
Changes in Ion Concentrations upon the Binding of Short Polyelectrolytes on Phospholipid Bilayers: Computer Study Addressing Interesting Physiological Consequences

Abstract
:
1. Introduction
2. Simulation Method
3. Model
4. Parameter Setting
5. Methodology of Simulations
6. Results and Discussion
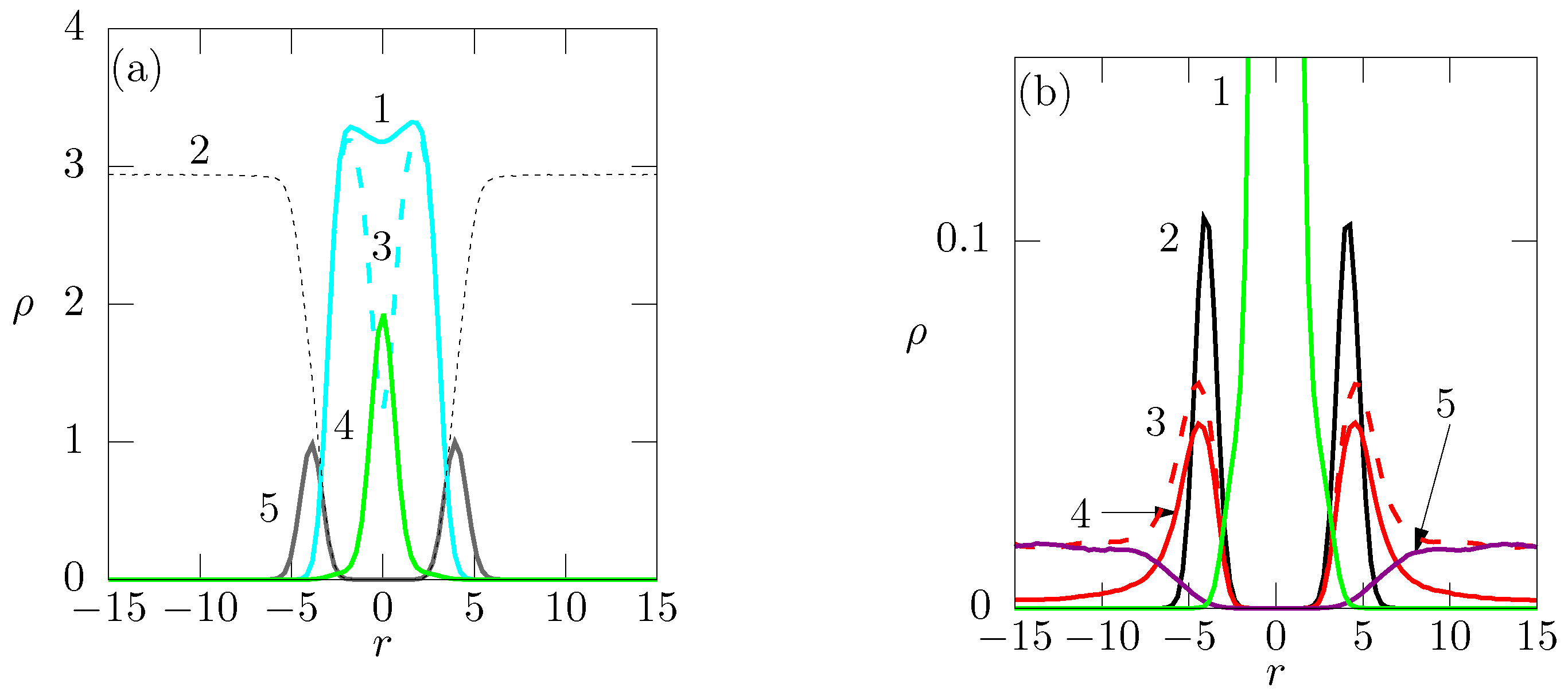
6.1. Partially Charged Membrane with Monovalent and Divalent Cations
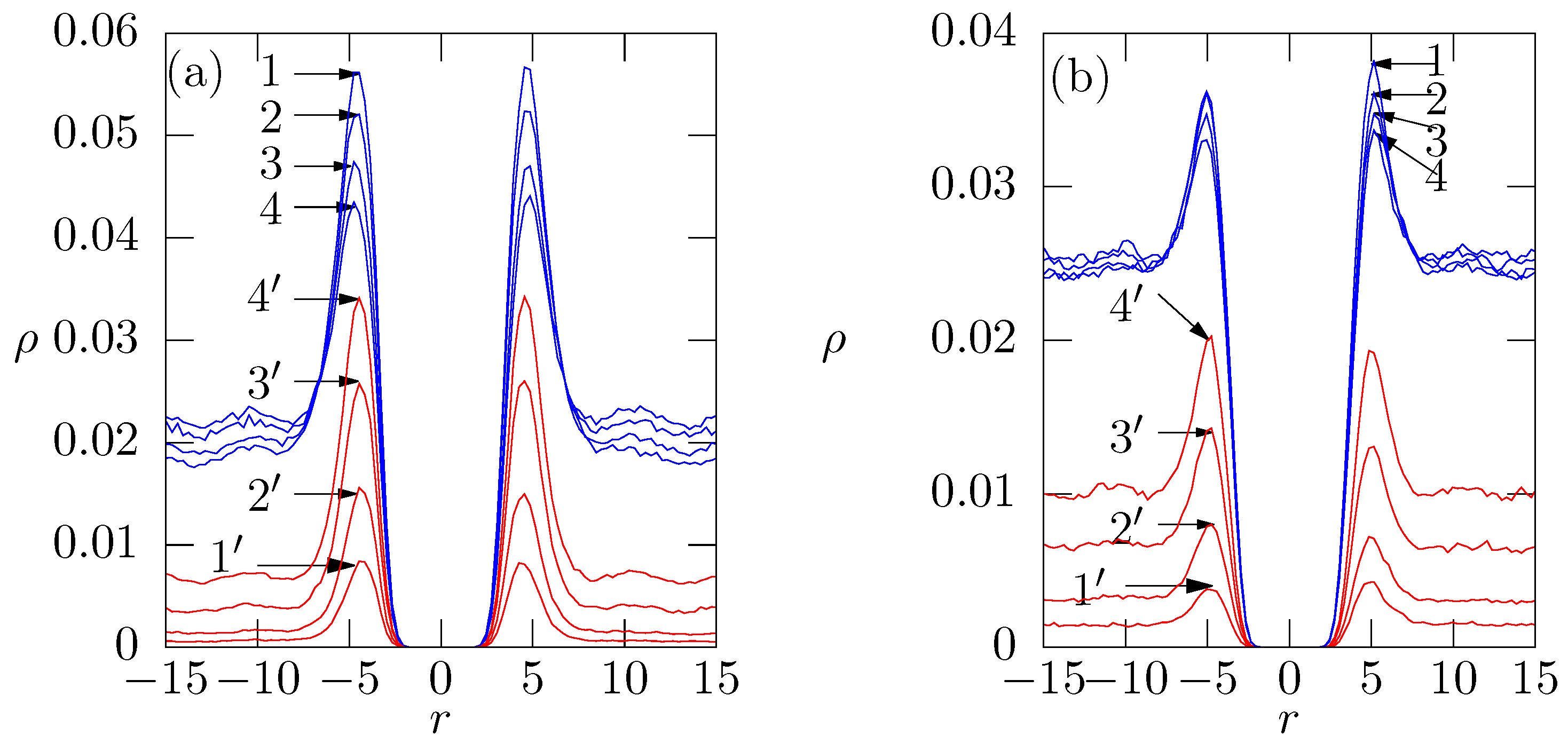
6.2. Effect of the Binding of Positively Charged Oligomers at the Membrane on the Distributions of Univalent and Divalent Ions
7. Summary and Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MDPI | Multidisciplinary Digital Publishing Institute |
| MRSA | Methicillin-resistant Staphylococcus aureus |
| DPD | Dissipative particle dynamics |
| FENE | Finitely extendible non-linear elastic |
| FH | Flory-Huggins |
| CP | Coulomb potential |
| IPEC | inter-polyelectrolyte complex |
| MD | Molecular Dynamics |
References
- Voet, D.; Voet, J.G. Biochemistry, 4th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2010; pp. 386–449. [Google Scholar]
- Nagarajan, R.; Ruckenstein, E. Theory of surfactant self-assembly—A predictive molecular thermodynamic approach. Langmuir 1991, 7, 2934–2969. [Google Scholar] [CrossRef]
- Nagarajan, R.; Ganesh, K. Block copolymer self-assembly in selective solvents—Spherical micelles with segregated cores. J. Chem. Phys. 1989, 90, 5843–5856. [Google Scholar] [CrossRef]
- Halperin, A.; Alexander, S. Polymeric micelles—Their relaxation kinetics. Macromolecules 1989, 22, 2403–2412. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef] [PubMed]
- Hiemenz, P.C.; Rajagopalan, R. Principles of Colloid and Surface Chemistry, Revised and Expanded; CRC Press: Boca Raton, FL, USA, 2016; pp. 4–5. [Google Scholar]
- Newton, A.C.; Bootman, M.D.; Scott, J.D. Second messengers. Cold Spring Harb. Perspect. Biol. 2016, 8, a005926. [Google Scholar] [CrossRef]
- Wu, G.; Ding, J.; Li, H.; Li, L.; Zhao, R.; Shen, Z.; Fan, X.; Xi, T. Effects of Cations and PH on Antimicrobial Activity of Thanatin and s-Thanatin Against Escherichia coli ATCC25922 and B-subtilis ATCC 21332. Curr. Microbiol. 2008, 57, 552–557. [Google Scholar] [CrossRef]
- Golshani-Hebroni, S. Mg++ requirement for MtHK binding, and Mg++ stabilization of mitochondrial membranes via activation of MtHK & MtCK and promotion of mitochondrial permeability transition pore closure: A hypothesis on mechanisms underlying Mg++’s antioxidant and cytoprotective effects. Gene 2016, 581, 1–13. [Google Scholar] [CrossRef]
