1. Introduction
Cytochrome
c oxidase, the terminal enzyme of the electron transport chain, is an indispensable part of mitochondrial machinery needed for ATP production in mammalian cells (OXPHOS). In addition to three mitochondria-encoded subunits, which are necessary for COX catalytic function, eleven nuclear-encoded subunits build up the COX enzyme and participate in the regulation of COX enzyme activity, as well as in the regulation of the whole OXPHOS system [
1]. The crucial position in the respiratory chain pathway, a large drop of Gibbs free energy during enzyme turnover, and relatively low in vivo reserve capacity [
2] predispose COX to serve as a mitochondrial OXPHOS regulator. This is indeed reflected by the emergence of numerous regulatory subunits during the evolution of individual eukaryotic lineages [
3], as well as by the discovery of numerous post-translational modifications signifying that COX has become a target of signaling pathways [
4]. In mammals, six COX subunits have isoforms with expression regulated in developmental, tissue-specific (COX6a, 7a, 8, and 6b), and environmental manners (COX4, NDUFA4). Subunits 6a, 7a, and 8 all exist in two variants, the L (liver) isoform is expressed ubiquitously, and the H (heart) isoform is expressed postnatally in heart and skeletal muscle [
5]. In contrast to the L isoforms, the triplet of H isoforms functions as an ATP/ADP sensor and modulates COX turnover and its H
+/e
− efficiency [
5]. An additional 7a variant, COX7a2L, is responsible for the association of complexes III and IV into supercomplexes [
6]. A second isoform of subunit 6b was discovered in mammalian testis (6b-2). Subunit 6b forms part of the cytochrome
c binding site, so the testis isoform was hypothesized to evolve in association with the testis isoform of cytochrome
c [
7].
The largest of the nuclear encoded subunits, COX4, is ideally predisposed to serve as a regulatory factor thanks to its interactions with other subunits throughout multiple parts of the COX enzyme. Its C-terminal part protrudes into the intermembrane space where it interacts with COX2 and helps shape the docking site for cytochrome c. The COX4 transmembrane helix is tightly associated with catalytic subunit COX1 and runs parallel alongside helices of nuclear-encoded subunits COX7b, COX8a, and COX7c. Moreover, the large membrane extrinsic domain of COX4 is located in the mitochondrial matrix and may function as a metabolic sensor. Indeed, COX4 has long been recognized as a target of allosteric ATP binding that decreases enzyme turnover, and this mechanism has been denoted as a second mechanism of respiratory control [
8]. Furthermore, it was reported that ATP binding is abolished upon phosphorylation of serine 58 in COX4-1 by the protein kinase A pathway, with all components located in the mitochondrial matrix, and is positively regulated by CO
2 and thus may serve to match the rates of the Krebs cycle with the respiratory chain [
9]. In yeast, two isoforms of the COX4 homolog COXV were discovered (COXVa and COXVb). Their ratio is exclusively regulated by oxygen concentration, with COXVb being expressed under hypoxia [
10]. The second isoform of mammalian COX4, COX4i2, discovered in 2001, was found to be predominantly expressed in the lung, and to a minor extent in the heart and brain [
11]. COX4i1 and COX4i2 are highly homologous in the C-terminal region; however, significant differences exist in the N-terminal part comprising the matrix domain. COX4i2 lacks the regulatory S58 residue, and in contrast to COX4i1, contains three cysteine residues that may form disulphide bridges under oxidative conditions and thus function as redox sensors [
11]. Importantly, and similarly to yeast, the mammalian COX4 isoform expression is regulated by oxygen availability [
12,
13,
14]. Interestingly, the gene duplication event giving rise to the isoforms occurred independently in both eukaryotic lineages. This suggests that the COX4 position in the enzyme enables the isoform switch to serve as a homeostatic response to optimize respiratory chain function according to oxygen availability. In support of that, studies on the mouse COX4i2 knockout model revealed that this protein is essential for acute hypoxic pulmonary vasoconstriction [
15,
16]. Later it was shown that COX4i2 is also likely involved in oxygen sensing by carotid bodies [
17,
18]. In human cells, the function of COX4 isoforms has been partially characterized in HeLa and HEK293 cells where COX4i2 expression under hypoxia increased enzyme activity and decreased ROS production from mitochondria [
13]. Increased activity of the COX4i2-containing enzyme was also found in astrocytes, associated with decreased sensitivity to allosteric inhibition by ATP [
19]. In contrast, a recent study in glioblastoma cells reported increased activity of the COX containing COX4i1 isoform [
20]. Some of these contradictory findings may be attributed to cellular models used in these studies, which were largely based on shRNA knock-down technology. Such an approach cannot completely abolish the expression of targeted proteins, and therefore, at least a minor expression of both isoforms is always present.
To unambiguously characterize the functional differences between COX4 isoforms, we constructed cellular models with exclusive knock-in of respective isoforms in the background of double CRISPR-Cas9-mediated COX4i1/2 knockout in HEK293 cells. We report that COX4 isoforms 1 and 2 are able to modulate COX oxygen affinity and can affect mitochondrial OXPHOS and the redox state even under normoxic conditions.
2. Materials and Methods
2.1. Cell Culture
HEK293 (human embryonic kidney) cells (ATCC® CRL-1573™) were cultivated under standard conditions (37 °C, 5% CO2 atmosphere) in nutrient-rich DMEM/F-12 (31331-028, Thermo Fischer Scientific, Waltham, MA, USA) supplemented with 10% (v/v) FBS (Thermo Fischer Scientific, 10270-106), 40 mM HEPES, 50 µM uridine, and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin, Thermo Fischer Scientific, 15140-122) to facilitate growth of highly glycolytic knock-out cells.
2.2. Preparation of HEK293 Cellular Knock-Out Models
Double COX4i1 and COX4i2 gene knock-out (KO) was introduced into HEK293 cells by CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats) knock-out technology employing Cas9-D10A paired nickase with two chimeric RNA duplexes [
21]. First, the COX4i2 KO HEK293 model was established and was subsequently used to introduce COX4i1 KO to obtain COX4i1/2 KO cells.
