related (ATR) kinase that triggers phosphorylation of the checkpoint kinase CHK1 that induces a cell-cycle arrest in the S/G2 phase by targeting CDC25 phosphatases [8]. Maintenance of the G2 checkpoint is then promoted by activation of the p53 pathway [9]. Recovery from the G2 checkpoint depends on protein phosphatase magnesium-dependent 1 delta (PPM1D, also called WIP1) that counteracts the activity of ATM at chromatin and prevents full activation of p53 [10,11,12,13]. Sustained activation of p53 during G2 can lead to premature activation of Anaphase-promoting complex/cyclosome and its coactivator Cdh1 followed by degradation of cyclin B1 and induction of senescence [14,15]. The depletion of PPM1D leads to abnormally high activation of p53 during G2 and to defects in homologous recombination, both contributing to the loss of recovery competence and induction of senescence [10,16,17]. In addition, senescence can result from sustained activation of p53 during G1 through the expression of downstream genes including CDKN1Ap21 [18,19]. PPM1D is not required for terminating the G1 checkpoint, as other phosphatases (including PP4) can suppress the p53 response after completion of DNA repair [20]. Importantly, the checkpoint pathway and oncogene-induced senescence represent an intrinsic barrier preventing genome instability and tumor development [21,22,23].
Amplification of the chromosomal locus 17q23.2 carrying the PPM1D is common in various cancer types and results in overexpression of enzymatically active PPM1D [24]. Amplification of PPM1D has been reported in about 10% of breast cancers, mainly those that retain a wild-type p53 status [24,25,26]. Data from PPM1D knock-out mice demonstrate that PPM1D promotes tumor growth by inhibiting p53 and p38/MAPK pathways [24,27,28]. In addition, high expression of PPM1D can also affect response to therapy as it reduces the sensitivity of cancer cells to doxorubicin and other chemotherapeutics [29,30]. Recently, we and others described new pathogenic mutations in exon 6 of the PPM1D that result in production of the C-terminally truncated PPM1D protein [31,32,33]. The deletion of the last 60 amino acids of PPM1D removes a degron regulating its rapid turnover and results in stabilization of the truncated PPM1D protein [31,34]. Importantly, deletion of the C-terminal tail leaves the catalytic domain of PPM1D intact and also preserves chromatin localization of the truncated protein [31]. Cancer cell lines (including U2OS and HCT116 cells) carrying heterozygous truncating mutations in PPM1D show G1 checkpoint override upon exposure to the mild level of IR [31]. Similarly, when we introduced truncating mutations in exon 6 of the PPM1D in human non-transformed retinal pigment epithelial (RPE1) cell lines using CRISPR/Cas9 technology, we observed decreased ability to induce the G1 checkpoint after exposure to IR [35]. However, whether the truncating PPM1D mutations contribute to tumorigenesis remains an open question. To experimentally address this, we have recently generated a mouse model in which we introduced a frame-shift mutation in the exon 6 of PPM1D using the Transcription activator-like effector nuclease (TALEN) technology [35]. We have found that the truncated PPM1DT allele protected intestinal stem cells from apoptosis by suppressing the p53 pathway [35]. In addition, PPM1DT/+ mice showed a higher amount of intestinal polyps and increased frequency of colon adenocarcinoma induced by constitutively active Wnt signaling in APCmin background [35]. Although the PPM1DT allele alone did not induce the formation of colon tumors, it significantly potentiated the phenotype and reduced the survival of APCmin mice [35].
T-cells differentiate in the thymus cortex from early progenitors by sequentially progressing through CD4−CD8− double-negative (DN) and CD4+CD8+ double-positive (DP) stages and leave the medulla as single positive CD4+ or CD8+ cells with a fully assembled T-cell receptor (TCR) [36,37]. Site-specific dsDNA breaks in TCR gene present in DN cells trigger the p53 response and are eventually repaired by V(D)J recombination allowing the transition to the DP stage [38,39,40]. Sustained activation of p53 in mice lacking PPM1D blocked the T-cell maturation at the DN stage [41]. In addition, PPM1D has recently been implicated in maturation of the medullary thymic epithelial cells as well as in the development of the B cells [42,43,44].
Here we used the knock-in Ppm1dT mouse model to study the impact of truncated PPM1D on cell survival and tumorigenesis in murine thymus. We find that thymocytes carrying truncated PPM1D escape apoptotic cell death and continue proliferation despite the presence of DNA damage. Although the truncated PPM1D did not significantly drive tumorigenesis upon exposure of mice to IR, it promoted the formation of thymic lymphoma in Trp53+/− heterozygotes. We propose that truncation of PPM1D prevents full activation of p53 upon genotoxic stress and promotes tumor formation in cells exhibiting partial loss of p53.
2. Materials and Methods
2.1. Animals
All animal experiments were approved by the ethical committee of the Institute of Molecular Genetics (c.j. 1/2016) and were performed in C57Bl/6 mice. The mouse strain carrying a Ppm1dT allele with a premature stop codon in exon 6 of the Ppm1d was described previously [35]. Trp53tm1Tyj/J mouse strain was obtained from the Jackson Laboratory (stock #002101) and was described previously [45]. Where indicated, mice were irradiated at age of 8–10 weeks using an X-RAD 225XL instrument equipped with a copper filter. Animal tissues were collected into a reagent for RNA isolation - RNA Blue (Top-Bio), homogenized using TissueLyser LT (Qiagen) and mixed with chloroform. RNA was isolated from the aqueous phase by precipitation with isopropanol. Proteins were isolated from the phenol-chloroform phase by precipitation with isopropanol, and after washing with 0.3 M guanine hydrochloride in 95% ethanol, proteins were dissolved in 2X Sodium Dodecyl Sulfate (SDS)-PAGE buffer.
2.2. Western Blotting
Protein concentration was measured by the Bicinchoninic Acid (BCA) protein assay (Thermo Fisher Scientific, Rockford, IL, USA). Proteins (30 μg) were separated on 4–20% gradient gels using SDS-PAGE and blotted onto a nitrocellulose membrane. Membranes were blocked in 3% milk in phosphate-buffer saline (PBS) for 2 h and incubated overnight with primary antibodies at 4 °C. The following antibodies were used for immunoblotting: rabbit monoclonal to PPM1D/WIP1 (Cell Signaling, #11901, clone D4F7), p53-pS15 (Cell Signaling, #12571), p21 (Abcam, #109199), KAP1-pS824 (Novus Biologicals, #NB100-2350), γH2AX-pS319 (Cell Signaling, #9718), p53 (Leica-CM5P-L), H2AX (Millipore, 07-627), Cleaved Caspase-3-Asp175 (Cell Signaling, #9664, clone 5A1E), KAP1 (GeneTex, Irvine, CA, USA, #102227, clone N1N2) and Importin β (Santa Cruz, sc-137016). After the washing, membranes were incubated with HRP-conjugated secondary antibodies (BioRad) and signal was detected using Enhanced chemiluminiscence (ECL) Western Blotting Substrate (Thermo Fisher Scientific, Rockford, IL, USA).
2.3. Flow Cytometry
Analysis of the developing T-cells by flow cytometry was performed as described [37]. Briefly, thymi from three 8-week old mice per genotype were smashed, strained and washed with 3% FBS and 2 mM EDTA. The mixture was centrifuged (300 g, 10 min) and red blood cells were lysed with the Ammonium-Chloride-Potassium (ACK) buffer (containing 150 mM NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA) for 2 min. The lysis was stopped, the cell mixture was spun down and the pellet was stained with the following antibody cocktail: CD25-PE/Cy7 (Biolegend, San Diego, CA, USA, PC61 #102016), CD4-FITC (eBioscience GK1.5 # 11-0041-82), CD3-PerCP-Cy5.5 (Biolegend, San Diego, CA, USA, 17A2), CD8a-PE (Biolegend, San Diego, CA, USA, 53-6.7) and CD44-APC (Biolegend, San Diego, CA, USA, IM7) and Hoechst 33,258 (Sigma). Cells were gated using FSC/SSC, singlets were discriminated using SSC-W parameter following exclusion of dead cells positive for Hoechst staining. Developing T-cells were gated according to CD8 and CD4 markers, and the DN population was further separated based on the CD44 and CD25 positivity.