- Saris, N.; Mervaala, E.; Karppanen, H.; Khawaja, J.; Lewenstam, A. Magnesium—An update on physiological, clinical and analytical aspects. Clin. Chim. Acta 2000, 294, 1–26. [Google Scholar] [CrossRef]
- Kumar, M.; Srivastava, S. Effect of calcium and magnesium on the antimicrobial action of enterocin LR/6 produced by Enterococcus faecium LR/6. Int. J. Antimicrob. Agents 2011, 37, 572–575. [Google Scholar] [CrossRef]
- Zhekova, H.R.; Ngo, V.; da Silva, M.C.; Salahub, D.; Noskov, S. Selective ion binding and transport by membrane proteins—A computational perspective. Coord. Chem. Rev. 2017, 345, 108–136. [Google Scholar] [CrossRef]
- Meriney, S.D.; Umbach, J.A.; Gundersen, C.B. Fast, Ca2+-dependent exocytosis at nerve terminals: Shortcomings of SNARE-based models. Prog. Neurobiol. 2014, 121, 55–90. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Qi, F.; Chen, Q.; Zhang, Q.; Qiao, Z.; Zhang, S.; Wei, T.; Yu, Q.; Yu, S.; Mao, Z.; et al. Surface modified with a host defense peptide-mimicking β-peptide polymer kills bacteria on contact with high efficacy. ACS Appl. Mater. Interfaces 2018, 10, 15395–15400. [Google Scholar] [CrossRef] [PubMed]
- Hancock, R.E.; Sahl, H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, X.; Zhu, R.; Hu, K.; Lan, W.W.; Wu, F.; Yang, L. Acid-activated antimicrobial random copolymers: A mechanism-guided design of antimicrobial peptide mimics. Macromolecules 2013, 46, 3959–3964. [Google Scholar] [CrossRef]
- Kuroda, K.; DeGrado, W.F. Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 2005, 127, 4128–4129. [Google Scholar] [CrossRef]
- Lienkamp, K.; Madkour, A.E.; Musante, A.; Nelson, C.F.; Nusslein, K.; Tew, G.N. Antimicrobial polymers prepared by ROMP with unprecedented selectivity: A molecular construction kit approach. J. Am. Chem. Soc. 2008, 130, 9836–9843. [Google Scholar] [CrossRef]
- Nederberg, F.; Zhang, Y.; Tan, J.P.; Xu, K.; Wang, H.; Yang, C.; Gao, S.; Guo, X.D.; Fukushima, K.; Li, L.; et al. Biodegradable nanostructures with selective lysis of microbial membranes. Nat. Chem. 2011, 3, 409–414. [Google Scholar] [CrossRef]
- Palermo, E.F.; Sovadinova, I.; Kuroda, K. Structural determinants of antimicrobial activity and biocompatibility in membrane-disrupting methacrylamide random copolymers. Biomacromolecules 2009, 10, 3098–3107. [Google Scholar] [CrossRef]
