
De-Suppression of Mesenchymal Cell Identities and Variable Phenotypic Outcomes Associated with Knockout of Bbs1
 , , , , , , , , , and
, , , , , , , , , and 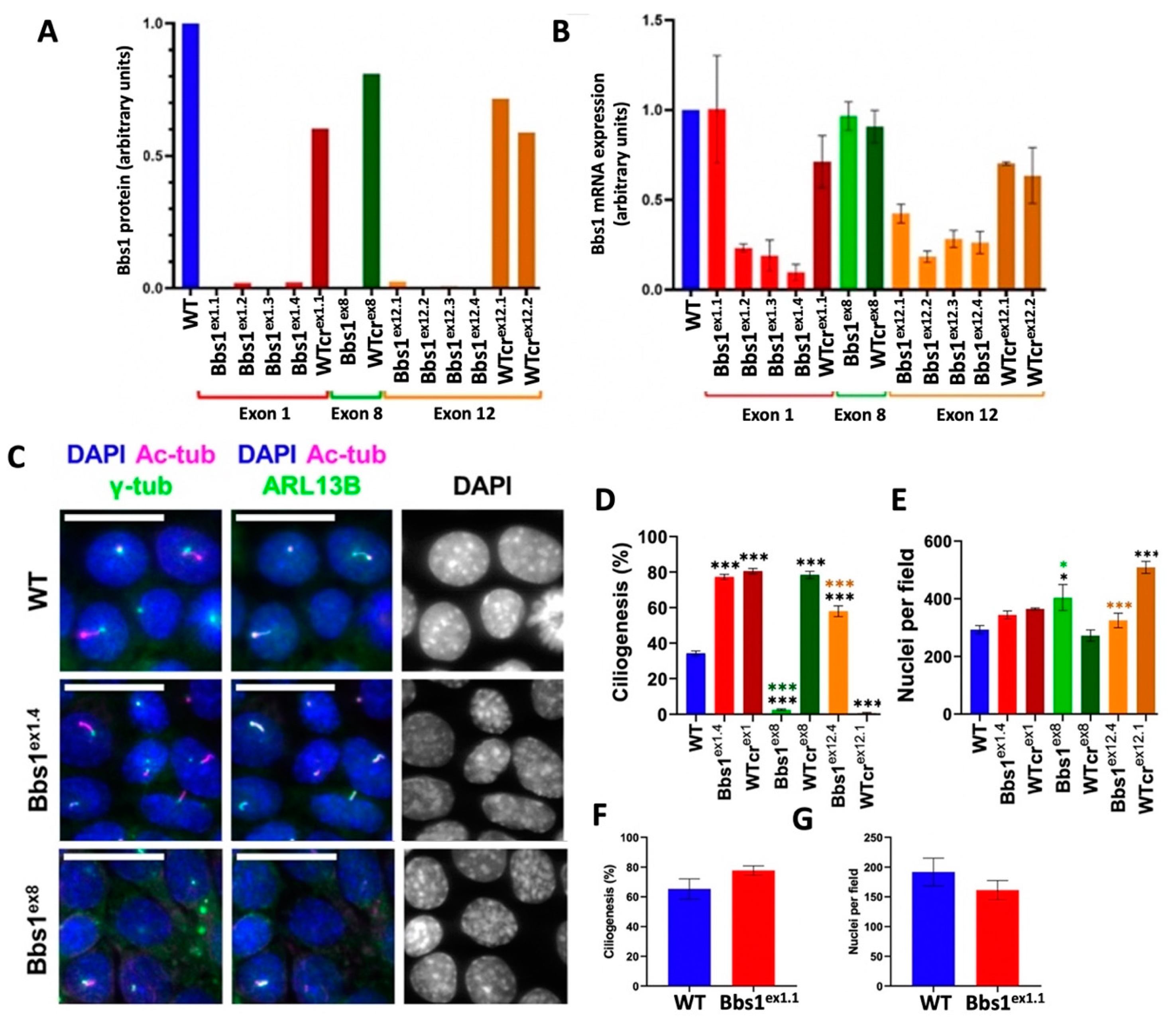{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights
- Loss of BBS1 is associated with differential expression of EMT hallmark genes, in particular representing enrichment for mesenchymal cell identities and variable loss of epithelial marker gene expression.
- This appeared to represent a failure to downregulate mesenchymal cell identities as cells aged through subsequent passages, as opposed to any kind of dedifferentiation.
- This did not correlate with morphological defects in ciliogenesis.
- This could have relevance to characteristic features of Bardet-Biedl syndrome such as fibrosis.