2.4. Immunohistochemistry
Tissues embedded in paraffin were sectioned on a Leica RM2255 microtome at a thickness of 3 μm. To examine the morphological changes in the tissues, the standard hematoxylin and eosin (H&E) staining protocol was followed. For staining with the Ki-67 proliferation marker (GeneTex, Irvine, CA, USA, #16667) and the Cleaved Caspase-3-Asp175 apoptotic marker (Cell Signaling, #9664, clone 5A1E), tissues were deparaffinized with xylene, isopropanol, and decreasing concentrations of ethanol. Heat-induced epitope retrieval was performed in 10 mM citrate buffer (pH 6.0). Samples were blocked with 0.3% hydrogen peroxide in methanol and in bovine-serum albumin (1%) with goat serum (5%) for 2h for Ki-67 staining or 5% BSA and 0.1% TX-100 in PBS for cleaved caspase staining. Incubation with the primary antibody was done overnight at 4 °C and with the biotin-conjugated secondary antibody for 1 h at room temperature. Signal was developed using VECTASTAIN ABC HRP kit (Vector Laboratories) and 3,3′-Diaminobenzidine (DAB). Apoptosis was detected using Click-iT™ Plus terminal deoxynucleotidyl transferase (TUNEL) Assay (Thermo Fisher Scientific, Rockford, IL, USA). Briefly, tissues were deparaffinized, fixed with 4% formaldehyde, and permeabilized with Proteinase K. The TdT reaction with Ethynyl-2′-Deoxyuridine, 5′-Triphosphate (EdUTP) incorporation into the dsDNA breaks was performed for 60 min at 37 °C. Click-iT™ Plus reaction for detecting EdUTP with Alexa Fluor 594 picolyl azide was performed for 30 min and nuclei were stained using 4′,6-Diamidino-2-Phenylindole, Dihydrochloride (DAPI). Slides were mounted using Q Path Coverquick mounting medium and visualized on a Leica DM6000 microscope (Leica) equipped with HC PL FLUOTAR 5X, and HC PLAN APO 20X and 40X objectives. Images were analyzed using ImageJ software [46]. After thresholding, total areas of positive and negative signal were measured in each image.
2.5. Real-Time Quantitative Reverse Transcriptase PCR (RT-qPCR)
cDNA was generated from 0.5 μg RNA using random hexamers and RevertAid Reverse Transcriptase (Thermo Fisher Scientific, Rockford, IL, USA). RT-qPCR was performed using LightCycler 480 SYBR Green I Master mix (Roche) as described [35]. The following primers were used for RT-qPCR: CDKN1A/p21 TGAGGAGGAGCATGAATGGAGACA and AACAGGTCGGACATCACCAGGATT; PUMA CCT-GGAGGGTCATGTACAATCT and TGCTACATGGT-GCAGAAAAAGT; PPM1D AGCCAGGAG-ACCTGTGTGAT and GGCATTACTGCGAACAAGGG; GAPDH ACAGCCGCATCTTCTTGTGCA-GTG and GGCCTTGACTGTGCCGTTGAATTT. The LightCycler480 software was used to determine Ct values. The data is shown as a ratio between the tested mRNA and GAPDH mRNA.
2.6. Statistical Analysis
Several animals used in each experimental group is indicated. Immunoblots were performed using tissues from at least 3 animals and representative images are shown. Statistical significance was evaluated using Graph Pad Prism 5.04 software. Unless stated otherwise, two-tailed unpaired t-test was used and p values < 0.05 are considered statistically significant. Kaplan-Meier survival plot was evaluated using a log-rank test. Error bars indicate standard deviation.
3. Results
3.1. Mice with Truncated PPM1D Show Impaired Acute DNA-Damage Response (DDR) in the Thymus upon γ-Irradiation
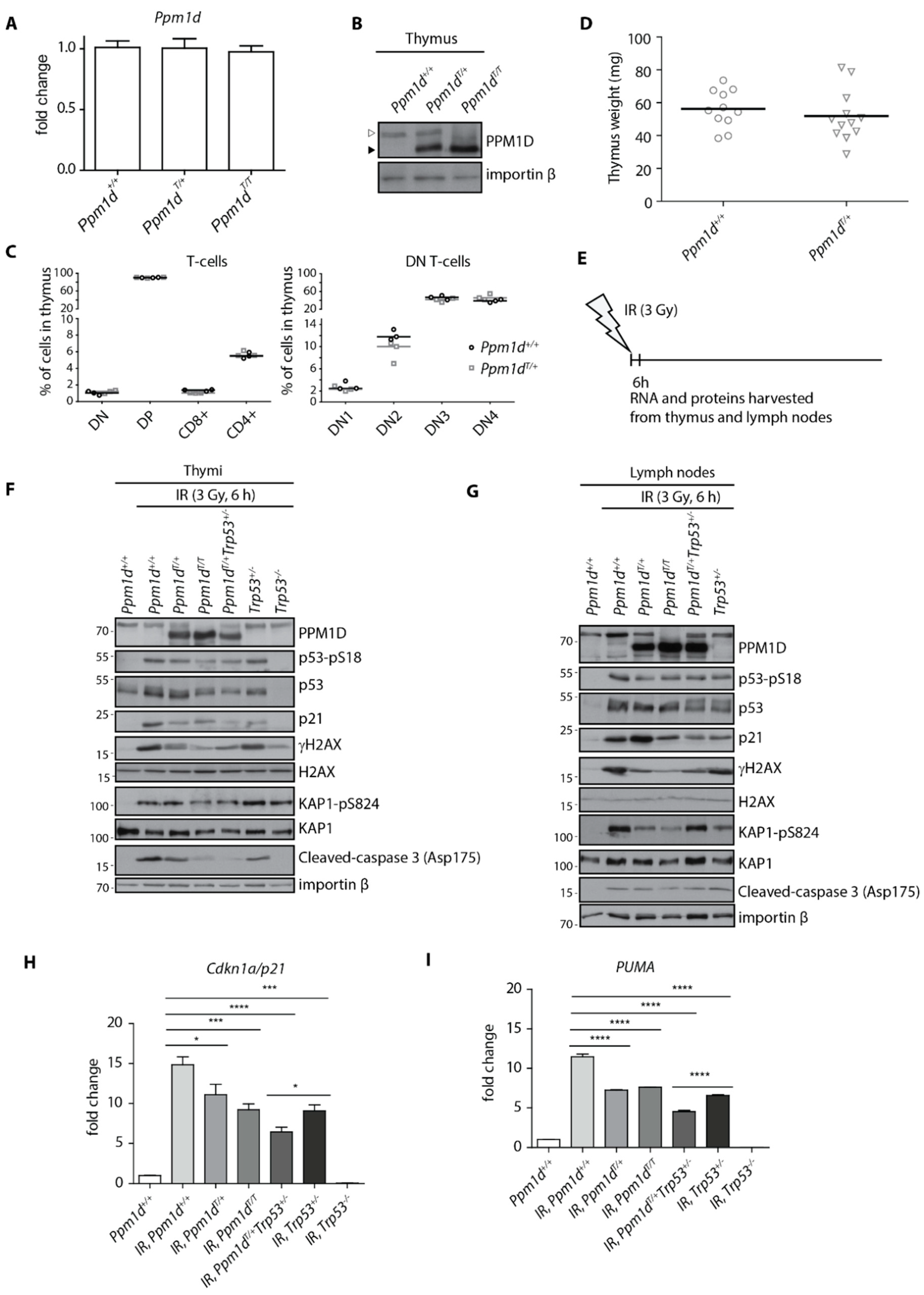
Previous reports described PPM1D expression in the developing thymocytes, fully differentiated T-cells and medullar thymic epithelial cells [41,42]. The loss of Ppm1d blocked progression of thymocytes from a DN to DP stage resulting in a decreased population of differentiated T-cells and in reduction of the thymus size in Ppm1d−/− mice [41]. As the block in T-cell development was associated with an increased activation of the tumor suppressor p53, we aimed here to evaluate the impact of PPM1D truncation on T-cell maturation using the PPM1DT knock-in mouse model [41]. First, we measured the expression of Ppm1d in mouse thymi using RT-qPCR and found that the wild-type Ppm1d and truncated Ppm1dT were expressed at comparable levels suggesting that introducing a stop codon in the last exon does not affect transcriptional control of Ppm1d mRNA (Figure 1A). As expected, the truncated Ppm1dT protein migrated faster on SDS-PAGE and was present at high levels due to stabilization of the truncated protein (Figure 1B) [31,35]. Using flow cytometry, we quantified individual populations of the developing T-cells in the thymi, but we did not find any significant differences between the wild-type and Ppm1dT/+ animals (Figure 1C). Consistent with this finding, we also did not observe any difference in the thymus size in the wild-type and Ppm1dT/+ animals (Figure 1D). These findings suggest that the truncated PPM1D does not affect the differentiation of the T-cells.
Next, we aimed to compare the response to genotoxic stress in lymphocytes carrying the wild-type PPM1D and the truncated PPM1DT. To this end, we exposed or not Ppm1d+/+, Ppm1dT/+ and Ppm1dT/T animals to a low level of IR (3 Gy) and collected the thymi and lymph nodes after 6 h (Figure 1E). Trp53−/− animals lacking the p53 and Trp53+/− heterozygotes were analyzed in parallel as controls. As expected, wild-type animals showed increased phosphorylation of p53 at S18 (corresponding to the human S15) and induction of the p53 transcriptional target CDKN1Ap21 after exposure to IR (Figure 1F). In contrast, PPM1DT/+ heterozygotes showed decreased level of p53.