- Qian, Y.X.; Zhang, D.F.; Wu, Y.M.; Chen, Q.; Liu, R.H. The design, synthesis and biological activity study of nylon-3 polymers as mimics of host defense peptides. Acta Polym. Sin. 2016, 10, 1300–1311. [Google Scholar]
- Sambhy, V.; Peterson, B.R.; Sen, A. Antibacterial and hemolytic activities of pyridinium polymers as a function of the spatial relationship between the positive charge and the pendant alkyl tail. Angew. Chem. 2008, 120, 1270–1274. [Google Scholar] [CrossRef]
- Song, A.; Walker, S.G.; Parker, K.A.; Sampson, N.S. Antibacterial studies of cationic polymers with alternating, random, and uniform backbones. ACS Chem. Biol. 2011, 6, 590–599. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Lee, M.W.; Mansbach, R.A.; Song, Z.; Bao, Y.; Peek, R.M.; Yao, C.; Chen, L.F.; Ferguson, A.L.; Wong, G.C.; et al. Helical antimicrobial polypeptides with radial amphiphilicity. Proc. Natl. Acad. Sci. USA 2015, 112, 13155–13160. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Lo, J.C.; Mei, Y.; Haney, E.F.; Siren, E.; Kalathottukaren, M.T.; Hancock, R.E.; Lange, D.; Kizhakkedathu, J.N. Toward infection-resistant surfaces: Achieving high antimicrobial peptide potency by modulating the functionality of polymer brush and peptide. ACS Appl. Mater. Interfaces 2015, 7, 28591–28605. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms: My perspective. Antimicrob. Pept. 2019, 1117, 3–6. [Google Scholar]
- Boman, H. Antibacterial peptides: Basic facts and emerging concepts. J. Intern. Med. 2003, 254, 197–215. [Google Scholar] [CrossRef] [PubMed]
- Gelman, M.A.; Weisblum, B.; Lynn, D.M.; Gellman, S.H. Biocidal activity of polystyrenes that are cationic by virtue of protonation. Org. Lett. 2004, 6, 557–560. [Google Scholar] [CrossRef]
- Sellenet, P.H.; Allison, B.; Applegate, B.M.; Youngblood, J.P. Synergistic activity of hydrophilic modification in antibiotic polymers. Biomacromolecules 2007, 8, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, X.; Falk, S.P.; Masters, K.S.; Weisblum, B.; Gellman, S.H. Nylon-3 polymers active against drug-resistant Candida albicans biofilms. J. Am. Chem. Soc. 2015, 137, 2183–2186. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Chen, X.; Gellman, S.H.; Masters, K.S. Nylon-3 polymers that enable selective culture of endothelial cells. J. Am. Chem. Soc. 2013, 135, 16296–16299. [Google Scholar] [CrossRef]
- Liu, R.; Chen, X.; Hayouka, Z.; Chakraborty, S.; Falk, S.P.; Weisblum, B.; Masters, K.S.; Gellman, S.H. Nylon-3 polymers with selective antifungal activity. J. Am. Chem. Soc. 2013, 135, 5270–5273. [Google Scholar] [CrossRef]