Abstract
:1. Introduction
2. Results
2.1. Generation and Characterisation of Bbs1-/- IMCD3 Cell Lines
2.2. Characterisation of Ciliogenesis in Bbs1-/- IMCD3 Cell Lines
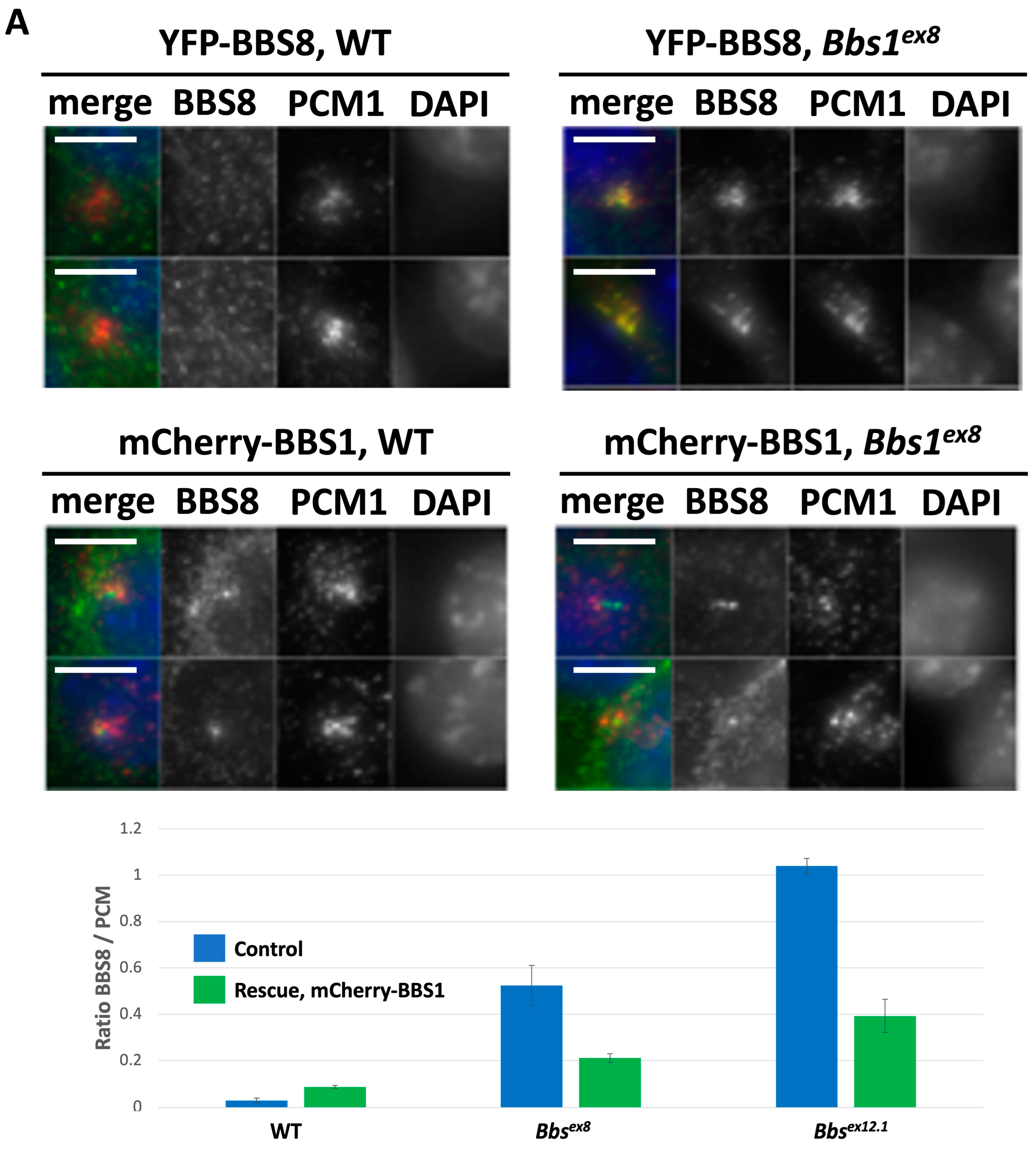
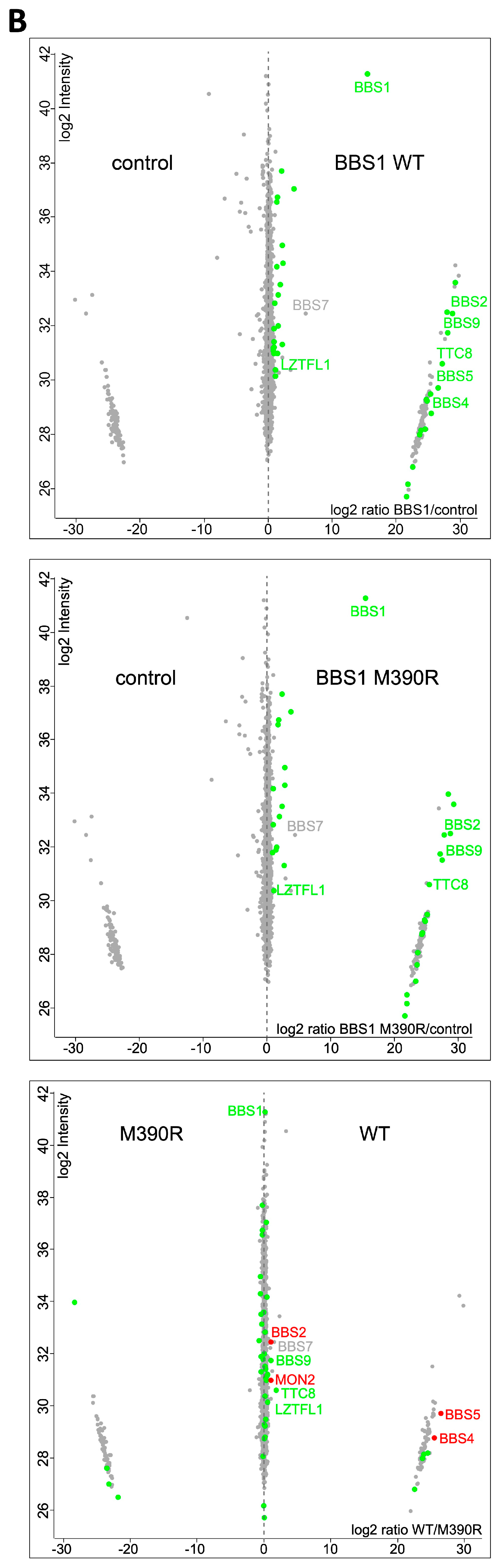
2.3. Impact of Loss of Bbs1 on the BBSome
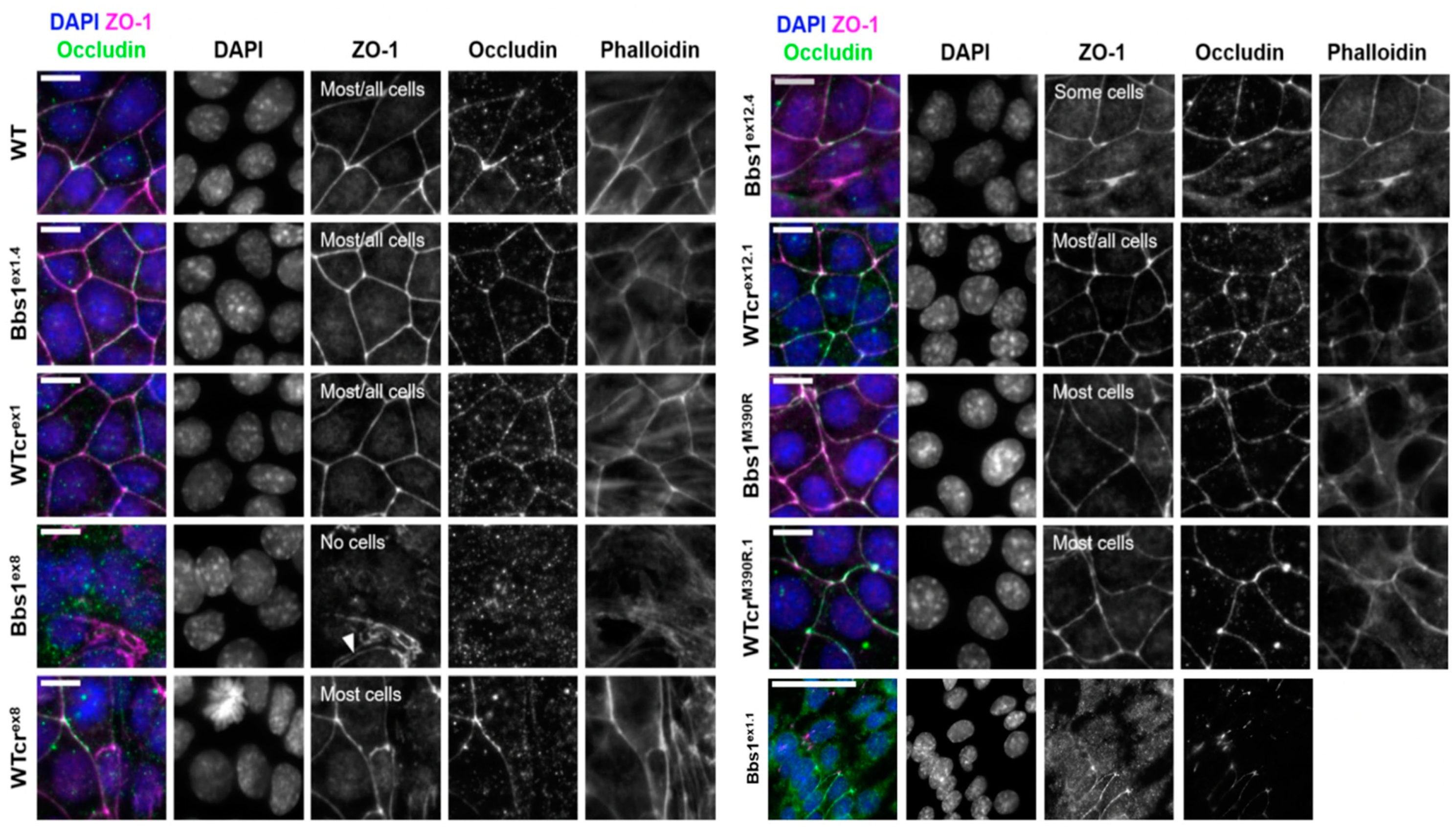
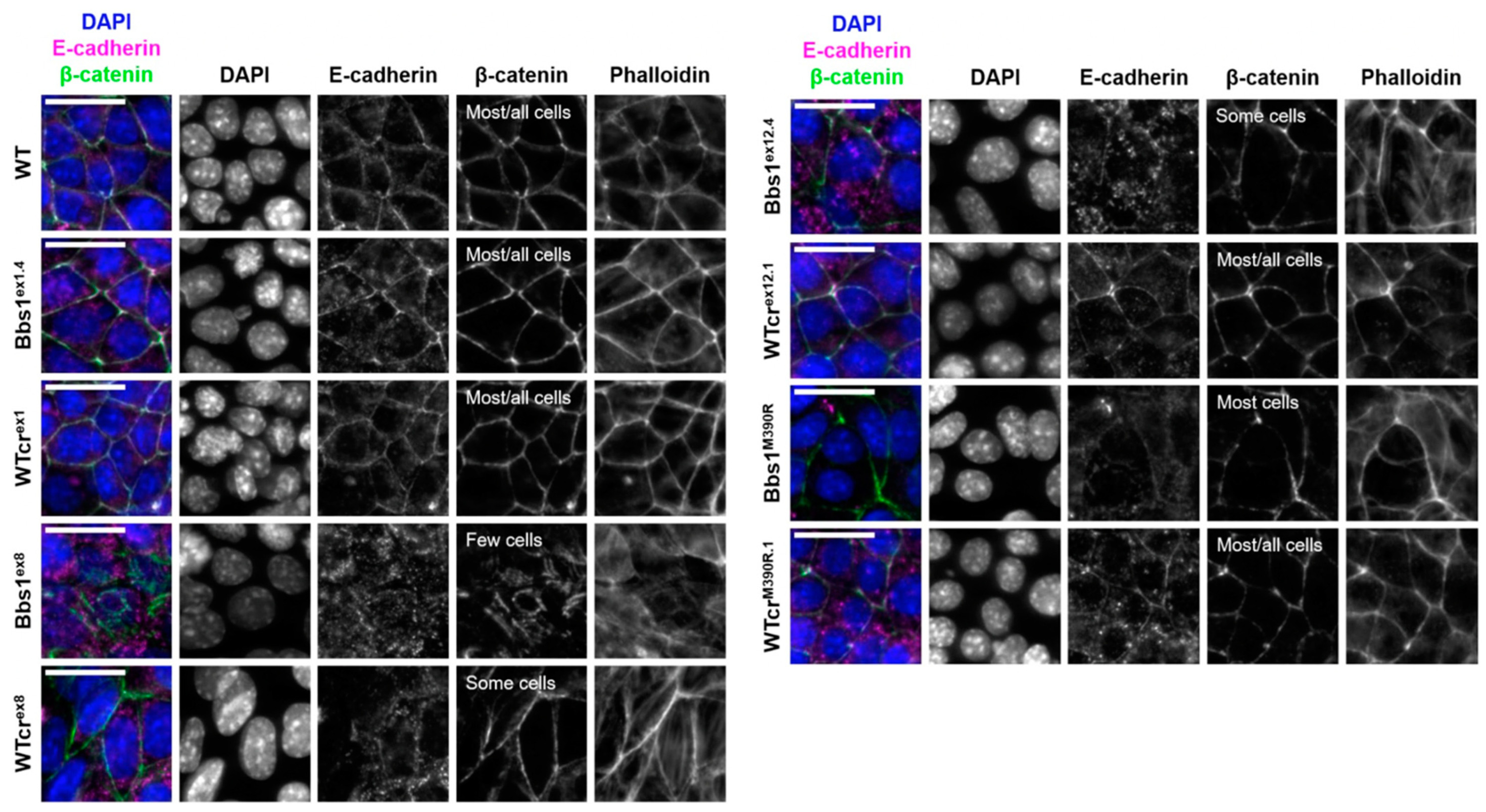
2.4. A Phenotypic Screen Reveals Clonal Variation in Cell–Cell Junction Formation in Bbs1-/- Clones
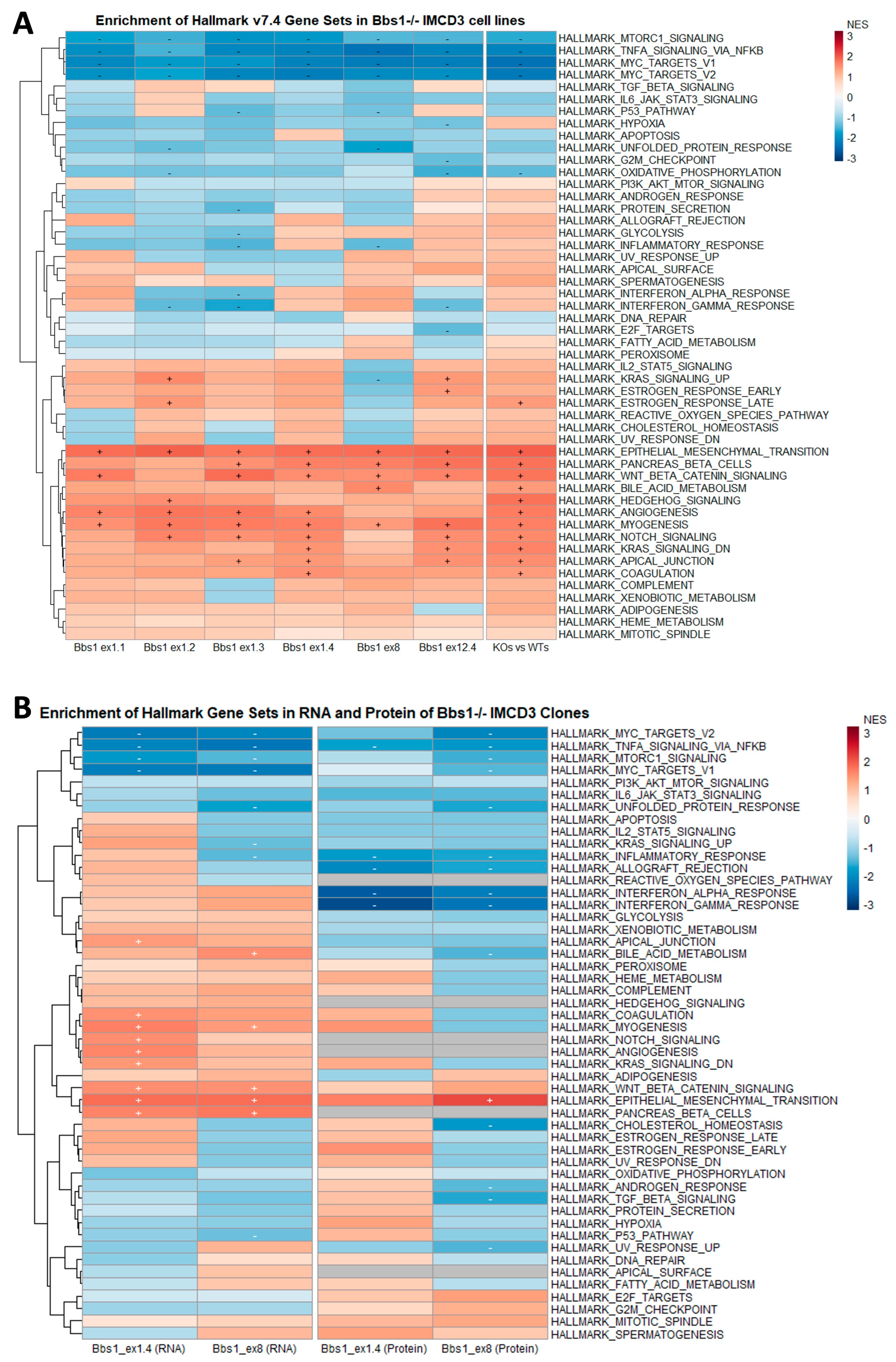
2.5. Enrichment of Mesenchymal Cell Identities in Bbs1-/- Clones Compared to Passage-Matched Controls
2.6. EMT Gene Set Enrichment Is a Feature of Diverse BBSome Transcriptomic Datasets
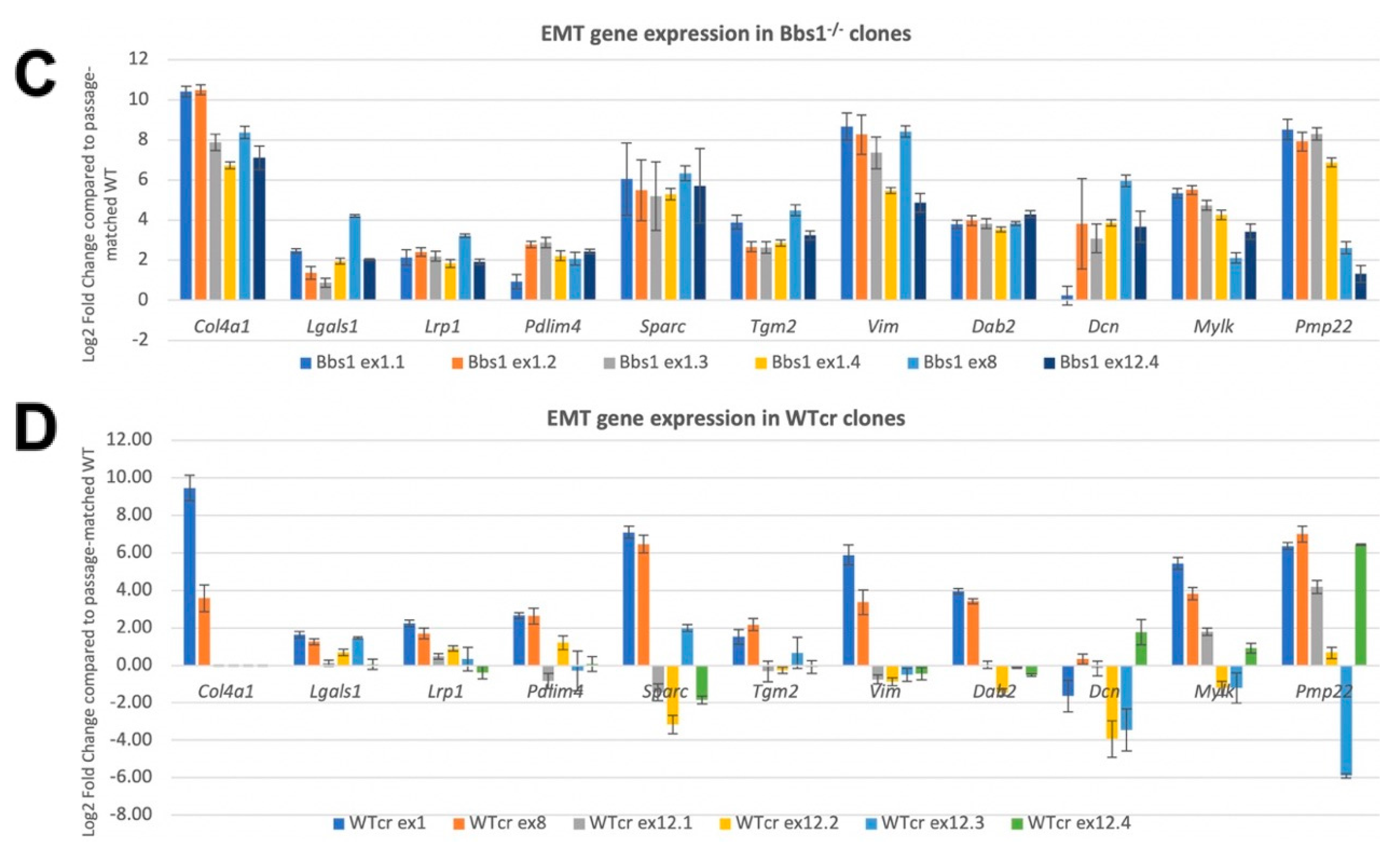
2.7. Confirmation of EMT DEGs and Analysis of Clonal Variation
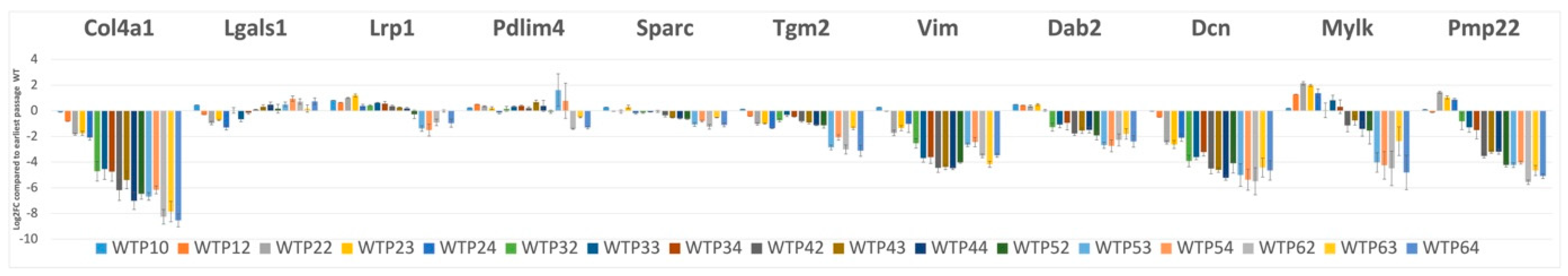
2.8. Suppression of Mesenchymal Cell Identities Is a Feature of Renal Epithelial Cell Maturation
3. Discussion
4. Materials and Methods
4.1. Ethics
4.2. Statistical Tools
4.3. Mass Spectrometry
4.4. Flow Cytometry
4.5. RNA Extraction for RNA-Seq
4.6. RNA Sequencing and Alignment
4.7. Differential Gene Expression Analysis
4.8. Gene Set Enrichment Analysis Using g:profiler
4.9. Gene Set Enrichment Analysis Using the GSEA Application
4.10. Cell Culture
4.11. Antibodies
4.12. Virus and Cell Line Preparation
4.13. Immunofluorescence
4.14. Fluorescence Microscopy
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hernandez-Hernandez, V.; Jenkins, D. Advances in the understanding of the BBSome complex structure and function. Res. Rep. Biol. 2015, 6, 191–201. [Google Scholar] [CrossRef]
- Nawaz, H.; Mujahid; Alam Khan, S.; Bibi, F.; Waqas, A.; Bari, A.; Fardous; Khan, N.; Muhammad, N.; Khan, A.; et al. Biallelic Variants in Seven Different Genes Associated with Clinically Suspected Bardet–Biedl Syndrome. Genes 2023, 14, 1113. [Google Scholar] [CrossRef] [PubMed]
