
Analysis of 5-Azacytidine Resistance Models Reveals a Set of Targetable Pathways

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
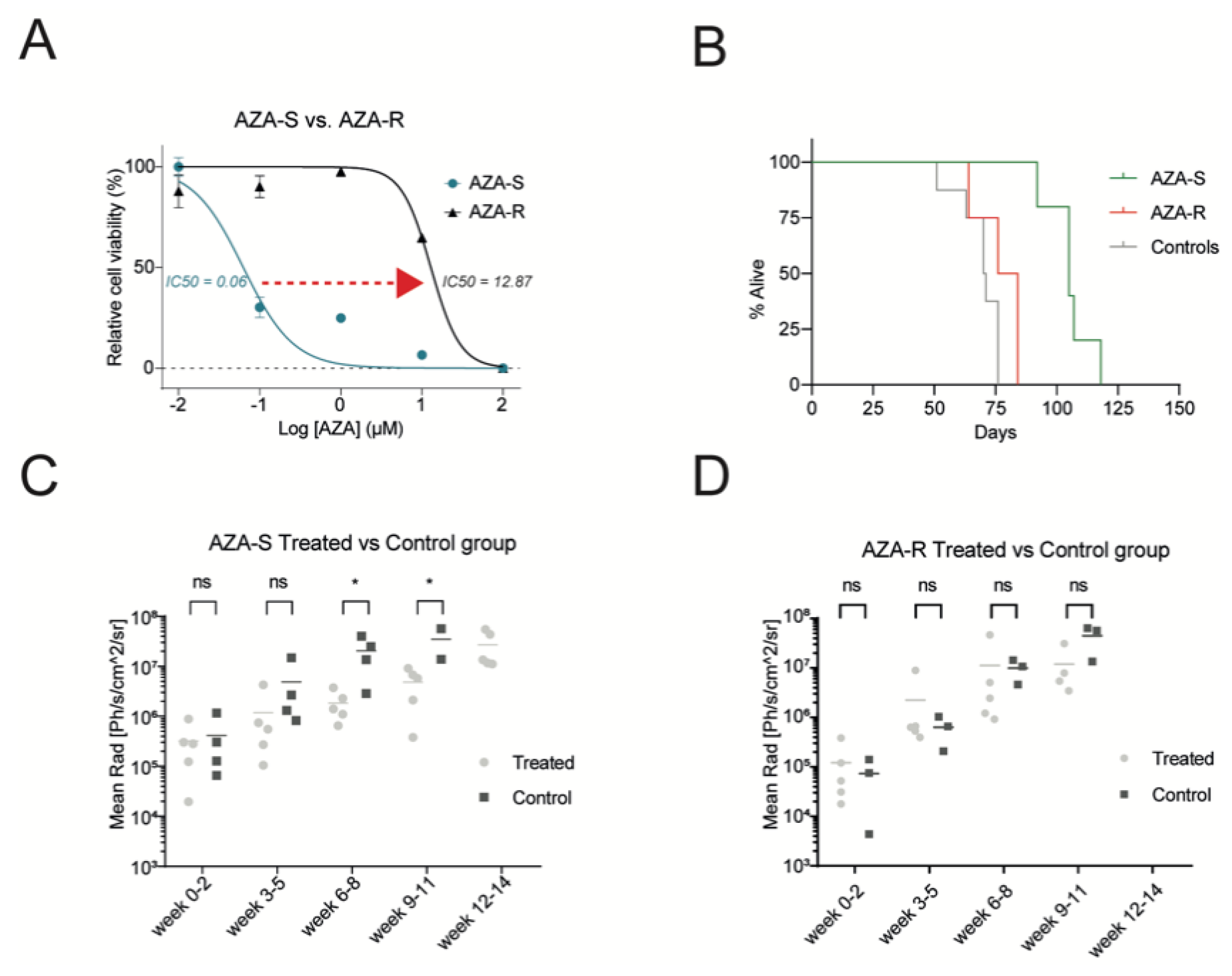
3.1. Development of a Cellular Model for AZA Resistance
3.2. Identification of Pathogenic AZA-R DNA Variants
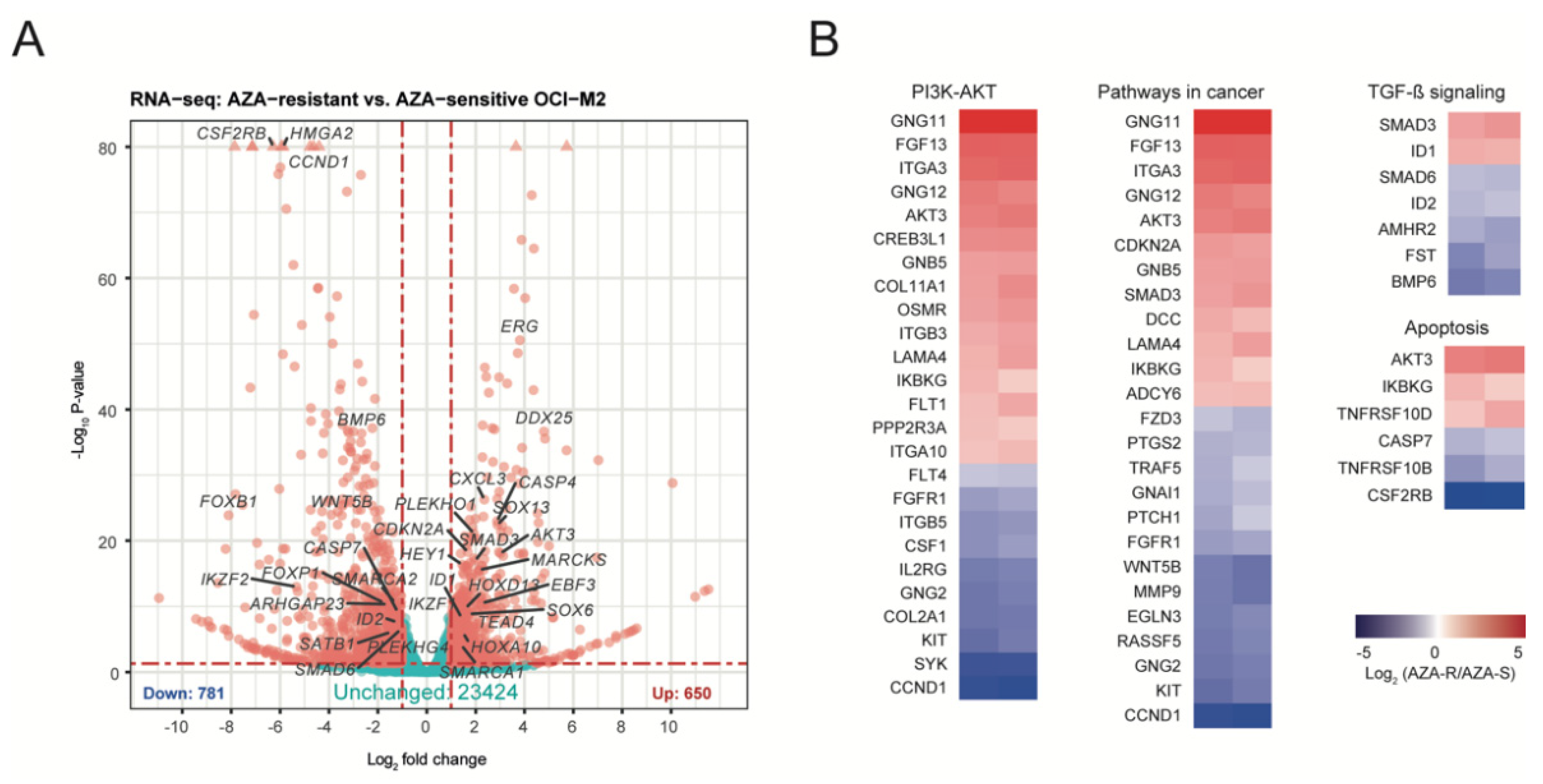
3.3. Identification of Molecular Patterns for AZA Resistance
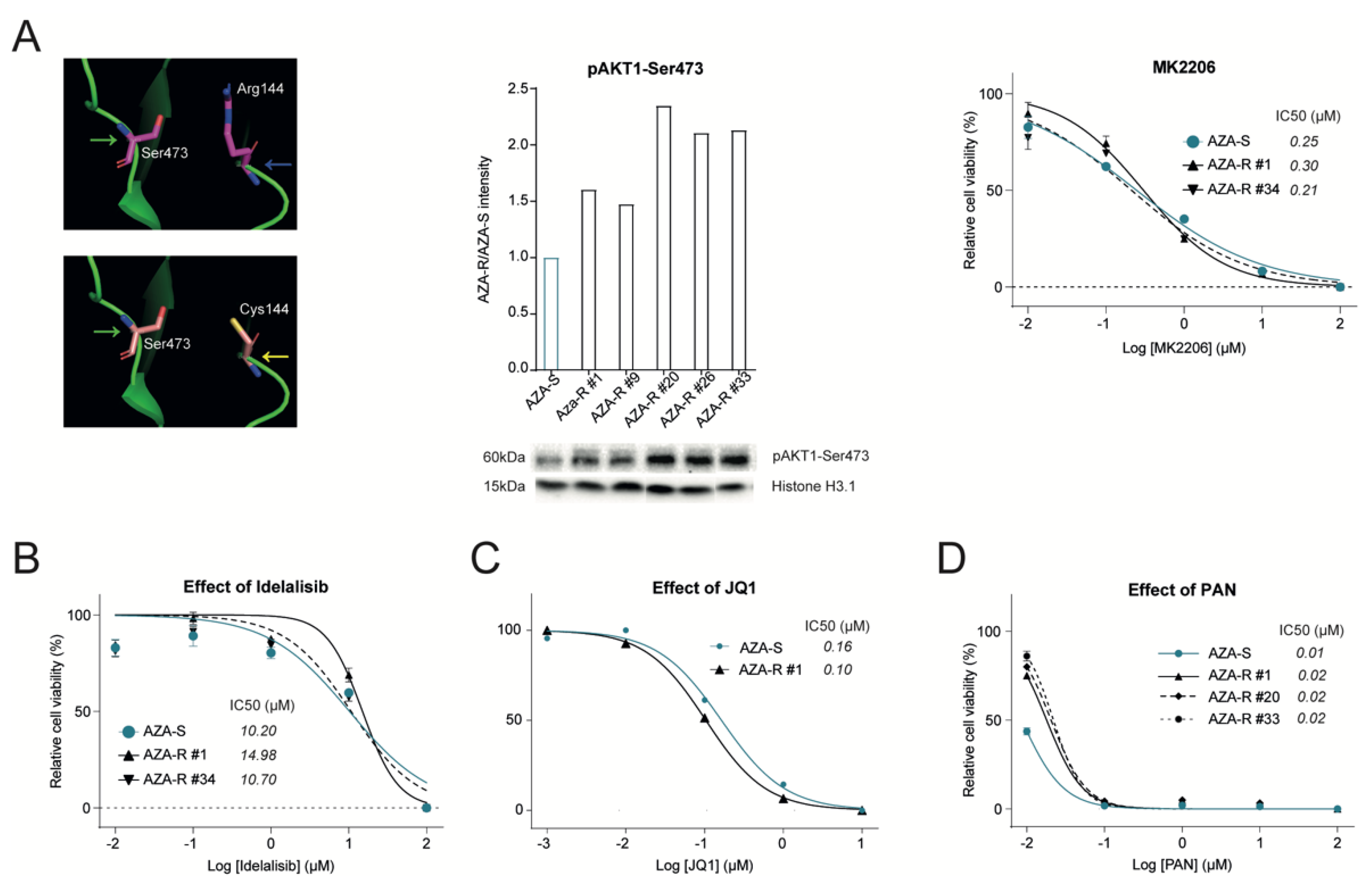
3.4. Dysregulation of the PI3K-AKT Pathway in AZA-R Cells
3.5. Validation of Other Signaling Pathways Deregulated in AZA-R Cells
3.6. Addressing Effect of Selected Signaling Pathway Inhibitors on AZA Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Finelli, C.; Giagounidis, A.; Schoch, R.; Gattermann, N.; Sanz, G.; List, A.; et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: A randomised, open-label, phase III study. Lancet Oncol. 2009, 10, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Fenaux, P.; Mufti, G.J.; Hellstrom-Lindberg, E.; Santini, V.; Gattermann, N.; Germing, U.; Sanz, G.; List, A.F.; Gore, S.; Seymour, J.F.; et al. Azacitidine prolongs overall survival compared with conventional care regimens in elderly patients with low bone marrow blast count acute myeloid leukemia. J. Clin. Oncol. 2010, 28, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Ozbalak, M.; Cetiner, M.; Bekoz, H.; Atesoglu, E.B.; Ar, C.; Salihoglu, A.; Tuzuner, N.; Ferhanoglu, B. Azacitidine has limited activity in ‘real life’ patients with MDS and AML: A single centre experience. Hematol. Oncol. 2012, 30, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Maurillo, L.; Venditti, A.; Spagnoli, A.; Gaidano, G.; Ferrero, D.; Oliva, E.; Lunghi, M.; D’Arco, A.M.; Levis, A.; Pastore, D.; et al. Azacitidine for the treatment of patients with acute myeloid leukemia: Report of 82 patients enrolled in an Italian Compassionate Program. Cancer 2012, 118, 1014–1022. [Google Scholar] [CrossRef] [Green Version]