- Liu, R.; Chen, X.; Chakraborty, S.; Lemke, J.J.; Hayouka, Z.; Chow, C.; Welch, R.A.; Weisblum, B.; Masters, K.S.; Gellman, S.H. Tuning the biological activity profile of antibacterial polymers via subunit substitution pattern. J. Am. Chem. Soc. 2014, 136, 4410–4418. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Masters, K.S.; Gellman, S.H. Polymer chain length effects on fibroblast attachment on nylon-3-modified surfaces. Biomacromolecules 2012, 13, 1100–1105. [Google Scholar] [CrossRef] [Green Version]
- Mowery, B.P.; Lee, S.E.; Kissounko, D.A.; Epand, R.F.; Epand, R.M.; Weisblum, B.; Stahl, S.S.; Gellman, S.H. Mimicry of antimicrobial host-defense peptides by random copolymers. J. Am. Chem. Soc. 2007, 129, 15474–15476. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Huo, D.; Nimmagadda, A.; Wu, J.; She, F.; Su, M.; Lin, X.; Yan, J.; Cao, A.; Xi, C.; et al. Small antimicrobial agents based on acylated reduced amide scaffold. J. Med. Chem. 2016, 59, 7877–7887. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Chakraborty, S.; Liu, R.; Gellman, S.H.; Weisshaar, J.C. Single-cell, time-resolved antimicrobial effects of a highly cationic, random nylon-3 copolymer on live Escherichia coli. ACS Chem. Biol. 2016, 11, 113–120. [Google Scholar] [CrossRef]
- Xue, R.; Chu, X.; Yang, F.; Liu, Z.; Yin, L.; Tang, H. Imidazolium-Based Polypeptide Coating with a Synergistic Antibacterial Effect and a Biofilm-Responsive Property. ACS Macro Lett. 2022, 11, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Ishantha Senevirathne, S.; Hasan, J.; Mathew, A.; Jaggessar, A.; Yarlagadda, P.K. Trends in Bactericidal Nanostructured Surfaces: An Analytical Perspective. ACS Appl. Bio Mater. 2021, 4, 7626–7642. [Google Scholar] [CrossRef]
- Juffer, A.; Shepherd, C.; Vogel, H. Protein-membrane electrostatic interactions: Application of the Lekner summation technique. J. Chem. Phys. 2001, 114, 1892–1905. [Google Scholar] [CrossRef]
- Hoogerbrugge, P.; Koelman, J. Simulating microscopic hydrodynamic phenomena with dissipative particle dynamics. EPL Europhys. Lett. 1992, 19, 155–160. [Google Scholar] [CrossRef]
- Groot, R.D.; Warren, P.B. Dissipative particle dynamics: Bridging the gap between atomistic and mesoscopic simulation. J. Chem. Phys. 1997, 107, 4423–4435. [Google Scholar] [CrossRef]
- Espanol, P.; Warren, P. Statistical mechanics of dissipative particle dynamics. EPL Europhys. Lett. 1995, 30, 191. [Google Scholar] [CrossRef]