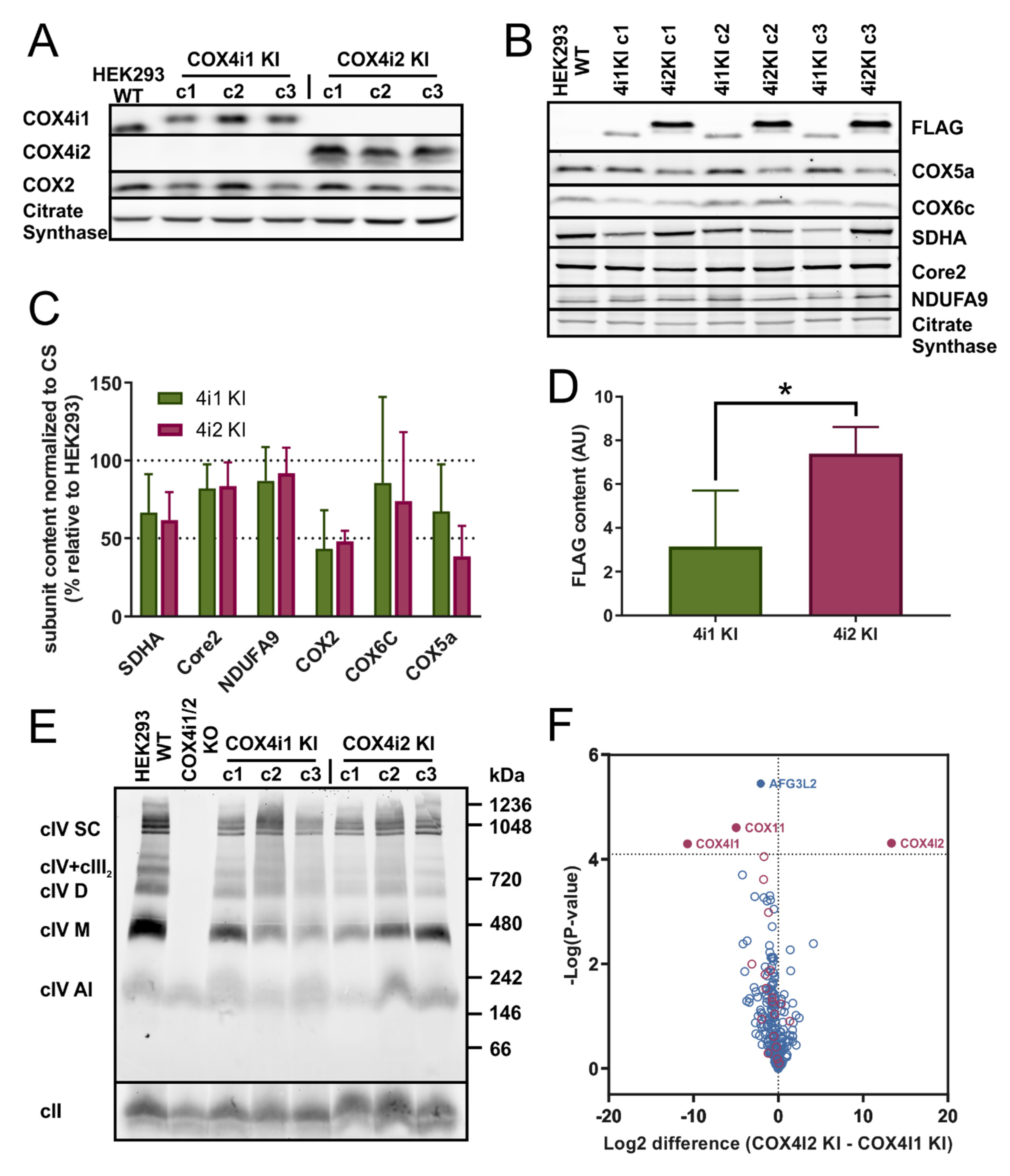
gRNAs for COX4i1 and COX4i2 obtained from a commercially available library (Merck, Darmstadt, Germany) were used for knock-out preparation. The HSR0002687616/HSL0002687619 gRNA pair was chosen to generate COX4i2 KO and the HSL0001377215/ HSR0001377224 gRNA pair for COX4i1 KO. gRNAs were received in the form of the expression plasmid (U6-gRNA, expressed by the U6 promoter), along with the expression plasmid for Cas9-D10A nickase fused with GFP (Merck). The efficiency of different combinations of gRNA pairs for the Cas9-D10A nickase system was determined by SURVEYOR assay (Integrated DNA Technologies, Coralville, IA, USA). Cells were transfected with relevant plasmids using Metafectene Pro (Biontex Laboratories GmbH, München, Germany), incubated for 72 h, trypsinized, and then diluted to a concentration of 20 cells/mL. This suspension was aliquoted (100 µL) into 96-well plates. Wells containing single-cell colonies were identified and further cultivated. Confluent cells in 6-well plates were harvested and used for selection of a knock-out clone by sequencing of CRISPR-targeted sites to identify nonsense mutations resulting in premature stop codons and by SDS-PAGE WB to confirm the complete absence of COX4 isoforms at the protein level.
2.3. COX4 Isoform Overexpression
COX4i1 (CCDS10955.1) and COX4i2 (CCDS13187.1) isoforms of the human COX4 subunit full-length cDNAs including the 3′ end sequence encoding the FLAG tag (DYKDDDDK) were obtained as synthetized DNA strands with 5′ and 3′ overhangs containing restriction sites used for insertion into the pcDNA3.1+ mammalian expression vector (Thermo Fischer Scientific). COX4i1-FLAG or COX4i2-FLAG constructs were transfected into COX4i1/2 KO cells using Metafectene Pro (Biontex Laboratories GmbH); the selection of stably-transfected cells was started 48 h post-transfection with 1 mg/mLG418. Single-cell clones were later established from G418-resistant cells, and three clones of each COX4i1 and COX4i2 knock-in cells were used for the following experiments.