- Pleyer, L.; Germing, U.; Sperr, W.R.; Linkesch, W.; Burgstaller, S.; Stauder, R.; Girschikofsky, M.; Schreder, M.; Pfeilstocker, M.; Lang, A.; et al. Azacitidine in CMML: Matched-pair analyses of daily-life patients reveal modest effects on clinical course and survival. Leuk. Res. 2014, 38, 475–483. [Google Scholar] [CrossRef] [Green Version]
- Prebet, T.; Gore, S.D.; Esterni, B.; Gardin, C.; Itzykson, R.; Thepot, S.; Dreyfus, F.; Rauzy, O.B.; Recher, C.; Ades, L.; et al. Outcome of high-risk myelodysplastic syndrome after azacitidine treatment failure. J. Clin. Oncol. 2011, 29, 3322–3327. [Google Scholar] [CrossRef]
- Harel, S.; Cherait, A.; Berthon, C.; Willekens, C.; Park, S.; Rigal, M.; Brechignac, S.; Thepot, S.; Quesnel, B.; Gardin, C.; et al. Outcome of patients with high risk Myelodysplastic Syndrome (MDS) and advanced Chronic Myelomonocytic Leukemia (CMML) treated with decitabine after azacitidine failure. Leuk. Res. 2015, 39, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Nazha, A.; Sekeres, M.A.; Bejar, R.; Rauh, M.J.; Othus, M.; Komrokji, R.S.; Barnard, J.; Hilton, C.B.; Kerr, C.M.; Steensma, D.P.; et al. Genomic Biomarkers to Predict Resistance to Hypomethylating Agents in Patients with Myelodysplastic Syndromes Using Artificial Intelligence. JCO Precis. Oncol. 2019, 3, 1–11. [Google Scholar] [CrossRef]
- Drusbosky, L.M.; Singh, N.K.; Hawkins, K.E.; Salan, C.; Turcotte, M.; Wise, E.A.; Meacham, A.; Vijay, V.; Anderson, G.G.; Kim, C.C.; et al. A genomics-informed computational biology platform prospectively predicts treatment responses in AML and MDS patients. Blood Adv. 2019, 3, 1837–1847. [Google Scholar] [CrossRef]
- Leonhardt, H.; Page, A.W.; Weier, H.U.; Bestor, T.H. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell 1992, 71, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Stomper, J.; Rotondo, J.C.; Greve, G.; Lubbert, M. Hypomethylating agents (HMA) for the treatment of acute myeloid leukemia and myelodysplastic syndromes: Mechanisms of resistance and novel HMA-based therapies. Leukemia 2021, 35, 1873–1889. [Google Scholar] [CrossRef]