- Niederlova, V.; Modrak, M.; Tsyklauri, O.; Huranova, M.; Stepanek, O. Meta-analysis of genotype-phenotype associations in Bardet-Biedl syndrome uncovers differences among causative genes. Hum. Mutat. 2019, 40, 2068–2087. [Google Scholar] [CrossRef] [PubMed]
- JJin, H.; White, S.R.; Shida, T.; Schulz, S.; Aguiar, M.; Gygi, S.P.; Bazan, J.F.; Nachury, M.V. The Conserved Bardet-Biedl Syndrome Proteins Assemble a Coat that Traffics Membrane Proteins to Cilia. Cell 2010, 141, 1208–1219. [Google Scholar] [CrossRef]
- Mourão, A.; Nager, A.R.; Nachury, M.V.; Lorentzen, E. Structural basis for membrane targeting of the BBSome by ARL6. Nat. Struct. Mol. Biol. 2014, 21, 1035–1041. [Google Scholar] [CrossRef]
- Hernandez-Hernandez, V.; Pravincumar, P.; Diaz-Font, A.; May-Simera, H.; Jenkins, D.; Knight, M.; Beales, P.L. Bardet–Biedl syndrome proteins control the cilia length through regulation of actin polymerization. Hum. Mol. Genet. 2013, 22, 3858–3868. [Google Scholar] [CrossRef]
- Ewerling, A.; Maissl, V.; Wickstead, B.; May-Simera, H.L. Neofunctionalization of ciliary BBS proteins to nuclear roles is likely a frequent innovation across eukaryotes. iScience 2023, 26, 106410. [Google Scholar] [CrossRef]
- Baldari, C.T.; Rosenbaum, J. Intraflagellar transport: It’s not just for cilia anymore. Curr. Opin. Cell Biol. 2009, 22, 75–80. [Google Scholar] [CrossRef]
- Taschner, M.; Lorentzen, A.; Mourão, A.; Collins, T.; Freke, G.M.; Moulding, D.; Basquin, J.; Jenkins, D.; Lorentzen, E. Crystal structure of intraflagellar transport protein 80 reveals a homo-dimer required for ciliogenesis. eLife 2018, 7, e33067. [Google Scholar] [CrossRef]
- Perretta-Tejedor, N.; Freke, G.; Seda, M.; Long, D.A.; Jenkins, D. Generating Mutant Renal Cell Lines Using CRISPR Technologies. Methods Mol. Biol. 2020, 2067, 323–340. [Google Scholar] [CrossRef]