2.4. COX4i2 Site-Directed Mutagenesis
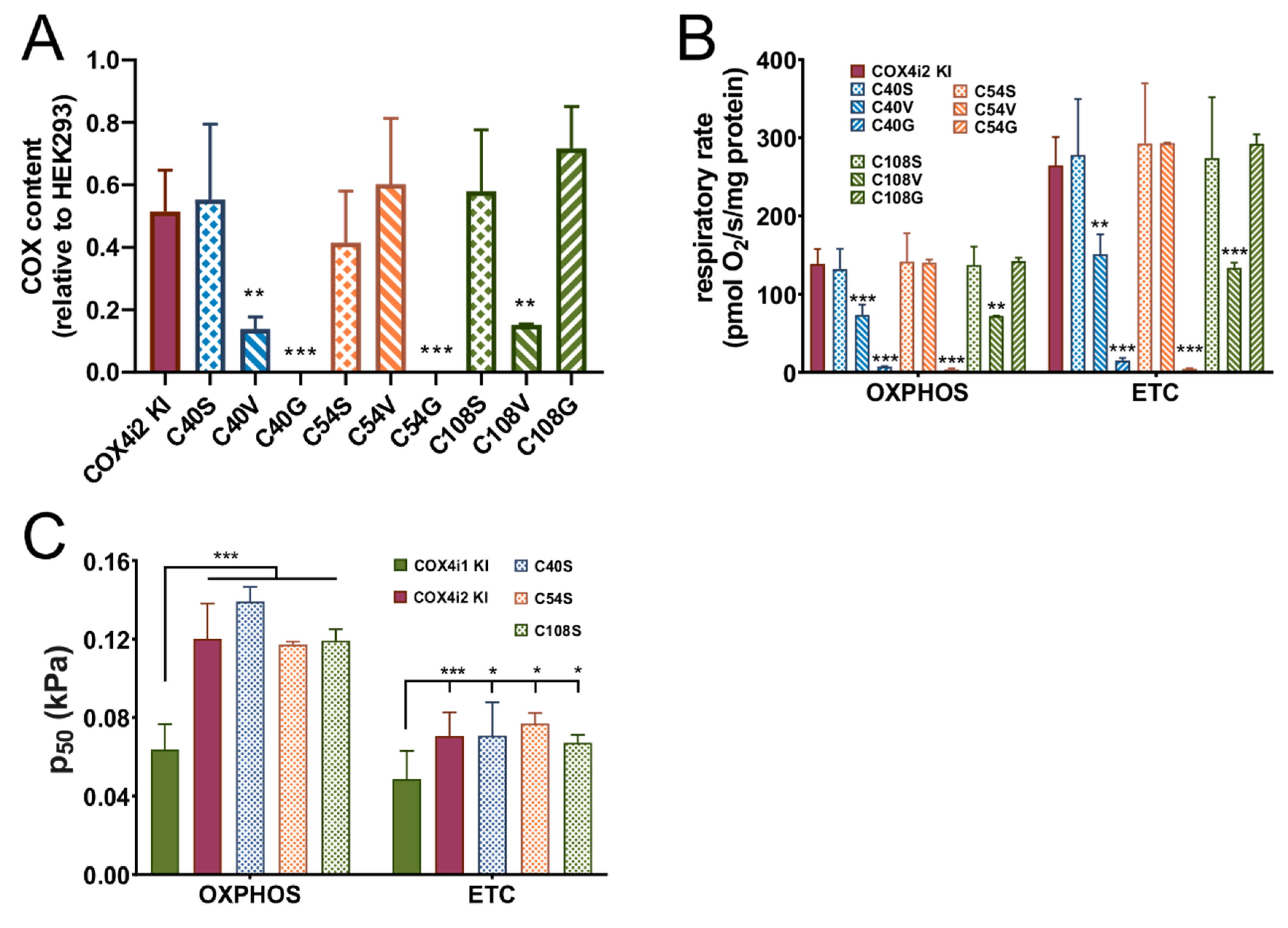
The COX4i2-FLAG pcDNA3.1+ construct was used to introduce mutations leading to single amino acid (AA) substitutions of three individual cysteine residues at positions 40, 54, and 108. Each cysteine was replaced by three AA variants, serine, representing a sterically similar amino acid where only sulphur is replaced by oxygen, or glycine and valine, which represent amino acids present at homologous positions in COX4i1. Mutations were introduced using the QuikChange Lightning kit (Agilent, Santa Clara, CA, USA), using primers suggested by the manufacturer’s on-line primer design tool. Mutagenesis was confirmed by sequencing, and mutated constructs were used for knock-in into COX4i1/2 KO cells as was the wild-type COX4i2-FLAG vector.
2.5. Electrophoretic Analyses
Sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE), specifically tricine SDS-PAGE, was used for separation of proteins according to their molecular weight under denaturing conditions [
22]. Frozen aliquots of harvested cellular pellets were suspended in SLB buffer (sample lysis buffer; 2% (
v/
v) 2-mercaptoethanol, 4% (
w/
v) SDS, 50 mM Tris (pH 7,0), 10% (
v/
v) glycerol, 0,02% Coomassie Brilliant Blue R-250) to a final concentration of 2–5 mg protein/mL. Samples were sonicated and then incubated for 20 min at 40 °C. Samples (10–50 µg of protein as indicated) were separated on 12% gel using a Mini-PROTEAN III apparatus (Bio-Rad, Hercules, CA, USA).
Blue-native (BN-PAGE) electrophoresis [
23] was used for analysis of native protein complexes. For protein complex solubilization, mild non-ionic detergent digitonin was used, as it maintains OXPHOS complexes and their supercomplexes and enables their detection.
Mitochondria from HEK293 cells were released by hypotonic shock and isolated using differential centrifugation as described [
24] and then stored as dry pellets at −80° C. Mitochondrial pellets were resuspended in solubilization buffer (50 mM NaCl, 50 mM imidazole/HCl, 2 mM 6-aminocaproic acid, 1 mM EDTA; pH 7,0) at a protein concentration 10 mg/mL. Digitonin (20% (
w/
v) stock solution) was added at 6 g/g protein, and solubilization proceeded on ice for 10 min. Samples were centrifuged at 30,000×
g (20 min, 4 °C), and supernatant containing released membrane proteins was collected. Then, glycerol was added to a final 5% (
v/
v) concentration, and Coomassie Brilliant Blue G-250 (CBB) was supplemented according to a detergent/dye ratio of 8:1 (
w/
w). Samples were separated on 5–16% gradient polyacrylamide gel using a Mini-PROTEAN III apparatus (Bio-Rad).
2.6. Western Blot and Immunodetection
Proteins separated by electrophoresis were transferred onto PVDF (polyvinylidene difluoride) membrane (Immobilon FL 0.45 µm, Merck) by semi-dry electroblotting using a Transblot SD apparatus (Bio-Rad). After blotting, the PVDF membrane was washed for 5 min in TBS (150 mM Tris-HCl, 10 mM NaCl; pH 7,5) and blocked in 5% (w/v) fat-free dry milk diluted in TBS for 1 h. Then, the membrane was washed 2 × 10 min in TBST (TBS with 0,1% (v/v) detergent Tween-20). For immunodetection, the membrane was incubated for 2 h in primary antibody (diluted in TBST) at room temperature. The following primary antibodies were used in the study: FLAG (Merck F1804), complex IV (COX1: Abcam 14705, Cambridge, UK, COX2: Abcam 110258, COX4i1: Abcam 14744, COX4i2: Abnova H00084701-M01, Taipei City, Taiwan, COX5a: Abcam 110262, COX6c: Abcam 110267), complex I (NDUFA9: Abcam, 14713), complex II (SDHA: Abcam 14715), complex III (Core 2: Abcam 14745), and citrate synthase (Abcam 129095). For quantitative detection, the corresponding infra-red fluorescent secondary antibodies (Alexa Fluor 680, Thermo Fisher Scientific; IRDye 800, LI-COR Biosciences, Lincoln, NE, USA) were used. Detection was performed using the fluorescence scanner Odyssey (LI-COR Biosciences) and signals were quantified by ImageLab software (Bio-Rad).
2.7. Immunoprecipitation and Mass Spectrometry Analysis
Whole-cell digitonin solubilizates (2 g/g of protein) from COX4i1-FLAG KI, COX4i2-FLAG KI, and COX4i1/2 KO (background control not containing FLAG-tagged protein) cells were immunoprecipitated with anti-FLAG M2 magnetic beads (Merck M8823). Washed beads with captured immunocomplexes were digested “on beads” with trypsin using the sodium deoxycholate procedure as described in [
25]. Desalted peptide digests were separated on a 50 cm C18 column using a 1 h elution gradient and were analyzed in an Orbitrap Fusion (Thermo Fisher Scientific) mass spectrometer. Resulting raw files were processed in MaxQuant (v. 1.6.6.0, maxquant.org) with the label-free relative quantification (LFQ) algorithm MaxLFQ [
26]. Downstream analysis and visualization were performed in Perseus (maxquant.org/perseus/) [
27].