Phosphorylation and a lower level of CDKN1Ap21 induction (Figure 1F). Levels of CDKN1Ap21 were comparable in Ppm1dT/+ and Trp53+/− whereas further reduction of CDKN1Ap21 was observed in the thymi of Ppm1dT/+Trp53+/− double heterozygotes suggesting that truncated PPM1D can promote the phenotype of a partial loss of the p53. Similarly, truncation of both alleles in Ppm1dT/T animals slightly enhanced dephosphorylation of p53-S18, γ-H2AX and KAP1-S824, all the established substrates of PPM1D, compared to Ppm1dT/+ but surprisingly we did not observe further reduction of p21 levels (Figure 1F, Supplementary Figure S1A,B). Probing for a cleaved caspase 3 suggested that even the mild dose of IR can lead to activation of the apoptotic pathway in wild-type thymocytes (Figure 1F). Importantly, activation of the apoptotic pathway was p53-dependent as cleavage of the caspase 3 did not occur in Trp53−/− animals. Notably, we observed a lower level of cleaved caspase 3 in the thymi from Ppm1dT/+ and Trp53+/− animals and further reduction in Ppm1dT/+Trp53+/− double heterozygotes (Figure 1F). The impact of the truncated PPM1D on the expression of CDKN1Ap21 and a pro-apoptotic gene PUMA was further confirmed by a RT-qPCR (Figure 1H,I). In good agreement with the results above, we observed a reduced expression of CDKN1A p21 and PUMA in the thymi from Ppm1dT/+Trp53+/− double heterozygotes compared to Ppm1dT/+ and Trp53+/− animals.
In parallel with the thymi, we analyzed the response to genotoxic stress in the lymph nodes (Figure 1G, Supplementary Figure S1C,D). As expected, we observed decreased level of γ-H2AX in mice carrying truncated PPM1D. Reduction of KAP1 phosphorylation was even more prominent in the lymph nodes than in the thymi confirming that a high activity of PPM1D was present in cells. Surprisingly, we did not observe any reduction of p21 and cleaved caspase 3 levels in mice carrying PPM1DT allele suggesting that its impact on the cell-cycle checkpoint and apoptosis may differ in various cell types and tissues.
We conclude that the acute response to genotoxic stress is partially suppressed by truncated PPM1D in the murine thymi, which is in good agreement with our recent observation of the impaired signaling in the intestinal stem cells in mice carrying Ppm1dT allele [35].
3.2. Truncated PPM1D Prevents Apoptosis and Provides a Proliferation Advantage after Genotoxic Stress
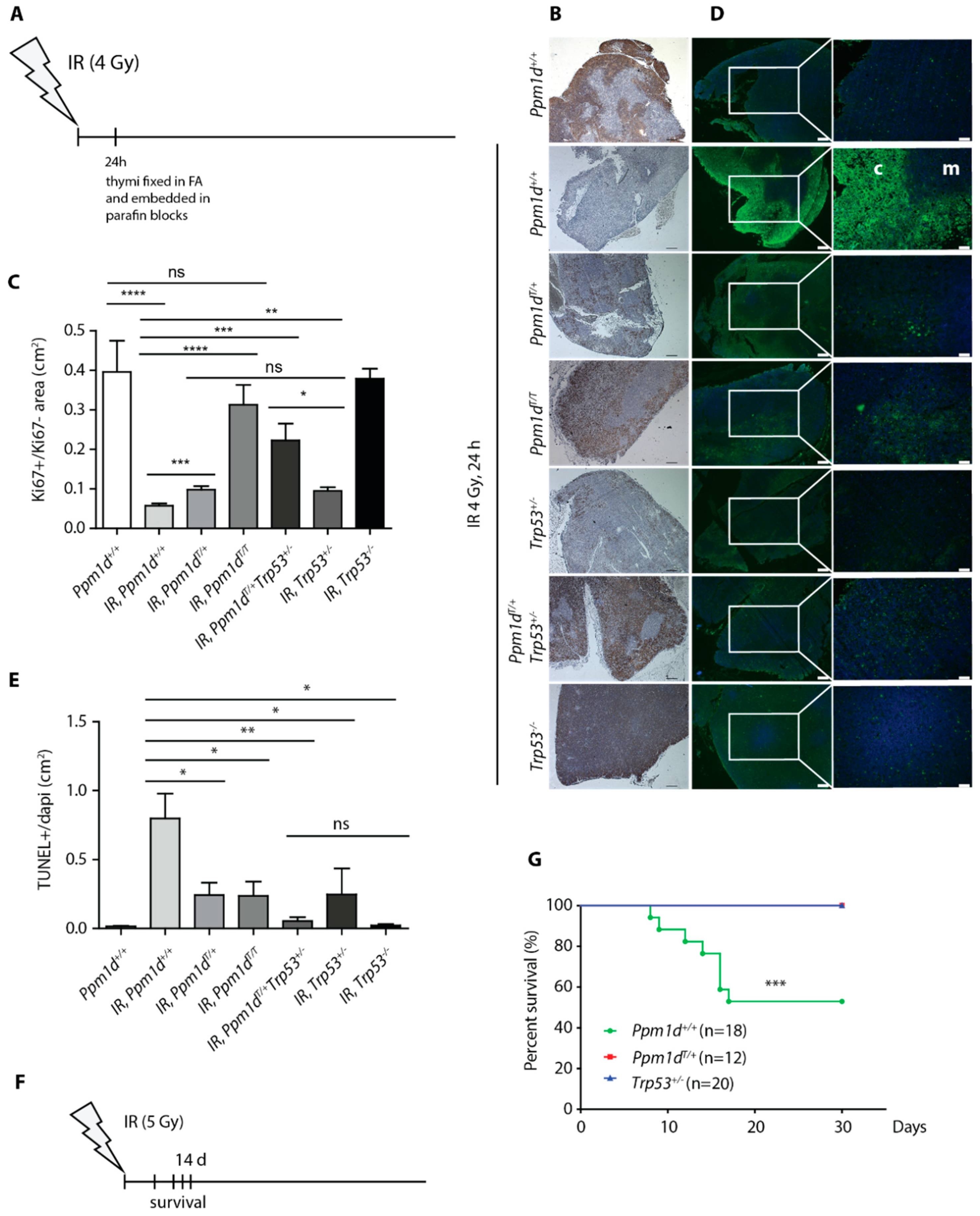
Decreased levels of CDKN1Ap21 and of cleaved caspase-3 in the thymi of the Ppm1dT/+ mice compared to the wild-type animals suggested that truncated PPM1D may impair activation of the cell-cycle checkpoint and induction of apoptosis after exposure to IR. Consistent with the cell-cycle arrest, we observed dramatically reduced signal of the proliferation marker Ki-67 in the thymi from the irradiated (4 Gy) wild-type mice (Figure 2A–C). Under the same conditions, Ppm1dT/+ mice showed significantly higher staining of Ki-67 marker upon irradiation to a level comparable with Trp53+/− heterozygotes (Figure 2B,C, Supplementary Figure S2A). Further increase in the proliferation was observed in Ppm1dT/+Trp53+/− double heterozygotes and in Ppm1dT/T animals exposed to IR and the Ki-67 signal in these mice was comparable to the control wild-type animals that were not exposed to genotoxic stress (Figure 2B,C). We conclude that the proliferation of thymocytes in Ppm1dT/+ animals may continue despite the presence of DNA damage. This defect in the checkpoint is further increased in Ppm1dT/T homozygotes suggesting that higher level of PPM1D may be needed to fully suppress the p53 pathway. Moreover, the intermediate phenotype observed in p53 heterozygotes is enhanced by truncation of one allele of the Ppm1d.
Next, we analyzed the same set of samples using TUNEL assay for DNA fragmentation, which is a hallmark of apoptotic cells [47,48]. Opposite to the Ki-67 signal, we observed high induction of the TUNEL+ signal in the thymic cortex of the wild-type animals 24 h after exposure to IR (Figure 2D,E, Supplementary Figure S2B). Notably, TUNEL+ signal was lower in Ppm1dT/+ and Ppm1dT/T mice and was further reduced in Ppm1dT/+Trp53+/− double heterozygotes to the level comparable to Trp53−/− knock-out animals (Figure 2D,E). To confirm that occurrence of the TUNEL signal corresponded to induction of apoptosis, we stained the sections from the thymi for the cleaved caspase 3 (Supplementary Figure S3A,B). Indeed, we observed a strong activation of the caspase 3 in the wild-type mice at 6 h after irradiation, whereas the staining was reduced in Ppm1dT/+ animals suggesting that the truncated PPM1D impaired induction of the apoptosis (Supplementary Figure S3A,B). Finally, we analyzed the thymi from the control and irradiated mice by flow cytometry (Supplementary Figure S3C). Although the DP CD4+/CD8+ cells represented a major population in the thymi of the control mice, we observed a strong reduction of the CD4+/CD8+ cells upon irradiation. On the other hand, reduction of the CD4+/CD8+ population was significantly lower in Ppm1dT/+ animals suggesting that truncated PPM1D protected these cells from programmed cell death. Overall, our data are consistent with impaired induction of apoptosis in the thymi of mice carrying truncated PPM1D although we have not formally excluded the possibility that the T-cells escape cell death by reverting the late stage of apoptosis through a process called anastasis [49,50].