- DiNardo, C.D.; Jonas, B.A.; Pullarkat, V.; Thirman, M.J.; Garcia, J.S.; Wei, A.H.; Konopleva, M.; Dohner, H.; Letai, A.; Fenaux, P.; et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N. Engl. J. Med. 2020, 383, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Fenaux, P.; Al-Kali, A.; Baer, M.R.; Sekeres, M.A.; Roboz, G.J.; Gaidano, G.; Scott, B.L.; Greenberg, P.; Platzbecker, U.; et al. Rigosertib versus best supportive care for patients with high-risk myelodysplastic syndromes after failure of hypomethylating drugs (ONTIME): A randomised, controlled, phase 3 trial. Lancet Oncol. 2016, 17, 496–508. [Google Scholar] [CrossRef]
- Sekeres, M.A.; Watts, J.; Radinoff, A.; Sangerman, M.A.; Cerrano, M.; Lopez, P.F.; Zeidner, J.F.; Campelo, M.D.; Graux, C.; Liesveld, J.; et al. Randomized phase 2 trial of pevonedistat plus azacitidine versus azacitidine for higher-risk MDS/CMML or low-blast AML. Leukemia 2021, 35, 2119–2124. [Google Scholar] [CrossRef] [PubMed]
- Janotka, L.; Messingerova, L.; Simonicova, K.; Kavcova, H.; Elefantova, K.; Sulova, Z.; Breier, A. Changes in Apoptotic Pathways in MOLM-13 Cell Lines after Induction of Resistance to Hypomethylating Agents. Int. J. Mol. Sci. 2021, 22, 2076. [Google Scholar] [CrossRef]
- Medyouf, H.; Mossner, M.; Jann, J.C.; Nolte, F.; Raffel, S.; Herrmann, C.; Lier, A.; Eisen, C.; Nowak, V.; Zens, B.; et al. Myelodysplastic cells in patients reprogram mesenchymal stromal cells to establish a transplantable stem cell niche disease unit. Cell Stem Cell 2014, 14, 824–837. [Google Scholar] [CrossRef] [Green Version]
- Ebinger, S.; Ozdemir, E.Z.; Ziegenhain, C.; Tiedt, S.; Castro Alves, C.; Grunert, M.; Dworzak, M.; Lutz, C.; Turati, V.A.; Enver, T.; et al. Characterization of Rare, Dormant, and Therapy-Resistant Cells in Acute Lymphoblastic Leukemia. Cancer Cell 2016, 30, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Rabbani, B.; Tekin, M.; Mahdieh, N. The promise of whole-exome sequencing in medical genetics. J. Hum. Genet. 2014, 59, 5–15. [Google Scholar] [CrossRef]