2.8. Cytochrome c Oxidase Activity
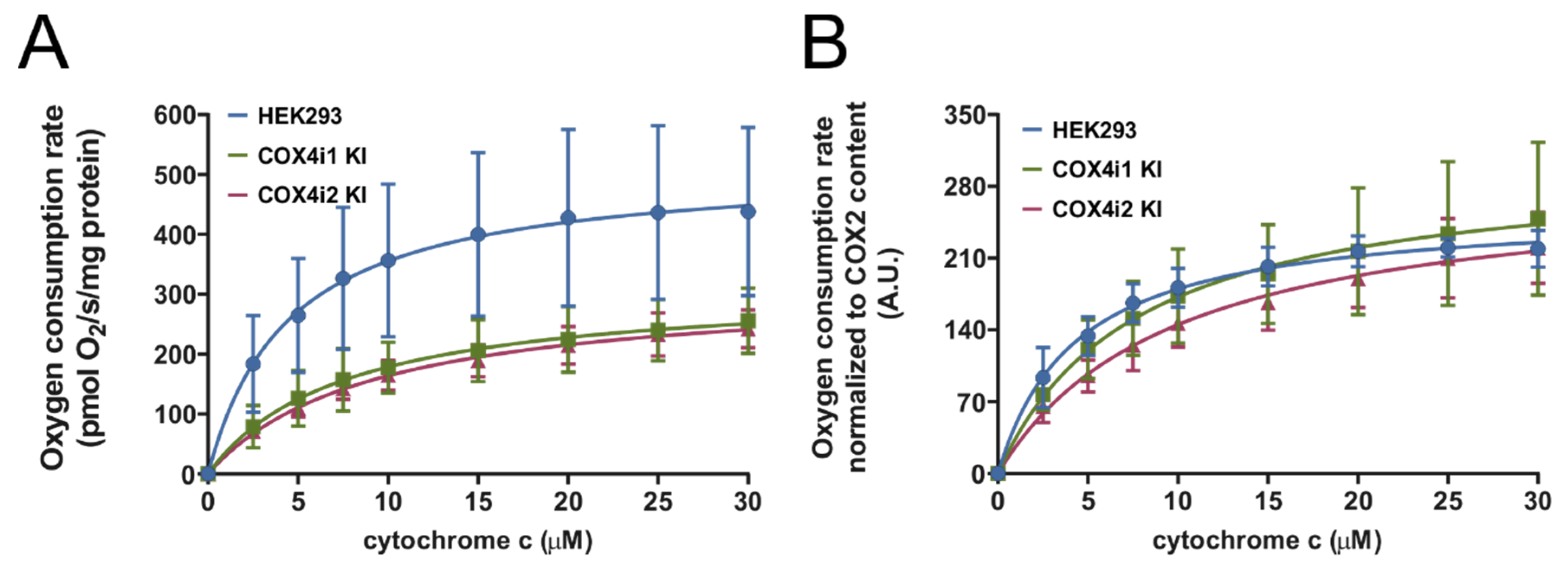
COX activity was analyzed using Oxygraph-2k (Oroboros, Innsbruck, Austria) in mitochondrial extracts essentially as described [
28]. Mitochondria isolated from cells were suspended in buffer composed of 10 mM K-HEPES (pH 7.4), 40 mM KCl, 2 mM EGTA, 1% Tween 20, and protease inhibitor cocktail (Merck), sonicated, and centrifuged at 30,000×
g for 2 min. Then, 200 µg protein of supernatants was added to 2 mL oxygraph chambers. COX activity was followed as the rate of oxygen consumption in the presence of 20 mM ascorbate serving as a reductant for cytochrome
c (bovine, Merck) titrated sequentially to reach final concentrations of 2.5–30 µM. Parallel measurement with COX activity fully inhibited by 0.5 mM KCN was always performed to subtract non-specific oxygen consumption due to autoxidation of ascorbate. A hyperbolic Michaelis-Menten function was fitted to experimental data using Prism 8 software (GraphPad, San Diego, CA, USA).
2.9. High-Resolution Respirometry
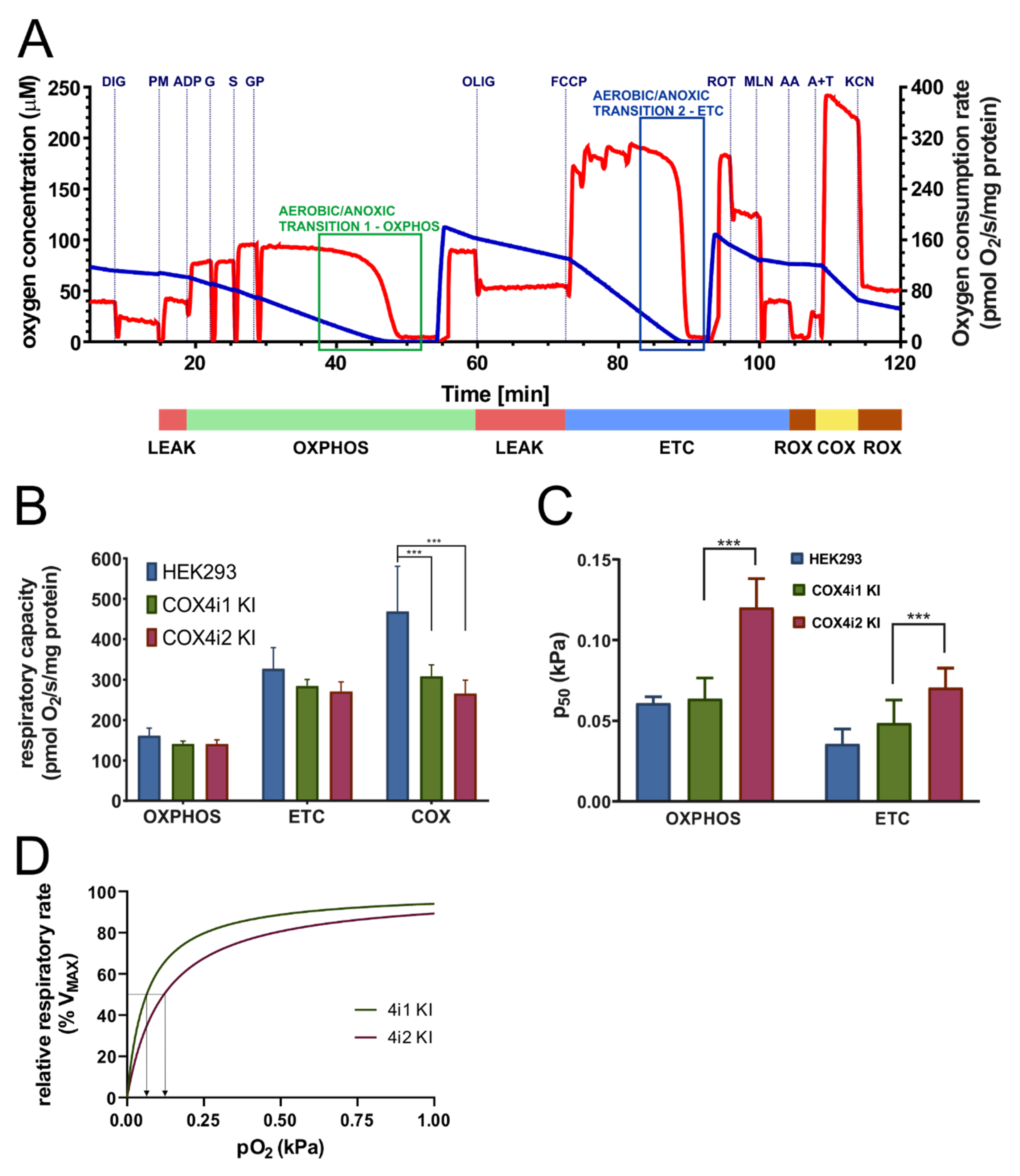
Mitochondrial respiration was measured at 30 °C as described in [
29] using Oxygraph-2k (Oroboros, Innsbruck, Austria). Freshly harvested cells (0.6 mg protein) were suspended in 2 mL of KCl medium (80 mM KCl, 3 mM MgCl
2, 1 mM EDTA, 5 mM K-Pi, 10 mM Tris-HCl; pH 7.4), and digitonin (0.05 g/g) protein was used to permeabilize the plasma membrane. For measurements, the following substrates and inhibitors were used: 2 mM malate, 10 mM pyruvate, 10 mM glutamate, 10 mM succinate, 10 mM glycerol 3-phosphate, 1 mM ADP, 0.5 µM oligomycin, 100–300 nM FCCP, 0.5 µM rotenone, 10 mM malonate, 0.25 µM antimycin A, 2 mM ascorbate, 1 mM TMPD, 0.5 mM KCN. The oxygen consumption was expressed in pmol O
2/s/mg protein.
Affinity to oxygen was determined by the p
50 value, the partial oxygen pressure (pO
2), at which the cellular respiratory rate is half-maximal, as described before [
30]. Briefly, the volume-specific rate of oxygen consumption (oxygen flux) was calculated as the negative slope of the oxygen concentration recorded at 1 s time intervals. The signal was deconvoluted with the exponential time constant of the oxygen sensor (3 to 5 s) and corrected for the instrumental background, which is a linear function of experimental pO
2 and results from oxygen consumption by the sensor and oxygen back-diffusion from low-capacity oxygen reservoirs. The p
50 parameter was obtained from a hyperbolic function, J
02 = (J
MAX. pO
2)/(p
50 + pO
2), fitted over the low oxygen range of 0 to 1.1 kPa. All calculations were performed using routine functions of Datlab 2 software (Oroboros) [
31].
2.10. XF Seahorse Bioenergetics Analyser Measurement
Parallel measurement of mitochondrial respiration and the glycolytic rate was performed using a Seahorse XFe24 analyzer (Agilent). One day prior to measurement, 3 × 10
4 cells were seeded in poly-L-lysine coated wells of the measuring plate and were cultivated in DMEM/F12 medium (see above). The next day, the microplate was washed with 1 mL of XF assay medium (modified DMEM, pH 7.4, 37 °C); 500 μL of the same medium with 0.2 % (
w/
w) BSA was pipetted, and the microplate was incubated at 37 °C for 30 min. Meanwhile, an XFe24 sensor cartridge was prepared by injection of substrates and inhibitors according to the described protocol [
32] to record metabolic rates with endogenous substrates before any addition (basal), and after subsequent additions reaching final concentrations of 10 mM glucose (Glu), 1 µM oligomycin (Oligo), 1 µM FCCP (FCCP), and inhibitor cocktail of 1 µM rotenone, 1 µg/mL antimycin A, and 100 mM 2-deoxyglucose (Rot+AA+2DG). For precise normalization of rates according to cell counts, immediately after the Seahorse run, cell nuclei were stained by Hoechst 33342 (final concentration 5 µg/mL). Images of whole wells were acquired by a Cytation 3 Cell Imaging Reader (BioTek, Winooski, VT, USA) and were analyzed using Gen5 software (BioTek) to obtain cell counts for each well.