- van Dam, T.J.; Wheway, G.; Slaats, G.G.; SYSCILIA Study Group; Huynen, M.A.; Giles, R.H. The SYSCILIA gold standard (SCGSv1) of known ciliary components and its applications within a systems biology consortium. Cilia 2013, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- van Dam, T.J.P.; Kennedy, J.; van der Lee, R.; de Vrieze, E.; Wunderlich, K.A.; Rix, S.; Dougherty, G.W.; Lambacher, N.J.; Li, C.; Jensen, V.L.; et al. CiliaCarta: An integrated and validated compendium of ciliary genes. PLoS ONE 2019, 14, e0216705. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- The Gene Ontology Consortium. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, S.; Katoh, Y.; Kobayashi, T.; Nakayama, K. BBS1 is involved in retrograde trafficking of ciliary GPCRs in the context of the BBSome complex. PLoS ONE 2018, 13, e01950055. [Google Scholar] [CrossRef] [PubMed]
- Prasai, A.; Cernohorska, M.S.; Ruppova, K.; Niederlova, V.; Andelova, M.; Draber, P.; Stepanek, O.; Huranova, M. The BBSome assembly is spatially controlled by BBS1 and BBS4 in human cells. J. Biol. Chem. 2020, 295, 14279–14290. [Google Scholar] [CrossRef]
- Yang, S.; Bahl, K.; Chou, H.-T.; Woodsmith, J.; Stelzl, U.; Walz, T.; Nachury, M.V. Near-atomic structures of the BBSome reveal the basis for BBSome activation and binding to GPCR cargoes. eLife 2020, 9, e55954. [Google Scholar] [CrossRef]
- Szaszi, K.; Amoozadeh, Y. New insights into functions, regulation, and pathological roles of tight junctions in kidney tubular epithelium. Int. Rev. Cell Mol. Biol. 2014, 308, 205–271. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Liberzon, A.; Subramanian, A.; Pinchback, R.; Thorvaldsdóttir, H.; Tamayo, P.; Mesirov, J.P. Molecular signatures database (MSigDB) 3.0. Bioinformatics 2011, 27, 1739–1740. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Gascue, C.; Tan, P.L.; Cardenas-Rodriguez, M.; Libisch, G.; Fernandez-Calero, T.; Liu, Y.P.; Astrada, S.; Robello, C.; Naya, H.; Katsanis, N.; et al. Direct role of Bardet–Biedl syndrome proteins in transcriptional regulation. J. Cell Sci. 2012, 125, 362–375. [Google Scholar] [CrossRef] [PubMed]