- Curik, N.; Burda, P.; Vargova, K.; Pospisil, V.; Belickova, M.; Vlckova, P.; Savvulidi, F.; Necas, E.; Hajkova, H.; Haskovec, C.; et al. 5-azacitidine in aggressive myelodysplastic syndromes regulates chromatin structure at PU.1 gene and cell differentiation capacity. Leukemia 2012, 26, 1804–1811. [Google Scholar] [CrossRef]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Sherman, B.T.; Da Huang, W.; Tan, Q.; Guo, Y.; Bour, S.; Liu, D.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID Knowledgebase: A gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinform. 2007, 8, 426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Huang, W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Traina, F.; Visconte, V.; Elson, P.; Tabarroki, A.; Jankowska, A.M.; Hasrouni, E.; Sugimoto, Y.; Szpurka, H.; Makishima, H.; O’Keefe, C.L.; et al. Impact of molecular mutations on treatment response to DNMT inhibitors in myelodysplasia and related neoplasms. Leukemia 2014, 28, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Polgarova, K.; Vargova, K.; Kulvait, V.; Dusilkova, N.; Minarik, L.; Zemanova, Z.; Pesta, M.; Jonasova, A.; Stopka, T. Somatic mutation dynamics in MDS patients treated with azacitidine indicate clonal selection in patients-responders. Oncotarget 2017, 8, 111966–111978. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.; Park, S.; Choi, H.; Kim, H.J.; Kwon, Y.R.; Ryu, D.; Kim, M.; Kim, T.M.; Kim, Y.J. Gene expression signatures associated with sensitivity to azacitidine in myelodysplastic syndromes. Sci. Rep. 2020, 10, 19555. [Google Scholar] [CrossRef] [PubMed]
- Pericole, F.V.; Lazarini, M.; de Paiva, L.B.; Duarte, A.; Vieira Ferro, K.P.; Niemann, F.S.; Roversi, F.M.; Olalla Saad, S.T. BRD4 Inhibition Enhances Azacitidine Efficacy in Acute Myeloid Leukemia and Myelodysplastic Syndromes. Front. Oncol. 2019, 9, 16. [Google Scholar] [CrossRef] [Green Version]
- Bertacchini, J.; Guida, M.; Accordi, B.; Mediani, L.; Martelli, A.M.; Barozzi, P.; Petricoin, E., 3rd; Liotta, L.; Milani, G.; Giordan, M.; et al. Feedbacks and adaptive capabilities of the PI3K/Akt/mTOR axis in acute myeloid leukemia revealed by pathway selective inhibition and phosphoproteome analysis. Leukemia 2014, 28, 2197–2205. [Google Scholar] [CrossRef]