2.11. ROS Production
ROS production was estimated as 1 μM CM-H
2DCFDA (chloromethyl derivative of 2′,7′-dichlorodihydrofluorescein diacetate, Thermo Fisher Scientific) fluorescence as in [
33]. Cells (3.x10
5) were seeded in poly-L-lysine coated 24-well plates, and the next day, cells were incubated for 2 h at 37 °C with CM-H
2DCFDA. The increase in the fluorescence was recorded with an Infinite M200 plate reader (Tecan Group Ltd., Männedorf, Switzerland) at 495/525 nm.
2.12. NAD+/NADH Ratio
Redox balance in terms of NAD
+/NADH was measured in cell lines using the NAD
+/NADH-Glo assay (Promega) essentially as described [
34]. Briefly, 5x10
3 cells were seeded into 96-well plates one day prior to measurement. NAD
+ and NADH concentrations in cell extracts were determined according to the manufacturer’s protocol, and the luminescence signal was recorded with an Infinite M200 plate reader (Tecan Group Ltd.).
4. Discussion
Energetic metabolism of mammalian organisms is vitally dependent on aerobic respiration, with 80–90% of inhaled oxygen being utilized by cytochrome
c oxidase, the terminal enzyme of the mitochondrial respiratory chain. The apparent K
m for oxygen ranges between 0.01 and 0.1 kPa, meaning that mitochondrial respiration is usually not limited by oxygen availability [
35]. However, even with sophisticated oxygen delivery by the cardiovascular system, local pO
2 can drop to levels as low as 0.3 kPa in some tissues. Therefore, oxygen reduction to water by cytochrome
c oxidase occurs under conditions of oxygen availability varying by two orders of magnitude [
36], which under extreme cases may result in incomplete COX saturation. Oxygen affinity of COX can be further diminished by allosteric factors such as NO [
37], as part of mechanisms that can redirect oxygen utilization from respiration to other oxygen-handling enzymes or non-enzymatic reactions. The significance of oxygen availability for COX function was strongly supported by the discovery of oxygen-regulated expression of COX4i2, the second isoform of regulatory, nuclear-encoded subunit COX4. Oxygen regulation of COX4i2 expression is exerted by several mechanisms, either through classical HIF-1 regulation [
13] or a novel oxygen response element (ORE) [
14], where transcription is regulated by the interplay of three proteins [
12]. Environmental conditions thus add yet another mode of COX regulation to complement nuclear-encoded subunit isoforms that are expressed in tissue or a developmental stage-dependent manner [
38]. Induction of COX4i1 expression under hypoxia was reported in multiple cell lines, and it was proposed that the COX4 isoform switch represents adaptation to decreased oxygen availability by improving efficiency of mitochondrial respiration [
13]. However, huge variation of Cox4i2 expression in mouse tissues in vivo [
14,
15] suggested that regulation of its content is complicated beyond just oxygen concentration, and the presence of the second isoform of COX4 may imply its specialized function. Indeed, high expression of Cox4i2 in pulmonary artery smooth muscle cells (PASMCs) of the mouse lung was associated with regulation of pulmonary hypoxic vasoconstriction (HPV) [
16]. More recently, increased expression of Cox4i2, along with two atypical COX subunits—COX8b and NDUFA4l2 triggered by HIF-2α—was observed in glomus cells of carotid bodies in mice [
17]. Such atypical COX complex present in these specialized cells was suggested as a hypothetical sensor involved in acute oxygen sensing [
18].