- Kilpinen, H.; Goncalves, A.; Leha, A.; Afzal, V.; Alasoo, K.; Ashford, S.; Bala, S.; Bensaddek, D.; Casale, F.P.; Culley, O.J.; et al. Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 2017, 546, 370–375, Erratum in Nature 2017, 546, 686. [Google Scholar] [CrossRef]
- Gröger, C.J.; Grubinger, M.; Waldhör, T.; Vierlinger, K.; Mikulits, W. Meta-Analysis of Gene Expression Signatures Defining the Epithelial to Mesenchymal Transition during Cancer Progression. PLoS ONE 2012, 7, e51136. [Google Scholar] [CrossRef]
- Iyer, V.; Boroviak, K.; Thomas, M.; Doe, B.; Riva, L.; Ryder, E.; Adams, D.J. No unexpected CRISPR-Cas9 off-target activity revealed by trio sequencing of gene-edited mice. PLoS Genet. 2018, 14, e1007503. [Google Scholar] [CrossRef]
- Forbes, T.A.; Howden, S.E.; Lawlor, K.; Phipson, B.; Maksimovic, J.; Hale, L.; Wilson, S.; Quinlan, C.; Ho, G.; Holman, K.; et al. Patient-iPSC-Derived Kidney Organoids Show Functional Validation of a Ciliopathic Renal Phenotype and Reveal Underlying Pathogenetic Mechanisms. Am. J. Hum. Genet. 2018, 102, 816–831. [Google Scholar] [CrossRef]
- May-Simera, H.L.; Wan, Q.; Jha, B.S.; Hartford, J.; Khristov, V.; Dejene, R.; Chang, J.; Patnaik, S.; Lu, Q.; Banerjee, P.; et al. Primary Cilium-Mediated Retinal Pigment Epithelium Maturation Is Disrupted in Ciliopathy Patient Cells. Cell Rep. 2018, 22, 189–205. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial–mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Aschauer, L.; Gruber, L.N.; Pfaller, W.; Limonciel, A.; Athersuch, T.J.; Cavill, R.; Khan, A.; Gstraunthaler, G.; Grillari, J.; Grillari, R.; et al. Delineation of the Key Aspects in the Regulation of Epithelial Monolayer Formation. Mol. Cell. Biol. 2013, 33, 2535–2550. [Google Scholar] [CrossRef]