Depending on the dose of IR, cell death occurs in various tissues and a loss of the stem cells in the bone marrow and the gastrointestinal tract can lead to a fatal failure of these organs. As we observed decreased level of apoptosis in the thymi of the Ppm1dT/+ animals we hypothesized that these animals could be more resistant to IR. Indeed, we observed that about half of the wild-type mice died within 2 weeks upon exposure to a single dose (5 Gy) of whole-body irradiation (Figure 2F,G). In striking contrast, none of the Ppm1dT/+ mice died under these conditions confirming that truncated PPM1D efficiently suppresses cell death in various tissues (Figure 2G). Interestingly, Trp53+/− mice were also resistant to IR suggesting that the effect of truncated Ppm1d could be mediated by partial suppression of p53 function.
3.3. Truncated PPM1D Promotes Formation of the Ionizing Radiation-Induced Lymphoma
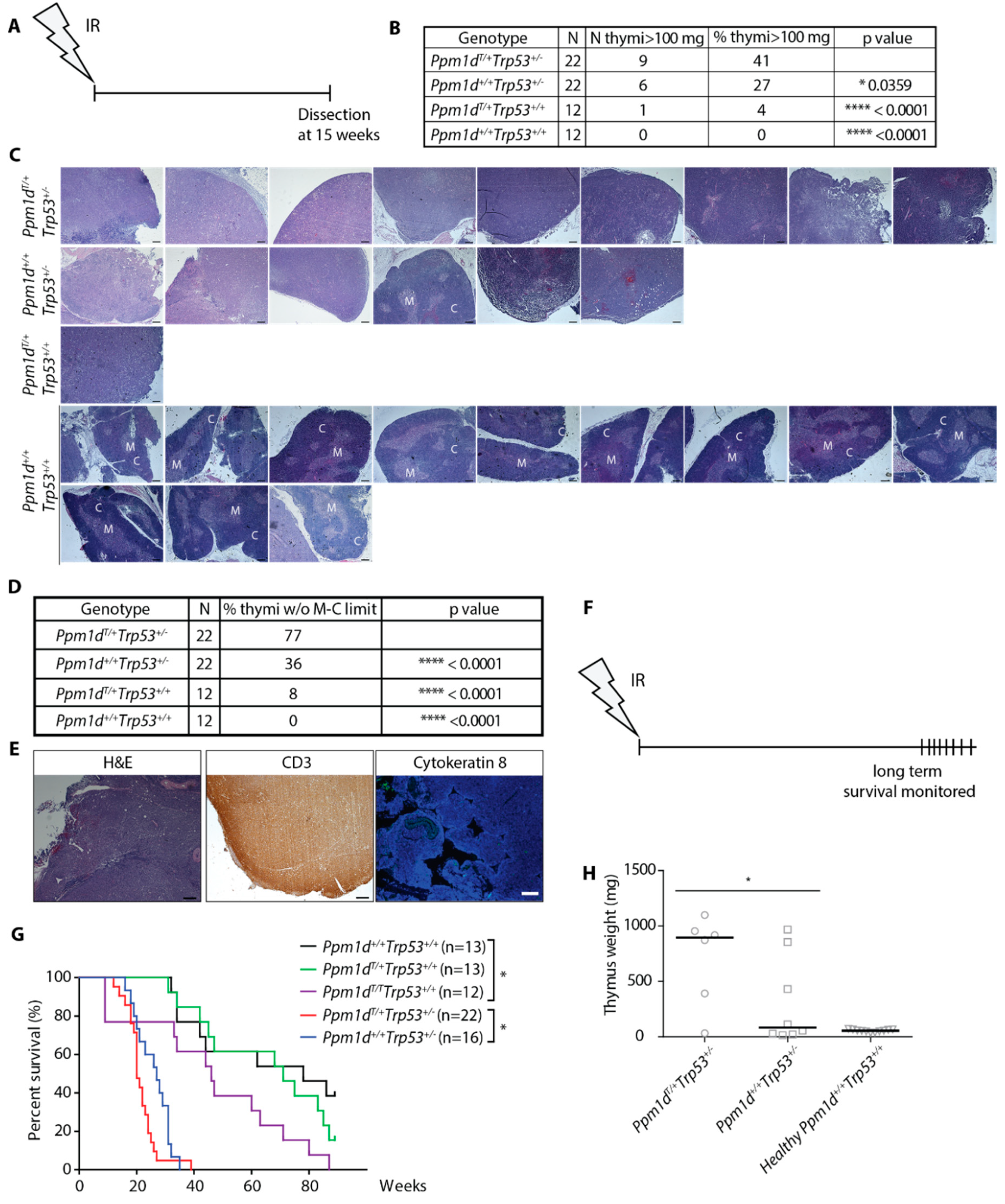
Encouraged by a striking difference in the survival of Ppm1dT/+ and Ppm1d+/+ animals after exposure to a high dose of IR, we aimed to investigate the long-term consequences of the exposure to a sublethal dose. We hypothesized that continued proliferation in the presence of genotoxic stress may lead to genome instability and promote cancer development in animals carrying truncated Ppm1d. To test this possibility we employed a model of IR-induced thymic lymphoma that develops in mice with suppressed p53 pathway upon single exposure to a sublethal dose of ionizing radiation [51,52]. As expected, all wild-type animals (12 out of 12) showed normal thymus size (Figure 3A,B, Supplementary Figure S4A) and morphology defined by the medullo-cortical delimitation (Figure 3C,D) 15 weeks after exposure to 4 Gy. As expected, about one fourth (6/22) of the Trp53+/− mice showed increased thymus weight due to lymphoma development (Figure 3B, Supplementary Figure S4A).
Under the same conditions, one out of 12 Ppm1dT/+ animals had an enlarged thymus (Figure 3B, Supplementary Figure S4A) and showed impaired delineation between thymic medulla and cortex (Figure 3C,D) indicative of the tumor growth [53]. Notably, higher penetrance of the phenotype was observed in Ppm1dT/+Trp53+/− double heterozygotes out of which 41% (9/22) developed lymphoma (Figure 3B, Supplementary Figure S3A). Similarly, the loss of medulla/cortex delineation increased in the Ppm1dT/+Trp53+/− double heterozygotes compared to Trp53+/− mice (Figure 3C,D). Immunohistochemical staining for CD3 (a T-cell marker) and cytokeratin 8 (a marker of thymic epithelial cells, [54]) revealed that the thymic tumors were T-cell lymphomas (Figure 3E). In addition to the T-cell lymphoma, one of the Ppm1dT/+Trp53+/− mice with a normal thymus size developed a fibrosarcoma (Supplementary Figure S4B). Another Ppm1dT/+Trp53+/− mouse showed impaired medulla/cortex delineation in a thymus of a border size (80.2 mg vs. 62.8 mg median thymus weight in the wild-type animals) and enlarged kidney also referred to as hydronephros) that has previously been linked with T-cell lymphomas [55] (Supplementary Figure S4C). In summary, our data suggest that truncation of Ppm1d might slightly increase frequency of the thymic lymphoma development (1/12 compared to 0/12 tumors in the wild-type animals) although considerably larger animal groups would be required to evaluate the statistical significance. On the other hand, we observed a significantly increased number of the tumors in Ppm1dT/+Trp53+/− double heterozygotes compared to Trp53+/− indicating that truncated Ppm1d may promote tumorigenesis in cells with a partial loss of p53 function.
Finally, we compared survival of mice after single exposure to a sublethal dose of IR. We found that median survival of the wild-type mice was about 70 weeks and was reduced to 30 weeks in Trp53+/− heterozygotes (Figure 3F,G). Significant reduction of the survival to 20 weeks was observed in Ppm1dT/+Trp53+/− double heterozygotes which is consistent with the increased frequency of the tumors described above (Figure 3G). Under the same conditions, overall survival of Ppm1dT/+ was indistinguishable from survival of the wild-type animals. However, survival of Ppm1dT/T homozygotes was reduced to 50 weeks which is consistent with a more pronounced defect in DNA damage response in Ppm1dT/T homozygotes compared to Ppm1dT/+ heterozygotes (Figure 3G). Moribund mice were sacrificed, and subsequent autopsy confirmed the presence of a T-cell lymphoma in 83% of Ppm1dT/+Trp53+/− mice compared to 37% in Trp53+/− mice (Figure 3H). We conclude that truncated Ppm1d reduces the survival of mice with a partial loss of p53 due to the increased frequency of the T-cell lymphoma formation.