In the present study, we aimed to identify how the switch between COX4i1 and COX4i2 isoforms is able to modulate COX function. In order to achieve this in a well-defined model, we generated knock-in cell lines of these isoforms independently in the background of COX4i1/2 double knock-out in HEK293 cells to obtain cells with exclusive expression of either isoform. Electrophoretic and MS analyses revealed that COX4i1 and COX4i2 knock-in cell lines were able to restore comparable levels of the COX complex with otherwise identical subunit composition, representing an ideal tool for unbiased characterization of isoform effects. Previously, contradicting results regarding the effect of the COX4 isoform on enzyme activity were reported. COX isolated from mouse lung containing COX4i2 had a higher turnover number than liver COX containing COX4i1 [
15]; similarly, transient overexpression of COX4i2 in HEK293T cells mildly augmented COX activity and cellular respiration [
13]. In contrast, the isoform switch from COX4i2 to COX4i1 in glioma cells was associated with increased COX activity and respiration [
20]. In our KI cell lines, no differences between COX4i1 and COX4i2 KI were observed in the case of COX activity, affinity to cytochrome
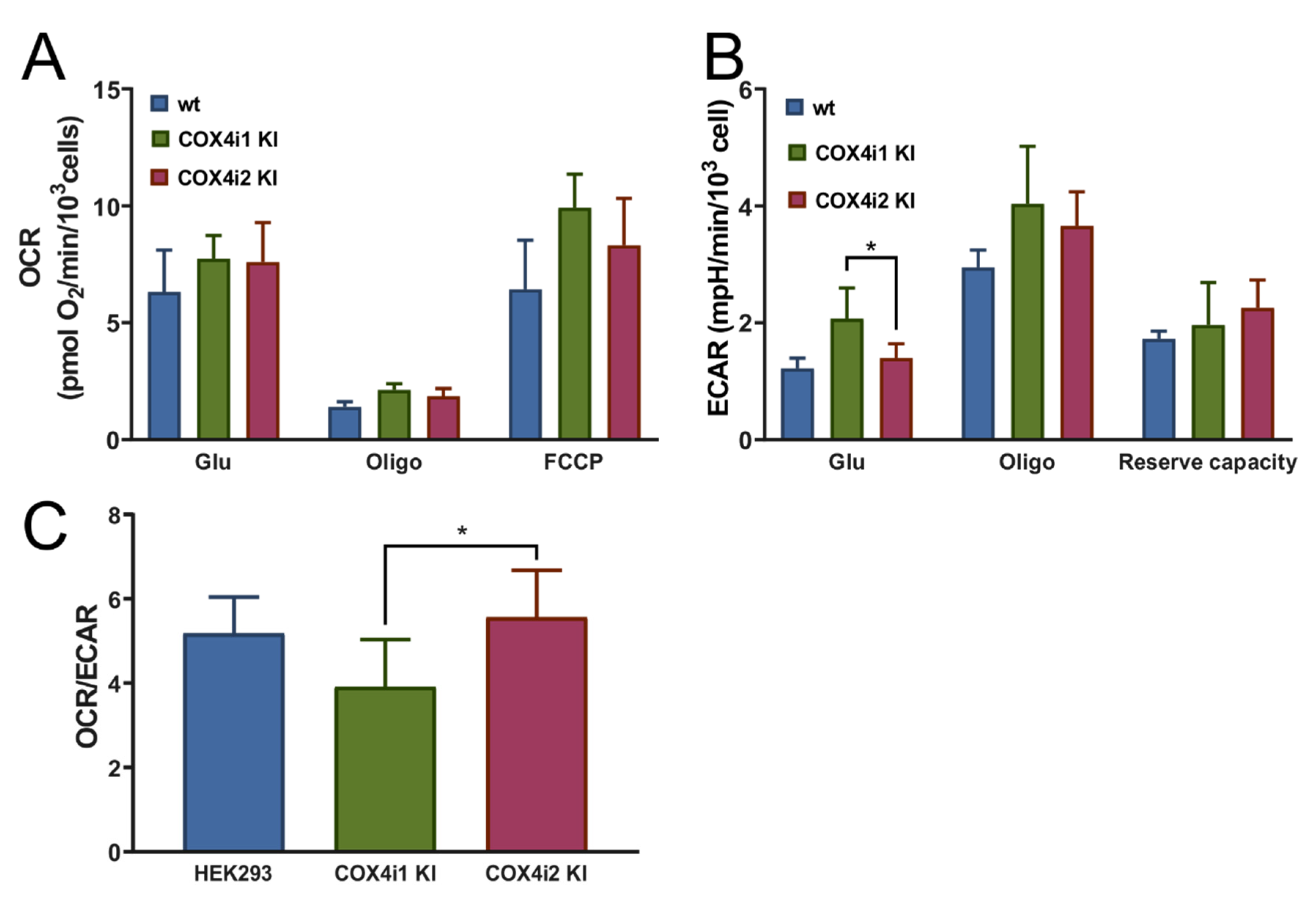
c or respiratory capacities, which suggests that previously published functional modulations may have been associated with regulatory mechanisms dependent on COX4 isoforms but emanating from levels of complexity beyond isoform exchange itself. Despite identical COX activities and respiratory capacities in our KI cell lines, analysis of energetic metabolism in intact cells revealed increased OCR/ECAR ratios in COX4i2 KI cell lines, indicative of the preference for mitochondrial ATP production rather than by anaerobic glycolysis. Both modes of energy provision are utilized by the majority of cultured cell lines, and under conditions of non-limiting substrate (glucose) supply and routine energy demand, mitochondrial OXPHOS activity may be downregulated.