- Delous, M.; Hellman, N.E.; Gaudé, H.-M.; Silbermann, F.; Le Bivic, A.; Salomon, R.; Antignac, C.; Saunier, S. Nephrocystin-1 and nephrocystin-4 are required for epithelial morphogenesis and associate with PALS1/PATJ and Par6. Hum. Mol. Genet. 2009, 18, 4711–4723. [Google Scholar] [CrossRef] [PubMed]
- Sang, L.; Miller, J.J.; Corbit, K.C.; Giles, R.H.; Brauer, M.J.; Otto, E.A.; Baye, L.M.; Wen, X.; Scales, S.J.; Kwong, M.; et al. Mapping the NPHP-JBTS-MKS Protein Network Reveals Ciliopathy Disease Genes and Pathways. Cell 2011, 145, 513–528. [Google Scholar] [CrossRef] [PubMed]
- Tobin, J.L.; Di Franco, M.; Eichers, E.; May-Simera, H.; Garcia, M.; Yan, J.; Quinlan, R.; Justice, M.J.; Hennekam, R.C.; Briscoe, J.; et al. Inhibition of neural crest migration underlies craniofacial dysmorphology and Hirschsprung’s disease in Bardet–Biedl syndrome. Proc. Natl. Acad. Sci. USA 2008, 105, 6714–6719. [Google Scholar] [CrossRef] [PubMed]
- Colla, T.T.C.S.; Dixon, J.; Edwards, S.J.; Gladwin, A.J.; Dixon, M.J.; Loftus, S.K.; Bonner, C.A.; Koprivnikar, K.; Wasmuth, J.J. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat. Genet. 1996, 12, 130–136. [Google Scholar] [CrossRef]
- Gál, Z.; Nieto, B.; Boukoura, S.; Rasmussen, A.V.; Larsen, D.H. Treacle Sticks the Nucleolar Responses to DNA Damage Together. Front. Cell Dev. Biol. 2022, 10, 892006. [Google Scholar] [CrossRef]
- Ng, S.B.; Buckingham, K.J.; Lee, C.; Bigham, A.W.; Tabor, H.K.; Dent, K.M.; Huff, C.D.; Shannon, P.T.; Jabs, E.W.; Nickerson, D.A.; et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010, 42, 30–35. [Google Scholar] [CrossRef]