4. Discussion
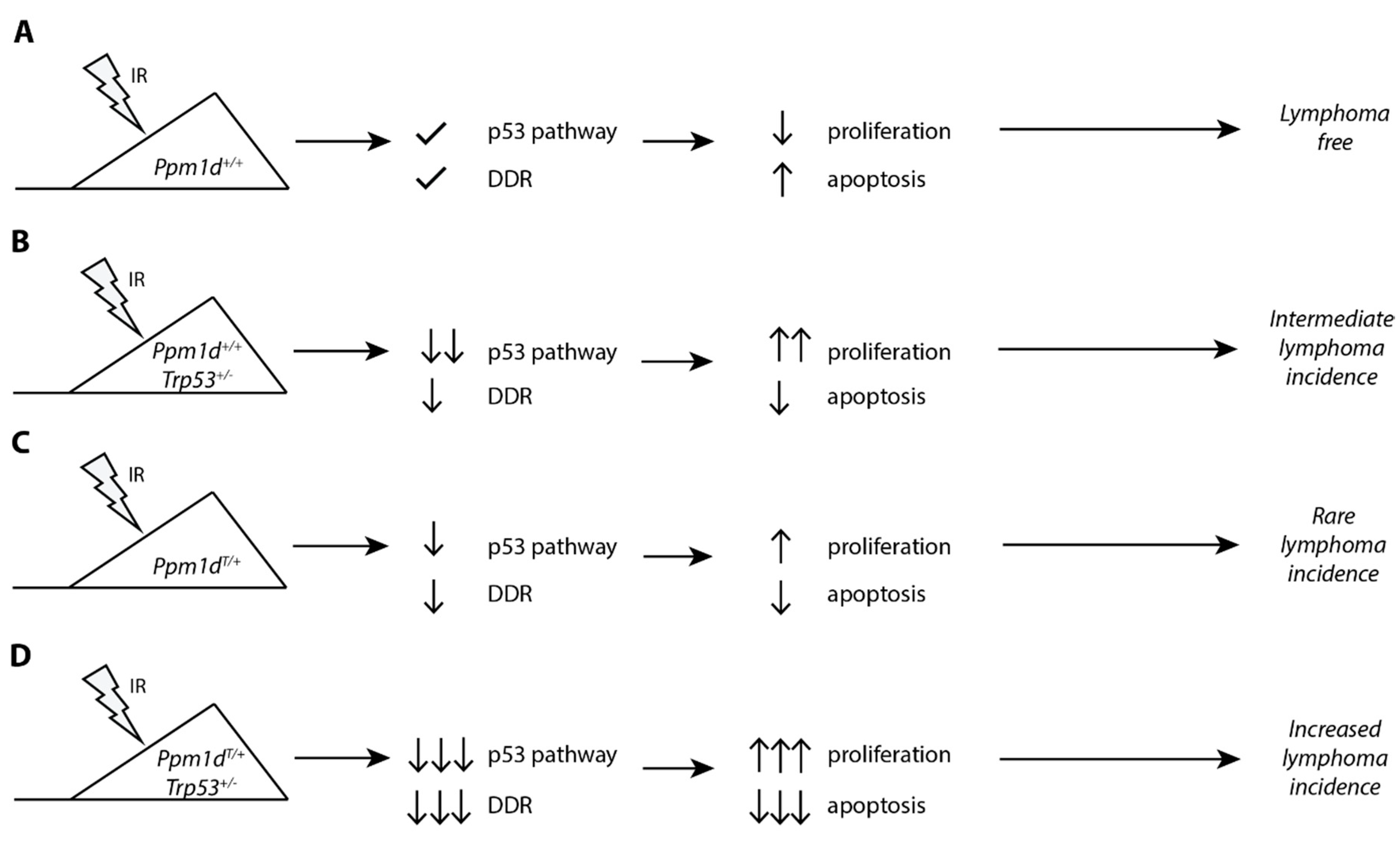
In this study, we exploited our recently generated transgenic model of the truncated Ppm1d to study its impact on DNA damage response in T lymphocytes and to evaluate its role in tumorigenesis (Figure 4). As with the mouse intestinal stem cells, we observe that the truncated Ppm1d accumulates in the T-cells at high quantities and impairs the cell response to genotoxic stress. Upon exposure to IR, cells carrying truncated Ppm1d express lower amount of CDKN1Ap21 and PUMA. As a result, the proliferation of the thymocytes in Ppm1dT/+ mice is not completely suppressed after exposure to IR which is consistent with a deficient checkpoint arrest that we previously reported in Human cells [31]. Moreover, clearance of damaged cells by a programmed cell death is also suppressed in the thymi of Ppm1dT/+ mice. Continuous cell proliferation and suppressed cell death under the condition of genotoxic stress are even more prominent in Ppm1dT/+Trp53+/− mice suggesting that truncation of Ppm1d suppresses p53 function only partially. In addition, we observed that the Ppm1dT/T homozygote showed a stronger phenotype than Ppm1dT/+ heterozygote in terms of the suppression of p53 function. This finding is surprising considering that a homozygous PPM1D mutation has not been reported in human tumors so far. Cancer cell lines carrying truncated PPM1D (including U2OS and HCT116 cells) are heterozygotes, and also colon carcinoma samples do not contain both mutant alleles [31,35]. Multiple nonsense or frame-shift mutations in the PPM1D observed in Acute Myeloid Leukemia (AML) patients spread throughout the whole exon 6 which results in many variants of the PPM1D protein with differently a sized C-terminal tail [34,56]. When performing CRISPR/Cas9 mutagenesis at various positions in exon 6 of the PPM1D we observed that some of the truncated PPM1D variants exhibited higher protein stability than others (data not shown). It is plausible that a similar situation occurs in human tumors and that mutations of PPM1D resulting in higher protein stability may allow stronger suppression of p53 than mutations leading to lower PPM1D levels. Although the stability of Ppm1d in the mouse model is clearly increased and the truncated Ppm1d is present at a higher level than the full-length Ppm1d, we observe just a partial effect on suppression of p53 resulting in a relatively weak cancer phenotype. Slight increase of Ppm1d activity in the Ppm1dT/T homozygote could suppress p53 function more efficiently leading to increased probability of tumor formation. We speculate that another Ppm1d truncation leading to higher protein stability could possibly reach the threshold required for efficient suppression of p53 already in a heterozygous state. It will be interesting to address this possibility in the future by generating other mouse models with nonsense mutations at various positions in exon 6 of the Ppm1d.
Amplification of the PPM1D locus leading to high expression levels of the full-length PPM1D protein is relatively common in human breast cancer and was observed mainly in tumors retaining the wild-type p53 [24,57]. Data from the transgenic mouse model used in this study suggest that the truncated Ppm1d has the potential to increase the pathogenicity of a partial loss of p53. Future research is needed to experimentally validate this possibility in clinically relevant human cancers.
Supplementary Materials
The following are available online at https://www.mdpi.com/2073-4409/9/9/2068/s1, Figure S1: Truncated PPM1D impairs DNA damage response, Figure S2: Image thresholding for quantification of proliferation and apoptosis in thymus, Figure S3: Truncated PPM1D prevents activation of caspase 3 after genotoxic stress, Figure S4: Truncated PPM1D promotes tumorigenesis in p53+/− background.
Author Contributions
Conceptualization, L.M.; investigation, A.S.M., M.B.; validation, M.S.; writing—original draft preparation, A.S.M. and L.M.; writing—review and editing, L.M.; funding acquisition, L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Czech Science Foundation (20-11931S) and the Czech Academy of Sciences (RVO 68378050). Animal work was partially supported by Czech Centre for Phenogenomics (project no. LM2015040), OP RDI CZ.1.05/2.1.00/19.0395 (project: Higher quality and capacity for transgenic models) and Biotechnology and Biomedicine Centre of the Academy of Sciences and Charles University (CZ.1.05/1.1.00/02.0109). Imaging was performed in Light Microscopy Core Facility of IMG supported by MEYS (LM2015062, CZ.02.1.01/0.0/0.0/16_013/0001775), OPPK (CZ.2.16/3.1.00/21547) and RVO: 68378050-KAV-NPUI. All experimental work was performed at IMG. ASM was partially supported by Grant Agency of the Charles University (140421).