COX has been identified as a target for multiple regulatory mechanisms, including posttranslational modifications and allosteric binding of adenine nucleotides enabling regulation based on the ATP/ADP ratio [
4,
5]. Indeed, the adenine nucleotide binding site on the matrix-facing domain as well as in vivo phosphorylation sites was identified in the COX4 subunit [
4]. In this regard, differential responses of COX4 isoforms to such regulations have been previously described. In astrocytes exposed to hypoxia, robust expression of COX4i2 was identified in contrast to neuronal cells, which retain mostly the COX4i1 isoform [
19]. In this study, activity measurements of COX solubilized from astrocytes displayed significantly attenuated allosteric inhibition by ATP, suggesting that COX4i2 is less prone to such modulation than COX4i1 [
19]. Further, it has been reported that allosteric ATP inhibition is modulated by the phosphorylation status of Ser58 in COX4i1 [
9]. Under conditions of increased flux of the TCA cycle, an increased level of carbonate activates the intramitochondrial PKA pathway, which leads to reversible phosphorylation of Ser58 and impairment of ATP binding through charge repulsion, thus enabling robust flux of electrons through the respiratory chain to match TCA cycle activity. In COX4i2, the corresponding phosphorylation site is missing; instead, negatively charged residues are present in the incriminated region. This suggests that COX4i2-harbouring COX resides are permanently in an activated state [
9], matching our observation of the increased OCR/ECAR ratio. Furthermore, this finding corresponds to an increased NAD
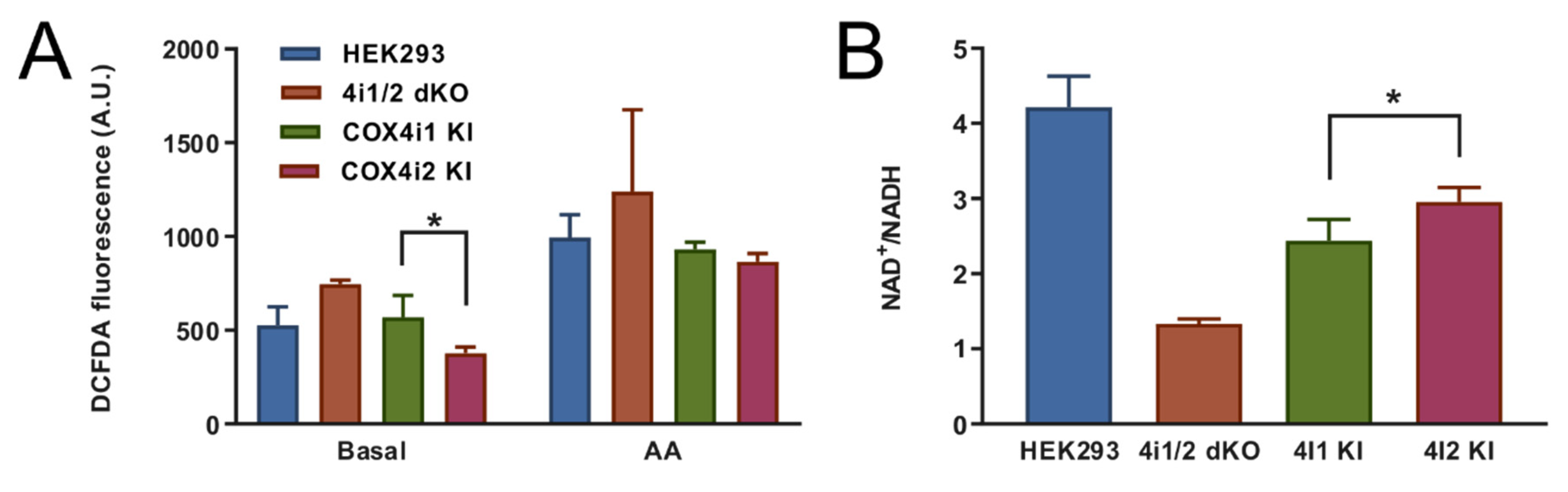
+/NADH ratio and decreased mitochondrial ROS production measured under the normoxic condition in COX4i2 KI cells in comparison with COX4i1 KI.
To enable functional alteration of COX under conditions of limited oxygen availability or to possess the ability to function as an oxygen sensor, COX4 isoforms should confer distinctive oxygen affinity to the enzyme. To the best of our knowledge, this aspect of the COX4 isoform switch has not been previously addressed before in sufficient detail. We analyzed the COX oxygen kinetics by high-resolution respirometry [
31]. This method yields the p
50 parameter, the “apparent K
m”, a measure of the sensitivity of cellular respiration to oxygen availability assayed in permeabilised cells with intact mitochondrial membranes, where the COX function is integrated in the respiratory chain. Our results consistently showed that oxygen affinity of the COX4i2-containing enzyme was significantly decreased compared to the COX4i1 counterpart. In our opinion, this represents the major finding of this study. The measured values of p
50 in parental HEK293 cells match previously reported values in isolated mitochondria and small cells [
35]. It should also be noted that the two-fold p
50 increase in COX4i2 versus COX4i1 KI can have significant in vivo implications. For example, similar alteration of COX oxygen affinity can be observed in cells with pathological mutations in the SURF1 protein, which lead to pathologic COX deficiency and is clinically associated with Leigh’s syndrome [
29,
30]. The mechanism of how COX4 isoform exchange modulates kinetics of oxygen binding is unclear, but up to three dioxygen diffusion areas with open channels relying on rapid and reversible conformational changes were proposed to operate simultaneously to sustain remarkably high affinity of COX to oxygen [
39]. Thus, any perturbation in the hydrophobic core may affect oxygen diffusion to the binuclear site within the COX1 subunit. This may be conferred by the transmembrane region of COX4 that runs parallel to the transmembrane helix 12 of COX1, but all available structural data were obtained with the COX4i1-containing enzyme, preventing evaluation of structural differences between isoforms. However, the COX4 subunit capability to modulate enzyme oxygen handling seems to be conserved in eukaryotes, as the expression of the isoform pair of the yeast COX4 homolog is also regulated by oxygen [
10]. COX isozymes containing the normoxic isoform COXVa (analogous to COX4i1) or hypoxia-induced COXVb (analogous to COX4i2) were analyzed by infrared spectroscopy in the CO-bound state (CO-IR) as a sensitive way to assess the effects of COXV isoforms on the binuclear reaction center environment [
40]. Absorption properties of COXVa-containing COX were similar to those of bovine enzyme and indicated the presence of multiple conformers capable of rapid interconversions. In contrast, narrower CO-IR band of the COXVb isozyme suggested a less dynamic enzyme with only one conformer [
40]. If oxygen diffusion to the binuclear center is indeed dependent on dynamic conformational changes, it would mean that the COXVa presence would facilitate it, while rigid conformation of the COXVb-containing enzyme may impose a diffusion barrier. If we speculate that an analogous situation occurs with mammalian COX4 isoforms, this could translate as an increase in p
50. This issue should, however, be resolved by direct structural comparison of mammalian COX isozymes. In fact, our cell models with the exclusive expression of individual COX4 isoforms may in future prove useful for such studies.