- White, R.M.; Cech, J.; Ratanasirintrawoot, S.; Lin, C.Y.; Rahl, P.B.; Burke, C.J.; Langdon, E.; Tomlinson, M.L.; Mosher, J.T.; Kaufman, C.K.; et al. DHODH modulates transcriptional elongation in the neural crest and melanoma. Nature 2011, 471, 518–522. [Google Scholar] [CrossRef]
- Santos, F.; Moreira, C.; Nóbrega-Pereira, S.; de Jesus, B.B. New Insights into the Role of Epithelial–Mesenchymal Transition during Aging. Int. J. Mol. Sci. 2019, 20, 891. [Google Scholar] [CrossRef]
- Meyer, J.R.; Krentz, A.D.; Berg, R.L.; Richardson, J.G.; Pomeroy, J.; Hebbring, S.J.; Haws, R.M. Kidney failure in Bardet–Biedl syndrome. Clin. Genet. 2022, 101, 429–441. [Google Scholar] [CrossRef]
- Hemachandar, R. Bardet–Biedl syndrome: A rare cause of end stage renal disease. Int. J. Appl. Basic Med. Res. 2015, 5, 70–72. [Google Scholar] [CrossRef]
- Croft, J.B.; Swift, M. Obesity, hypertension, and renal disease in relatives of Bardet-Biedl syndrome sibs. Am. J. Med. Genet. 1990, 36, 37–42. [Google Scholar] [CrossRef]
- O’Dea, D.; Parfrey, P.S.; Harnett, J.D.; Hefferton, D.; Cramer, B.C.; Green, J. The importance of renal impairment in the natural history of bardet-biedl syndrome. Am. J. Kidney Dis. 1996, 27, 776–783. [Google Scholar] [CrossRef]
- Beales, P.L.; Elcioglu, N.; Woolf, A.S.; Parker, D.; Flinter, F.A. New criteria for improved diagnosis of Bardet-Biedl syndrome: Results of a population survey. J. Med. Genet. 1999, 36, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Sparks, K.; Hoskins, B.; Bagkeris, E.; McGowan, B.; Carroll, P.; Huda, M.; Mujahid, S.; Peters, C.; Barrett, T.; et al. Genetic predictors of cardiovascular morbidity in Bardet–Biedl syndrome. Clin. Genet. 2014, 87, 343–349, Erratum in Clin. Genet. 2016, 89, 636. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, E.; Beales, P.L. Bardet–Biedl syndrome. Eur. J. Hum. Genet. 2012, 21, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Putoux, A.; Attie-Bitach, T.; Martinovic, J.; Gubler, M.