Acknowledgments
Authors are grateful to Jozef Pavel and Zuzana Chalupova for technical assistance, Michaela Efenberkova (Institute of Molecular Genetics (IMG), Prague) for image analysis, Peter Makovicky (Biotechnology and Biomedicine Center of the Academy of Sciences and Charles University, Prague) for histological examination of the tumors, Martin Gregor (IMG, Prague) for providing reagents, Jan Dobes (IMG, Prague) for help with flow cytometry and Marketa Vancurova for text editing.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Medema, R.H.; Macůrek, L. Checkpoint control and cancer. Oncogene 2012, 31, 2601–2613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, M.; Raaijmakers, J.A.; Bakker, B.; Spierings, D.C.J.; Lansdorp, P.M.; Foijer, F.; Medema, R.H. p53 Prohibits Propagation of Chromosome Segregation Errors that Produce Structural Aneuploidies. Cell Rep. 2017, 19, 2423–2431. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.-Y.; Ikeda, M.; Taya, Y.; Prives, C. DNA Damage-Induced Phosphorylation of p53 Alleviates Inhibition by MDM2. Cell 1997, 91, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Shieh, S.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev. 2000, 14, 289–300. [Google Scholar]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Sørensen, C.S.; Syljuåsen, R.G.; Falck, J.; Schroeder, T.; Rönnstrand, L.; Khanna, K.K.; Zhou, B.B.; Bartek, J.; Lukas, J. Chk1 regulates the S phase checkpoint by coupling the physiological turnover and ionizing radiation-induced accelerated proteolysis of Cdc25A. Cancer Cell 2003, 3, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Bunz, F.; Dutriaux, A.; Lengauer, C.; Waldman, T.; Zhou, S.; Brown, J.P.; Sedivy, J.M.; Kinzler, K.W.; Vogelstein, B. Requirement for p53 and p21 to Sustain G2 Arrest after DNA Damage. Science 1998, 282, 1497–1501. [Google Scholar] [CrossRef]
- Lindqvist, A.; de Bruijn, M.; Macurek, L.; Bras, A.; Mensinga, A.; Bruinsma, W. Wip1 confers G2 checkpoint recovery competence by counteracting p53-dependent transcriptional repression. EMBO J. 2009, 28, 3196–3206. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, H.; Benada, J.; Müllers, E.; Akopyan, K.; Burdova, K.; Koolmeister, T.; Helleday, T.; Medema, R.H.; Macurek, L.; Lindqvist, A. ATM/Wip1 activities at chromatin control Plk1 re-activation to determine G2 checkpoint duration. EMBO J. 2017, 36, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Shreeram, S.; Demidov, O.N.; Hee, W.K.; Yamaguchi, H.; Onishi, N.; Kek, C.; Timofeev, O.N.; Dudgeon, C.; Fornace, A.J.; Anderson, C.W.; et al. Wip1 Phosphatase Modulates ATM-Dependent Signaling Pathways. Mol. Cell 2006, 23, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Fiscella, M.; Zhang, H.; Fan, S.; Sakaguchi, K.; Shen, S.; Mercer, W.E.; Vande Woude, G.F.; O’Connor, P.M.; Appella, E. Wip1, a novel human protein phosphatase that is induced in response to ionizing radiation in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 1997, 94, 6048–6053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krenning, L.; Feringa, F.M.; Shaltiel, I.A.; van den Berg, J.; Medema, R.H. Transient Activation of p53 in G2 Phase Is Sufficient to Induce Senescence. Mol. Cell 2014, 55, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müllers, E.; Cascales, H.S.; Jaiswal, H.; Saurin, A.T.; Lindqvist, A. Nuclear translocation of Cyclin B1 marks the restriction point for terminal cell cycle exit in G2 phase. Cell Cycle 2014, 13, 2733–2743. [Google Scholar] [CrossRef] [Green Version]
- Feringa, F.M.; Raaijmakers, J.A.; Hadders, M.A.; Vaarting, C.; Macurek, L.; Heitink, L.; Krenning, L.; Medema, R.H. Persistent repair intermediates induce senescence. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Burdova, K.; Storchova, R.; Palek, M.; Macurek, L. WIP1 Promotes Homologous Recombination and Modulates Sensitivity to PARP Inhibitors. Cells 2019, 8, 1258. [Google Scholar] [CrossRef] [Green Version]
- Purvis, J.E.; Karhohs, K.W.; Mock, C.; Batchelor, E.; Loewer, A.; Lahav, G. p53 dynamics control cell fate. Science 2012, 336, 1440–1444. [Google Scholar] [CrossRef] [Green Version]
- Barr, A.R.; Cooper, S.; Heldt, F.S.; Butera, F.; Stoy, H.; Mansfeld, J.; Novák, B.; Bakal, C. DNA damage during S-phase mediates the proliferation-quiescence decision in the subsequent G1 via p21 expression. Nat. Commun. 2017, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Shaltiel, I.A.; Aprelia, M.; Saurin, A.T.; Chowdhury, D.; Kops, G.J.P.L.; Voest, E.E.; Medema, R.H. Distinct phosphatases antagonize the p53 response in different phases of the cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 7313–7318. [Google Scholar] [CrossRef] [Green Version]
- Bartkova, J.; Horejsi, Z.; Koed, K.; Kramer, A.; Tort, F.; Zieger, K.; Guldberg, P.; Sehested, M.; Nesland, J.M.; Lukas, C.; et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature 2005, 434, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.-V.F.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef] [PubMed]
- Bulavin, D.V.; Demidov, O.N.; Saito, S.I.; Kauraniemi, P.; Phillips, C.; Amundson, S.A.; Ambrosino, C.; Sauter, G.; Nebreda, A.R.; Anderson, C.W.; et al. Amplification of PPM1D in human tumors abrogates p53 tumor-suppressor activity. Nat. Genet. 2002, 31, 210–215. [Google Scholar] [CrossRef]
- Emelyanov, A.; Bulavin, D.V. Wip1 phosphatase in breast cancer. Oncogene 2015, 34, 4429–4438. [Google Scholar] [CrossRef] [Green Version]
- Yu, E.; Ahn, Y.S.; Jang, S.J.; Kim, M.J.; Yoon, H.S.; Gong, G.; Choi, J. Overexpression of the wip1 gene abrogates the p38 MAPK/p53/Wip1 pathway and silences p16 expression in human breast cancers. Breast Cancer Res. Treat. 2007, 101, 269–278. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16Ink4a-p19Arf pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Demidov, O.N.; Kek, C.; Shreeram, S.; Timofeev, O.; Fornace, A.J.; Appella, E.; Bulavin, D.V. The role of the MKK6//p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene 2006, 26, 2502–2506. [Google Scholar] [CrossRef] [Green Version]
- Pechackova, S.; Burdova, K.; Benada, J.; Kleiblova, P.; Jenikova, G.; Macurek, L. Inhibition of WIP1 phosphatase sensitizes breast cancer cells to genotoxic stress and to MDM2 antagonist nutlin-3. Oncotarget 2016, 7, 14458–14475. [Google Scholar] [CrossRef] [Green Version]
- Pecháčková, S.; Burdová, K.; Macurek, L. WIP1 phosphatase as pharmacological target in cancer therapy. J. Mol. Med. 2017, 95, 589–599. [Google Scholar] [CrossRef] [Green Version]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Sevčík, J.; Pecháčková, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Chen, L.H.; Wan, H.; Yang, R.; Wang, Z.; Feng, J.; Yang, S.; Jones, S.; Wang, S.; Zhou, W.; et al. Exome sequencing identifies somatic gain-of-function PPM1D mutations in brainstem gliomas. Nat. Genet. 2014, 46, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kahn, J.D.; Miller, P.G.; Silver, A.J.; Sellar, R.S.; Bhatt, S.; Gibson, C.; McConkey, M.; Adams, D.; Mar, B.; Mertins, P.; et al. PPM1D truncating mutations confer resistance to chemotherapy and sensitivity to PPM1D inhibition in hematopoietic cells. Blood 2018, 132, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Burocziova, M.; Burdova, K.; Martinikova, A.S.; Kasparek, P.; Kleiblova, P.; Danielsen, S.A.; Borecka, M.; Jenikova, G.; Janečková, L.; Pavel, J.; et al. Truncated PPM1D impairs stem cell response to genotoxic stress and promotes growth of APC-deficient tumors in the mouse colon. Cell Death Dis. 2019, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Germain, R.N. T-cell development and the CD4–CD8 lineage decision. Nat. Rev. Immunol. 2002, 2, 309–322. [Google Scholar] [CrossRef]
- Ceredig, R.; Rolink, T. A positive look at double-negative thymocytes. Nat. Rev. Immunol. 2002, 2, 888–897. [Google Scholar] [CrossRef]
- Bogue, M.A.; Zhu, C.; Aguilar-Cordova, E.; Donehower, L.A.; Roth, D.B. p53 is required for both radiation-induced differentiation and rescue of V(D)J rearrangement in scid mouse thymocytes. Genes Dev. 1996, 10, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Guidos, C.J.; Williams, C.J.; Grandal, I.; Knowles, G.; Huang, M.T.; Danska, J.S. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev. 1996, 10, 2038–2054. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Lenardo, M.J.; Zúñiga-Pflücker, J.C. p53 prevents maturation to the CD4+CD8+ stage of thymocyte differentiation in the absence of T cell receptor rearrangement. J. Exp. Med. 1996, 183, 1923–1928. [Google Scholar] [CrossRef] [Green Version]