- Karimi-Varzaneh, H.A.; Van Der Vegt, N.F.; Müller-Plathe, F.; Carbone, P. How good are coarse-grained polymer models? A comparison for atactic polystyrene. ChemPhysChem 2012, 13, 3428–3439. [Google Scholar] [CrossRef] [PubMed]
- Procházka, K.; Limpouchová, Z.; Štěpánek, M.; Šindelka, K.; Lísal, M. DPD Modelling of the Self-and Co-Assembly of Polymers and Polyelectrolytes in Aqueous Media: Impact on Polymer Science. Polymers 2022, 14, 404. [Google Scholar] [CrossRef]
- Atkins, P.; Keeler, J.; de Paula, J. Atkins’ Physical Chemistry; Oxford University Press: Oxford, UK, 2014; pp. 600–601. [Google Scholar]
- Lee, M.T.; Vishnyakov, A.; Neimark, A.V. Coarse-grained model of water diffusion and proton conductivity in hydrated polyelectrolyte membrane. J. Chem. Phys. 2016, 144, 014902. [Google Scholar] [CrossRef]
- Lee, M.T.; Vishnyakov, A.; Neimark, A.V. Calculations of Critical Micelle Concentration by Dissipative Particle Dynamics Simulations: The Role of Chain Rigidity. J. Phys. Chem. B 2013, 117, 10304–10310. [Google Scholar] [CrossRef]
- Groot, R.D. Electrostatic interactions in dissipative particle dynamics—Simulation of polyelectrolytes and anionic surfactants. J. Chem. Phys. 2003, 118, 11265–11277. [Google Scholar] [CrossRef]
- Posel, Z.; Limpouchova, Z.; Sindelka, K.; Lisal, M.; Prochazka, K. Dissipative particle dynamics study of the pH-dependent behavior of poly (2-vinylpyridine)-block-poly (ethylene oxide) diblock copolymer in aqueous buffers. Macromolecules 2014, 47, 2503–2514. [Google Scholar] [CrossRef]
- Ibergay, C.; Malfreyt, P.; Tildesley, D.J. Electrostatic interactions in dissipative particle dynamics: Toward a mesoscale modeling of the polyelectrolyte brushes. J. Chem. Theory Comput. 2009, 5, 3245–3259. [Google Scholar] [CrossRef]
- González-Melchor, M.; Mayoral, E.; Velázquez, M.E.; Alejandre, J. Electrostatic interactions in dissipative particle dynamics using the Ewald sums. J. Chem. Phys. 2006, 125, 224107. [Google Scholar] [CrossRef]
- Procházka, K.; Šindelka, K.; Wang, X.; Limpouchová, Z.; Lísal, M. Self-assembly and co-assembly of block polyelectrolytes in aqueous solutions. Dissipative particle dynamics with explicit electrostatics. Mol. Phys. 2016, 114, 3077–3092. [Google Scholar] [CrossRef]
- Lahmar, F.; Tzoumanekas, C.; Theodorou, D.N.; Rousseau, B. Onset of Entanglements Revisited. Dynamical Analysis. Macromolecules 2009, 42, 7485–7494. [Google Scholar] [CrossRef]