- Bertacchini, J.; Frasson, C.; Chiarini, F.; D’Avella, D.; Accordi, B.; Anselmi, L.; Barozzi, P.; Forghieri, F.; Luppi, M.; Martelli, A.M.; et al. Dual inhibition of PI3K/mTOR signaling in chemoresistant AML primary cells. Adv. Biol. Regul. 2018, 68, 2–9. [Google Scholar] [CrossRef] [Green Version]
- Darici, S.; Alkhaldi, H.; Horne, G.; Jorgensen, H.G.; Marmiroli, S.; Huang, X. Targeting PI3K/Akt/mTOR in AML: Rationale and Clinical Evidence. J. Clin. Med. 2020, 9, 2934. [Google Scholar] [CrossRef]
- Nyakern, M.; Tazzari, P.L.; Finelli, C.; Bosi, C.; Follo, M.Y.; Grafone, T.; Piccaluga, P.P.; Martinelli, G.; Cocco, L.; Martelli, A.M. Frequent elevation of Akt kinase phosphorylation in blood marrow and peripheral blood mononuclear cells from high-risk myelodysplastic syndrome patients. Leukemia 2006, 20, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Xia, X.; Kong, Y.; Jia, F.; Yuan, B.; Li, R.; Li, Q.; Wang, Y.; Cui, M.; Dai, Z.; et al. Deregulation of tumor suppressive ASXL1-PTEN/AKT axis in myeloid malignancies. J. Mol. Cell Biol. 2020, 12, 688–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steelman, L.S.; Franklin, R.A.; Abrams, S.L.; Chappell, W.; Kempf, C.R.; Basecke, J.; Stivala, F.; Donia, M.; Fagone, P.; Nicoletti, F.; et al. Roles of the Ras/Raf/MEK/ERK pathway in leukemia therapy. Leukemia 2011, 25, 1080–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bazarbachi, A.; Labopin, M.; Battipaglia, G.; Djabali, A.; Passweg, J.; Socie, G.; Forcade, E.; Blaise, D.; Chevallier, P.; Orvain, C.; et al. Sorafenib improves survival of FLT3-mutated acute myeloid leukemia in relapse after allogeneic stem cell transplantation: A report of the EBMT Acute Leukemia Working Party. Haematologica 2019, 104, e398–e401. [Google Scholar] [CrossRef] [Green Version]