- Schito, M.; Demidov, O.; Saito, S.; Ashwell, J.; Appella, E. Wip1 phosphatase-deficient mice exhibit defective T cell maturation due to sustained p53 activation. J. Immunol. 2006, 176, 4818–4825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Li, H.; Luo, H.; Zhang, L.; Hu, X.; Yang, T.; Sun, C.; Chen, H.; Zhang, L.; Zhao, Y. Phosphatase Wip1 is essential for the maturation and homeostasis of medullary thymic epithelial cells in mice. J. Immunol. 2013, 191, 3210–3220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, W.; Hu, X.; Chen, Z.; Liu, L.; Tian, Y.; Chen, H.; Cong, Y.S.; Yang, F.; Zhang, L.; Rudolph, K.L.; et al. Phosphatase Wip1 controls antigen-independent B-cell development in a p53-dependent manner. Blood 2015, 126, 620–628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uyanik, B.; Grigorash, B.B.; Goloudina, A.R.; Demidov, O.N. DNA damage-induced phosphatase Wip1 in regulation of hematopoiesis, immune system and inflammation. Cell Death Discov. 2017, 3, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Jacks, T.; Remington, L.; Williams, B.O.; Schmitt, E.M.; Halachmi, S.; Bronson, R.T.; Weinberg, R.A. Tumor spectrum analysis in p53-mutant mice. Curr. Biol. 1994, 4, 1–7. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Enari, M.; Sakahira, H.; Yokoyama, H.; Okawa, K.; Iwamatsu, A.; Nagata, S. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 1998, 391, 43–50. [Google Scholar] [CrossRef]
- Gorczyca, W.; Bruno, S.; Darzynkiewicz, R.; Gong, J.; Darzynkiewicz, Z. DNA strand breaks occurring during apoptosis—Their early insitu detection by the terminal deoxynucleotidyl transferase and nick translation assays and prevention by serine protease inhibitors. Int. J. Oncol. 1992, 1, 639–648. [Google Scholar] [CrossRef]
- Zhao, R.; Kaakati, R.; Lee, A.K.; Liu, X.; Li, F.; Li, C.Y. Novel roles of apoptotic caspases in tumor repopulation, epigenetic reprogramming, carcinogenesis, and beyond. Cancer Metastasis Rev. 2018, 37, 227–236. [Google Scholar] [CrossRef]
- Tang, H.L.; Tang, H.M.; Mak, K.H.; Hu, S.; Wang, S.S.; Wong, K.M.; Wong, C.S.; Wu, H.Y.; Law, H.T.; Liu, K.; et al. Cell survival, DNA damage, and oncogenic transformation after a transient and reversible apoptotic response. Mol. Biol. Cell 2012, 23, 2240–2252. [Google Scholar] [CrossRef]
- Kemp, C.J.; Wheldon, T.; Balmain, A. p53-deficient mice are extremely susceptible to radiation-induced tumorigenesis. Nat. Genet. 1994, 8, 66–69. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Ströbel, P.; Hartmann, E.; Rosenwald, A.; Kalla, J.; Ott, G.; Friedel, G.; Schalke, B.; Kasahara, M.; Tomaru, U.; Marx, A. Corticomedullary differentiation and maturational arrest in thymomas. Histopathology 2014, 64, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Odaka, C.; Loranger, A.; Takizawa, K.; Ouellet, M.; Tremblay, M.J.; Murata, S.; Inoko, A.; Inagaki, M.; Marceau, N. Keratin 8 is required for the maintenance of architectural structure in thymus epithelium. PLoS ONE 2013, 8, e75101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauder, A.J.; Jolin, H.E.; Smith, P.; van den Berg, J.G.; Jones, A.; Wisden, W.; Smith, K.G.; Dasvarma, A.; Fallon, P.G.; McKenzie, A.N. Lymphomagenesis, hydronephrosis, and autoantibodies result from dysregulation of IL-9 and are differentially dependent on Th2 cytokines. J. Immunol. 2004, 173, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Hsu, J.I.; Dayaram, T.; Tovy, A.; De Braekeleer, E.; Jeong, M.; Wang, F.; Zhang, J.; Heffernan, T.P.; Gera, S.; Kovacs, J.J.; et al. PPM1D Mutations Drive Clonal Hematopoiesis in Response to Cytotoxic Chemotherapy. Cell Stem Cell 2018, 23, 700–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauta, J.; Alarmo, E.-L.; Kauraniemi, P.; Karhu, R.; Kuukasjärvi, T.; Kallioniemi, A. The serine-threonine protein phosphatase PPM1D is frequently activated through amplification in aggressive primary breast tumours. Breast Cancer Res. Treat. 2006, 95, 257–263. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Truncated PPM1D impairs DNA damage response in mouse thymus. Expression of PPM1D mRNA was analyzed by RT-qPCR in the thymi of Ppm1d+/+, Ppm1dT/+ and Ppm1dT/T mice and was normalized to GAPDH (n = 3) (A). Thymi from mice of indicated genotypes were lysed and proteins were separated by SDS-PAGE. Samples were probed with antibody against PPM1D and importin-β as a loading control. The empty and full arrowheads indicate the position of the full-length and the C-terminally truncated PPM1D, respectively. (B). Cells from thymi from Ppm1d+/+ and Ppm1dT/+ mice were analyzed by flow cytometry. Plotted are the counts of the indicated populations as follows: double-negative T-cells (DN and DN1, DN2, DN3, DN4), double-positive T-cells (DP), CD8-positive T-cells (CD8+) and CD4-positive T-cells (CD4+) (n = 3) (C). The median size of the thymus was determined in Ppm1d +/+ (n = 11) and Ppm1dT/+ (n = 12) mice (D). A scheme of the experimental setup in F-I. Mice were exposed or not to a low dose of IR (3 Gy), sacrificed after 6 h and thymi and lymph nodes were collected (E). Proteins isolated from thymi from mice of indicated genotypes exposed to mock or to IR were probed with the indicated antibodies by immunoblotting (F). Proteins isolated from inguinal lymph nodes from mice of indicated genotypes exposed to mock or to IR were probed with the indicated antibodies by immunoblotting (G). RNA isolated from thymi from mice in E was analyzed by RT-qPCR. The expression of CDKN1Ap21 mRNA was normalized to GAPDH. Statistical significance was evaluated by two-tailed t-test, error bars indicate SD, n = 5 (H). RNA isolated from thymi from mice in D was analyzed by RT-qPCR. The expression of PUMA mRNA was normalized to GAPDH. Statistical significance was evaluated by two-tailed t-test, error bars indicate SD, n = 5. * p <0.05; *** p < 0.0005; **** p < 0.0001 (I).
Figure 1.
Truncated PPM1D impairs DNA damage response in mouse thymus. Expression of PPM1D mRNA was analyzed by RT-qPCR in the thymi of Ppm1d+/+, Ppm1dT/+ and Ppm1dT/T mice and was normalized to GAPDH (n = 3) (A). Thymi from mice of indicated genotypes were lysed and proteins were separated by SDS-PAGE. Samples were probed with antibody against PPM1D and importin-β as a loading control. The empty and full arrowheads indicate the position of the full-length and the C-terminally truncated PPM1D, respectively. (B). Cells from thymi from Ppm1d+/+ and Ppm1dT/+ mice were analyzed by flow cytometry. Plotted are the counts of the indicated populations as follows: double-negative T-cells (DN and DN1, DN2, DN3, DN4), double-positive T-cells (DP), CD8-positive T-cells (CD8+) and CD4-positive T-cells (CD4+) (n = 3) (C). The median size of the thymus was determined in Ppm1d +/+ (n = 11) and Ppm1dT/+ (n = 12) mice (D). A scheme of the experimental setup in F-I. Mice were exposed or not to a low dose of IR (3 Gy), sacrificed after 6 h and thymi and lymph nodes were collected (E). Proteins isolated from thymi from mice of indicated genotypes exposed to mock or to IR were probed with the indicated antibodies by immunoblotting (F). Proteins isolated from inguinal lymph nodes from mice of indicated genotypes exposed to mock or to IR were probed with the indicated antibodies by immunoblotting (G). RNA isolated from thymi from mice in E was analyzed by RT-qPCR. The expression of CDKN1Ap21 mRNA was normalized to GAPDH. Statistical significance was evaluated by two-tailed t-test, error bars indicate SD, n = 5 (H). RNA isolated from thymi from mice in D was analyzed by RT-qPCR. The expression of PUMA mRNA was normalized to GAPDH. Statistical significance was evaluated by two-tailed t-test, error bars indicate SD, n = 5. * p <0.05; *** p < 0.0005; **** p < 0.0001 (I).

Figure 2.
Truncated PPM1D prevents apoptosis and promotes proliferation after genotoxic stress. A scheme of the experimental setup of B–E. Thymi were collected from mice sacrificed 24 h after exposure to mock or to IR (4 Gy) and analyzed by immunohistochemistry (A). Histology sections of thymi from mice of indicated genotypes exposed to mock or to IR (4 Gy) were probed with antibody against Ki-67 (proliferation marker). Representative images are shown. Magnification 5×, bars indicate 200 μm (B). Quantification of Ki-67 signal from B. At least 3 sections from 3 mice per genotype were quantified. Error bars indicate SD. Statistical significance was evaluated by two-tailed t-test, * p < 0.05; ** p < 0.01; *** p < 0.0005; **** p < 0.0001. (C). Histology sections of thymi from mice of indicated genotypes exposed to mock or to IR were subjected to TUNEL assay (apoptosis marker). Note high TUNEL+ signal in cortex (c) but not in medulla (m) of the Ppm1d+/+Trp53+/+ mice. Representative images are shown. Magnification 5× and 20×, bars indicate 200 μm and 50 μm, respectively (D). Quantification of TUNEL+ signal from D. At least 3 sections from 3 mice per genotype were quantified. Error bars indicate SD. Statistical significance was evaluated by two-tailed t-test, * p < 0.05; ** p < 0.01 (E). A scheme of the experimental setup in G (F). Wild-type Ppm1d+/+Trp53+/+, Ppm1dT/+Trp53+/+, and Ppm1d+/+Trp53+/− mice were exposed to 5 Gy of IR and their survival was monitored for subsequent 30 days. n values indicate the numbers of animals of each genotype. Statistical significance of the Kaplan-Meier survival plot was determined by a log-rank test, **** p < 0.005 (G).