The role of COX4i2 in HPV was associated with mitochondrial membrane hyperpolarization inducing increased ROS production at upstream respiratory complexes. Mitochondrial ROS serve as signaling molecules leading to inhibition of ion channels at the plasma membrane of PASMCs leading to its depolarization and Ca
2+ influx-triggering contraction [
16]. Alternatively, according to our findings in HEK293 KI cell lines, an increase in hypoxic ROS production at complex III that is critical for HPV [
41] could be caused by premature inhibition of COX4i2-containing COX due to its decreased oxygen affinity, while sufficient oxygen concentration would be available at ROS-producing sites which are generally more sensitive to oxygen availability than COX [
42]. Indeed, in the COX4 knock-out cell line mimicking enzyme inhibition, we observed robustly increased ROS production. Interestingly, p
50 in COX4i2 KI cells was not modulated by mutations of either of three cysteine residues, which arguably represent the most significant structural difference between COX4 isoforms. In contrast, increased hypoxic ROS production in COX4i2-overexpressing CMT cells was attenuated by cysteine replacement [
16]. This suggests that additional modification of COX4i2 may be necessary for affecting proper function as oxygen sensor. Indeed, in glomus cells of carotid bodies, HIF-2α induced expression of COX4i2, but also two other non-canonical COX subunits, namely, COX8b and NDUFA4l2 [
17,
18]. The latter harbors an extra cysteine residue compared to its ubiquitous isoform, which might hypothetically form a disulphide link with COX4i2. In our study, the immunoprecipitation analysis of HEK293 KI cell lines (
Figure 1F) showed COX 4i1 and 4i2 isozymes with otherwise identical isoform composition of other COX subunits. Thus, we can demonstrate that a change in oxygen affinity is solely associated with COX4 isoforms and does not involve modulation via cysteine residues. On the other hand, modulation of hypoxic ROS production may be a more complex phenomenon involving a switch in yet other subunits, present only in physiologically specialized cells that assemble the dedicated oxygen-sensing enzyme.
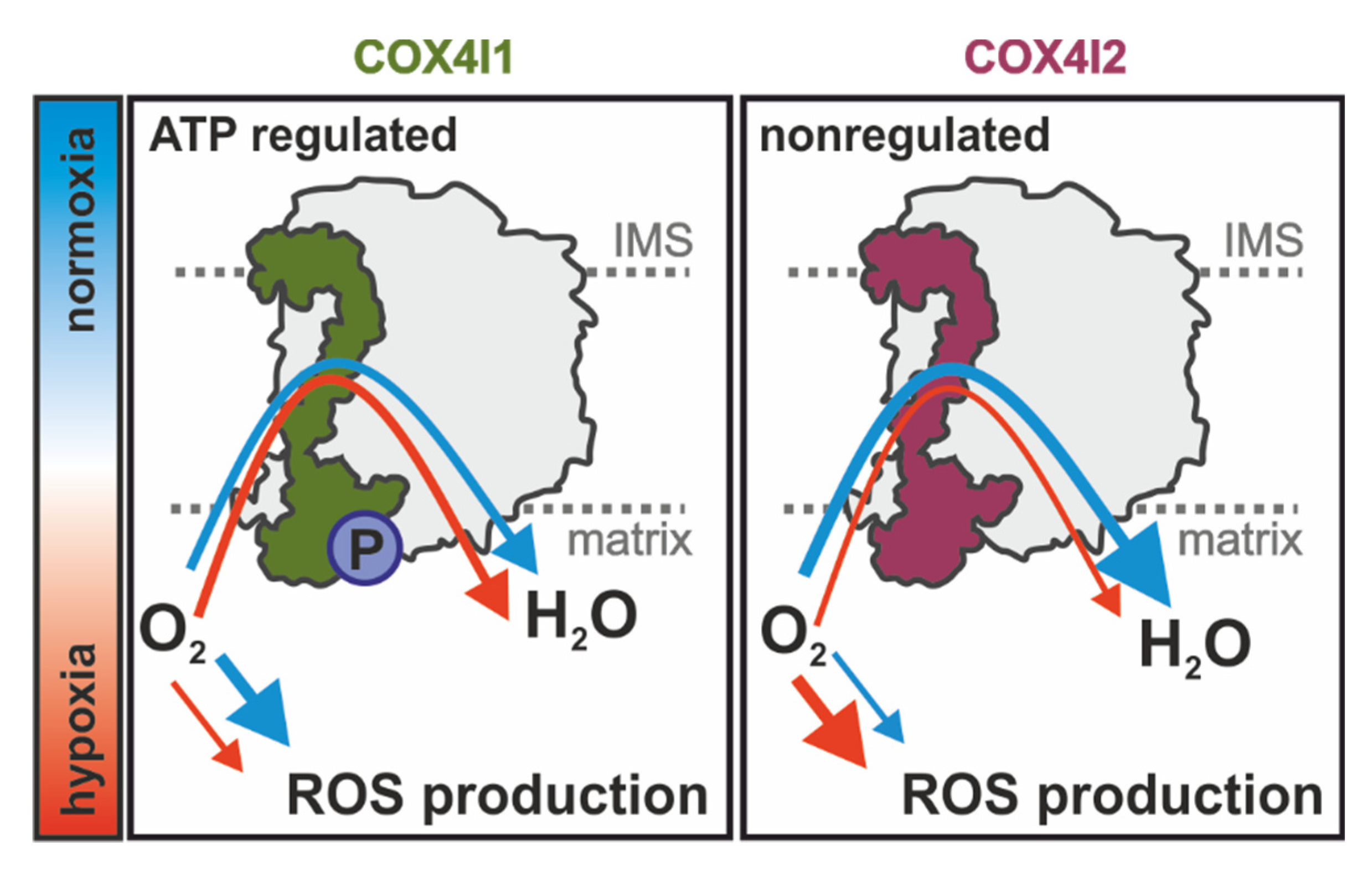
Overall, our study extends current knowledge on the regulatory role of COX4 isoforms and shows that besides affecting the ability of COX to respond to ATP levels [
9,
19], the COX4i2 subunit also facilitates a decrease in cytochrome
c oxidase affinity to oxygen. These findings thus further characterize unique properties of the COX4i2 isoform, in accordance with the view that the COX4i2 subunit represents an important effector in regulation of COX activity and respiratory chain energetic function under hypoxic conditions. According to previously published information and novel findings of the present study, the COX4 isoform exchange regulated by oxygen availability has consequences for cellular respiration as well as mitochondrial ROS production. Activity of the energy status-regulated COX4i1 isozyme is less sensitive to hypoxia; therefore, the enzyme can operate at extremely low oxygen concentrations, limiting non-respiratory oxygen consumption. In contrast, the hypoxia-sensitive COX4i2 isozyme becomes prematurely inhibited under oxygen-limiting conditions, leading to a reduction of upstream components of the respiratory chain, promoting increased ROS production (
Figure 7). Further studies are necessary to dissect the interplay of oxygen utilization, regulation of ROS production, and putative redox modification of COX4i2, as well as the role of other non-canonical COX subunits that may be involved in this remarkable physiological phenomenon of oxygen sensing.

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