- Voisset, E.; Brenet, F.; Lopez, S.; de Sepulveda, P. SRC-Family Kinases in Acute Myeloid Leukaemia and Mastocytosis. Cancers 2020, 12, 1996. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Manero, G.; Sekeres, M.A.; Egyed, M.; Breccia, M.; Graux, C.; Cavenagh, J.D.; Salman, H.; Illes, A.; Fenaux, P.; DeAngelo, D.J.; et al. A phase 1b/2b multicenter study of oral panobinostat plus azacitidine in adults with MDS, CMML or AML with 30% blasts. Leukemia 2017, 31, 2799–2806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Cai, T.; Lin, X.; Huang, X.; Bui, M.H.; Plotnik, J.P.; Bellin, R.J.; Faivre, E.J.; Kuruvilla, V.M.; Lam, L.T.; et al. Selective Inhibition of the Second Bromodomain of BET Family Proteins Results in Robust Antitumor Activity in Preclinical Models of Acute Myeloid Leukemia. Mol. Cancer Ther. 2021, 20, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Tool | Category (Count 1) | p-Value 2 | Genes 3 |
|---|---|---|---|
| GAD_dis. | cancer (232) | 9 × 10−6 | AKT1, AKT3, IKZF1, IKZF2, IKZF3, ERG, KIT, SMAD3, CCND1, CDKN2A, RUNX1 |
| metabolic (335) | 3 × 10−3 | KIT, KLF13, KLF6, PBX3, SMAD3, SMAD6, SOX13, CDH2, CTH, DPP10, EVI5, FGFR1, FLT1 | |
| hematologic (239) | 4 × 10−3 | ERG, SATB1, SMARCA2, CDKN2A, AKT3, ERCC1, JMJD1C, SYK, HGF | |
| UP_KEY | protein phosphorylation (534) | 6 × 10−21 | AKT1, AKT3, BCOR, BMX, ACVR1, FLT1, FLT4, FOXP1, FOXL2, PTPN6, PTPN13, ERG |
| alternative splicing (629) | 2 × 10−16 | ARL13B, EEF1A1, GFM1, PABPC1, PARG, PARP14, PARP3, PARP8, PARP9, PCBP2 | |
| KEGG | PI3K-AKT signaling (38) | 5 × 10−5 | AKT1, AKT3, CCND1, CSF1, PTK2, PIK3R3, FLT1, FLT4 |
| chemokine signaling (24) | 2 × 10−4 | CCR7, CXCL16, CXCL2, CXCL3, SHC1, PLCB3, VAV1, VAV2 | |
| Rap1 signaling (25) | 5 × 10−4 | CSF1, EGFR, FGF13, ID1, SKAP1, RGS14, HGF | |
| pathways in cancer (37) | 1 × 10−3 | BRCA2, KIT, SMAD3, CDKN2A, EGFR, FZD3, HDAC2, RUNX1 | |
| Ras signaling pathway (25) | 3 × 10−3 | ARF6, GNG11, GNG12, GNB5, GNG2, RASSF5, SHC1, KSR1 | |
| GO_BP | GTPase activity (57) | 4 × 10−6 | AGAP1, ASAP2, ASAP3, GDI1, RAP1GAP2, RALGPS2, ARHGAP23, RGS12, RGS14, RGS20, TRIO |
| regulation of proliferation (23) | 3 × 10−4 | BMX, TAL1, CDCA7, ERBB3, HOXD13, PTK2B, PTK2, KIT | |