-C. Phenotypic variability of Bardet-Biedl syndrome: Focusing on the kidney. Pediatr. Nephrol. 2012, 27, 7–15. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Boldt, K.; Schumacher, A.; Roepman, R.; Ueffing, M. A novel tandem affinity purification strategy for the efficient isolation and characterisation of native protein complexes. Proteomics 2007, 7, 4228–4234. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Boldt, K.; Schumacher, A.; Ueffing, M. Tandem affinity purification of protein complexes from mammalian cells by the Strep/FLAG (SF)-TAP tag. Methods Mol. Biol. 2009, 564, 359–372. [Google Scholar] [CrossRef]
- Gloeckner, C.J.; Boldt, K.; Ueffing, M. Strep/FLAG Tandem Affinity Purification (SF-TAP) to Study Protein Interactions. Curr. Protoc. Protein Sci. 2009, 57, 19.20.1–19.20.19. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. g:Profiler: A web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freke, G.M.; Martins, T.; Davies, R.J.; Beyer, T.; Seda, M.; Peskett, E.; Haq, N.; Prasai, A.; Otto, G.; Jeyabalan Srikaran, J.; et al. De-Suppression of Mesenchymal Cell Identities and Variable Phenotypic Outcomes Associated with Knockout of Bbs1. Cells 2023, 12, 2662. https://doi.org/10.3390/cells12222662
Freke GM, Martins T, Davies RJ, Beyer T, Seda M, Peskett E, Haq N, Prasai A, Otto G, Jeyabalan Srikaran J, et al. De-Suppression of Mesenchymal Cell Identities and Variable Phenotypic Outcomes Associated with Knockout of Bbs1. Cells. 2023; 12(22):2662. https://doi.org/10.3390/cells12222662
Chicago/Turabian StyleFreke, Grace Mercedes, Tiago Martins, Rosalind Jane Davies, Tina Beyer, Marian Seda, Emma Peskett, Naila Haq, Avishek Prasai, Georg Otto, Jeshmi Jeyabalan Srikaran, and et al. 2023. "De-Suppression of Mesenchymal Cell Identities and Variable Phenotypic Outcomes Associated with Knockout of Bbs1" Cells 12, no. 22: 2662. https://doi.org/10.3390/cells12222662