Figure 2.
Truncated PPM1D prevents apoptosis and promotes proliferation after genotoxic stress. A scheme of the experimental setup of B–E. Thymi were collected from mice sacrificed 24 h after exposure to mock or to IR (4 Gy) and analyzed by immunohistochemistry (A). Histology sections of thymi from mice of indicated genotypes exposed to mock or to IR (4 Gy) were probed with antibody against Ki-67 (proliferation marker). Representative images are shown. Magnification 5×, bars indicate 200 μm (B). Quantification of Ki-67 signal from B. At least 3 sections from 3 mice per genotype were quantified. Error bars indicate SD. Statistical significance was evaluated by two-tailed t-test, * p < 0.05; ** p < 0.01; *** p < 0.0005; **** p < 0.0001. (C). Histology sections of thymi from mice of indicated genotypes exposed to mock or to IR were subjected to TUNEL assay (apoptosis marker). Note high TUNEL+ signal in cortex (c) but not in medulla (m) of the Ppm1d+/+Trp53+/+ mice. Representative images are shown. Magnification 5× and 20×, bars indicate 200 μm and 50 μm, respectively (D). Quantification of TUNEL+ signal from D. At least 3 sections from 3 mice per genotype were quantified. Error bars indicate SD. Statistical significance was evaluated by two-tailed t-test, * p < 0.05; ** p < 0.01 (E). A scheme of the experimental setup in G (F). Wild-type Ppm1d+/+Trp53+/+, Ppm1dT/+Trp53+/+, and Ppm1d+/+Trp53+/− mice were exposed to 5 Gy of IR and their survival was monitored for subsequent 30 days. n values indicate the numbers of animals of each genotype. Statistical significance of the Kaplan-Meier survival plot was determined by a log-rank test, **** p < 0.005 (G).

Figure 3.
Truncated PPM1D promotes formation of IR-induced lymphoma in mice. Schematic of the experimental setup. At 8–10 weeks of age, mice were exposed to a single dose of ionizing radiation (4 Gy) and followed for another 15 weeks when they were sacrificed and subjected to autopsy. Thymi were evaluated by histology (A). Mice of indicated genotypes were treated as in A. Percentage of thymi of abnormal weight (>100 mg) was scored. n value indicates the number of animals in each experimental group. Statistical significance to the Ppm1dT/+Trp53+/− was determined by Fisher’s one-tailed exact test, * p < 0.05; **** p < 0.0001. (B). H&E staining of the enlarged thymi from mice in A and B. Representative images of thymi considered to be enlarged are shown for genotypes Ppm1d+/−, Trp53+/−, and Ppm1dT/+Trp53+/− mice and all thymi from the wild-type mice are shown. Regions of the cortex (C) and medulla (M) are indicated. Magnification 5×, scale bar indicates 200 µm (C). Quantification of delineation between cortex and medulla from the experiment in C. Multiple sections were evaluated to cover the whole thymus. Statistical significance to the Ppm1dT/+Trp53+/− was determined by Fisher’s one-tailed exact test, **** p < 0.0001. (D). Representative images of a thymic tumor from B are shown. Section of the thymus was stained with H&E, or an antibody against CD3 (T-cell marker) or cytokeratin 8 (stromal marker) (E). Schematic of the experimental setup in G-H (F). Mice of indicated genotypes were exposed to a single dose of IR (4 Gy) and their survival was followed in time. n values indicate the numbers of animals of each genotype. Statistical significance of the Kaplan-Meier survival plot was determined by a log-rank test, * p < 0.05. (G). Terminally moribund mice from G were sacrificed, dissected, and median size of thymi was determined. Statistical significance was determined by the one-tailed t-test (p = 0.045) (H).
Figure 3.
Truncated PPM1D promotes formation of IR-induced lymphoma in mice. Schematic of the experimental setup. At 8–10 weeks of age, mice were exposed to a single dose of ionizing radiation (4 Gy) and followed for another 15 weeks when they were sacrificed and subjected to autopsy. Thymi were evaluated by histology (A). Mice of indicated genotypes were treated as in A. Percentage of thymi of abnormal weight (>100 mg) was scored. n value indicates the number of animals in each experimental group. Statistical significance to the Ppm1dT/+Trp53+/− was determined by Fisher’s one-tailed exact test, * p < 0.05; **** p < 0.0001. (B). H&E staining of the enlarged thymi from mice in A and B. Representative images of thymi considered to be enlarged are shown for genotypes Ppm1d+/−, Trp53+/−, and Ppm1dT/+Trp53+/− mice and all thymi from the wild-type mice are shown. Regions of the cortex (C) and medulla (M) are indicated. Magnification 5×, scale bar indicates 200 µm (C). Quantification of delineation between cortex and medulla from the experiment in C. Multiple sections were evaluated to cover the whole thymus. Statistical significance to the Ppm1dT/+Trp53+/− was determined by Fisher’s one-tailed exact test, **** p < 0.0001. (D). Representative images of a thymic tumor from B are shown. Section of the thymus was stained with H&E, or an antibody against CD3 (T-cell marker) or cytokeratin 8 (stromal marker) (E). Schematic of the experimental setup in G-H (F). Mice of indicated genotypes were exposed to a single dose of IR (4 Gy) and their survival was followed in time. n values indicate the numbers of animals of each genotype. Statistical significance of the Kaplan-Meier survival plot was determined by a log-rank test, * p < 0.05. (G). Terminally moribund mice from G were sacrificed, dissected, and median size of thymi was determined. Statistical significance was determined by the one-tailed t-test (p = 0.045) (H).

Figure 4.
A proposed model for the role of truncated PPM1D in the ionizing radiation-induced lymphoma. Mice carrying wild-type Ppm1d and Tpr53 activate the p53 pathway and DNA damage response upon exposure to ionizing radiation leading to block of the cell proliferation or to apoptosis in the thymus. This protects genome integrity and prevents tumor development (A). A partial loss of the p53 function in Trp53+/− mice suppresses activation of the checkpoint induced by ionizing radiation. This allows continuous cell proliferation in the presence of DNA damage leading to accumulation of the genetic changes and lymphoma development (B). Mice carrying C-terminally truncated Ppm1d fail to fully activate p53 and DNA damage response after exposure to ionizing radiation. In most cases, residual p53 function is sufficient to suppress proliferation and tumor formation. However, a fraction of cells proliferates despite the existing DNA damage which may occasionally result in tumor development (C). Combination of a partial loss of p53 and suppression of its function in Ppm1dT/+Trp53+/− mice allows continuous proliferation in the presence of DNA damage and prevents clearance of damaged cells by apoptosis. As a result, thymic lymphoma develops more frequently in Ppm1dT/+Trp53+/− mice compared to Ppm1dT/+ or Trp53+/− alone (D).
Figure 4.
A proposed model for the role of truncated PPM1D in the ionizing radiation-induced lymphoma. Mice carrying wild-type Ppm1d and Tpr53 activate the p53 pathway and DNA damage response upon exposure to ionizing radiation leading to block of the cell proliferation or to apoptosis in the thymus. This protects genome integrity and prevents tumor development (A). A partial loss of the p53 function in Trp53+/− mice suppresses activation of the checkpoint induced by ionizing radiation. This allows continuous cell proliferation in the presence of DNA damage leading to accumulation of the genetic changes and lymphoma development (B). Mice carrying C-terminally truncated Ppm1d fail to fully activate p53 and DNA damage response after exposure to ionizing radiation. In most cases, residual p53 function is sufficient to suppress proliferation and tumor formation. However, a fraction of cells proliferates despite the existing DNA damage which may occasionally result in tumor development (C). Combination of a partial loss of p53 and suppression of its function in Ppm1dT/+Trp53+/− mice allows continuous proliferation in the presence of DNA damage and prevents clearance of damaged cells by apoptosis. As a result, thymic lymphoma develops more frequently in Ppm1dT/+Trp53+/− mice compared to Ppm1dT/+ or Trp53+/− alone (D).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Martinikova, A.S.; Burocziova, M.; Stoyanov, M.; Macurek, L. Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells 2020, 9, 2068. https://doi.org/10.3390/cells9092068
AMA Style
Martinikova AS, Burocziova M, Stoyanov M, Macurek L. Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma. Cells. 2020; 9(9):2068. https://doi.org/10.3390/cells9092068
Chicago/Turabian StyleMartinikova, Andra S., Monika Burocziova, Miroslav Stoyanov, and Libor Macurek. 2020. "Truncated PPM1D Prevents Apoptosis in the Murine Thymus and Promotes Ionizing Radiation-Induced Lymphoma" Cells 9, no. 9: 2068. https://doi.org/10.3390/cells9092068
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.