| protein kinase activity (10) | 7 × 10−4 | CCR7, RASSF2, CSF1, PTPRC | |
| signal transduction (86) | 1 × 10−3 | AKT1, AKT3, BMX, ERG, RIN3, SHC1, IL9R, ERBB3, CSF2RB, MOK, KIT | |
| GO_MF | protein binding (525) | 4 × 10−6 | ABCA1, BCOR, DNA2, EHD2, KLF6, SATB1, ACVR1, CCND1, CDKN2A, HOXA10, HDAC2, HDAC6, HGF, HMGA2 |
| GTPase activity (33) | 2 × 10−5 | AGAP1, ASAP2, ASAP3, CDC42EP1, DLC1, GDI1, RAP1GAP2, ARHGEF6, RIN3, RINL, RGS12, RGS14, RGS20 | |
| phospholipid binding (14) | 5 × 10−4 | SHC1, DAPP1, STAP1, AGAP1 | |
| protein kinase binding (36) | 7 × 10−4 | BCL10, CYLD, SMAD3, CCND1, CDKN2A, HCLS1, MAPK6, PTK2, PTPN6, PTPRC, PTPRK, SYK, SKAP1 | |
| INTERPRO | Pleckstrin-like domain (53) | 2 × 10−9 | AKT1, AKT3, AGAP1, BMX, RALGPS2, SHC1, PLEKHA4, PLEKHA6, PLEKHO1, PTK2B, PTK2, STAP1 |
| Pleckstrin domain (37) | 8 × 10−8 | VAV1, VAV2, TRIO, PLEKHG4, PLCL1, GRB14, DOK4, CDH2, ARHGEF6, ASAP2, ASAP3 | |
| Src Homology 2 domain (18) | 4 × 10−5 | BMX, RIN3, RINL, SLA, GRB14, PTPN6, SYK | |
| Ser-Thre/Tyr- kinase (19) | 2 × 10−4 | KIT, EGFR, FLT1, FLT4, KSR1, MAP3K7, PTK2B, PTK, ROR1, TIE1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Minařík, L.; Pimková, K.; Kokavec, J.; Schaffartziková, A.; Vellieux, F.; Kulvait, V.; Daumová, L.; Dusilková, N.; Jonášová, A.; Vargová, K.S.; et al. Analysis of 5-Azacytidine Resistance Models Reveals a Set of Targetable Pathways. Cells 2022, 11, 223. https://doi.org/10.3390/cells11020223
Minařík L, Pimková K, Kokavec J, Schaffartziková A, Vellieux F, Kulvait V, Daumová L, Dusilková N, Jonášová A, Vargová KS, et al. Analysis of 5-Azacytidine Resistance Models Reveals a Set of Targetable Pathways. Cells. 2022; 11(2):223. https://doi.org/10.3390/cells11020223
Chicago/Turabian StyleMinařík, Lubomír, Kristýna Pimková, Juraj Kokavec, Adéla Schaffartziková, Fréderic Vellieux, Vojtěch Kulvait, Lenka Daumová, Nina Dusilková, Anna Jonášová, Karina Savvulidi Vargová, and et al. 2022. "Analysis of 5-Azacytidine Resistance Models Reveals a Set of Targetable Pathways" Cells 11, no. 2: 223. https://doi.org/10.3390/cells11020223