- Warren, P.B.; Vlasov, A. Screening properties of four mesoscale smoothed charge models, with application to dissipative particle dynamics. J. Chem. Phys. 2014, 140, 084904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Šindelka, K.; Limpouchová, Z.; Lísal, M.; Procházka, K. Dissipative particle dynamics study of electrostatic self-assembly in aqueous mixtures of copolymers containing one neutral water-soluble block and one either positively or negatively charged polyelectrolyte block. Macromolecules 2014, 47, 6121–6134. [Google Scholar] [CrossRef]
- Šindelka, K.; Limpouchová, Z.; Lísal, M.; Procházka, K. The electrostatic co-assembly in non-stoichiometric aqueous mixtures of copolymers composed of one neutral water-soluble and one polyelectrolyte (either positively or negatively charged) block: A dissipative particle dynamics study. Phys. Chem. Chem. Phys. 2016, 18, 16137–16151. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Shillcock, J.C.; Lipowsky, R. Equilibrium structure and lateral stress distribution of amphiphilic bilayers from dissipative particle dynamics simulations. J. Chem. Phys. 2002, 117, 5048–5061. [Google Scholar] [CrossRef]
- Gao, L.; Shillcock, J.; Lipowsky, R. Improved dissipative particle dynamics simulations of lipid bilayers. J. Chem. Phys. 2007, 126, 01B602. [Google Scholar] [CrossRef]
- Groot, R.D.; Rabone, K. Mesoscopic simulation of cell membrane damage, morphology change and rupture by nonionic surfactants. Biophys. J. 2001, 81, 725–736. [Google Scholar] [CrossRef]
- Li, X.; Gao, L.; Fang, W. Dissipative particle dynamics simulations for phospholipid membranes based on a four-to-one coarse-grained mapping scheme. PLoS ONE 2016, 11, e0154568. [Google Scholar] [CrossRef]
- Jakobsen, A.F.; Mouritsen, O.G.; Besold, G. Artifacts in dynamical simulations of coarse-grained model lipid bilayers. J. Chem. Phys. 2005, 122, 204901. [Google Scholar] [CrossRef]
- Jakobsen, A.F. Constant-pressure and constant-surface tension simulations in dissipative particle dynamics. J. Chem. Phys. 2005, 122, 124901. [Google Scholar] [CrossRef] [PubMed]
- Kranenburg, M.; Nicolas, J.P.; Smit, B. Comparison of mesoscopic phospholipid–water models. Phys. Chem. Chem. Phys. 2004, 6, 4142–4151. [Google Scholar] [CrossRef] [Green Version]
- Šindelka, K.; Limpouchová, Z.; Procházka, K. Computer study of the solubilization of polymer chains in polyelectrolyte complex cores of polymeric nanoparticles in aqueous media. Phys. Chem. Chem. Phys. 2018, 20, 29876–29888. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
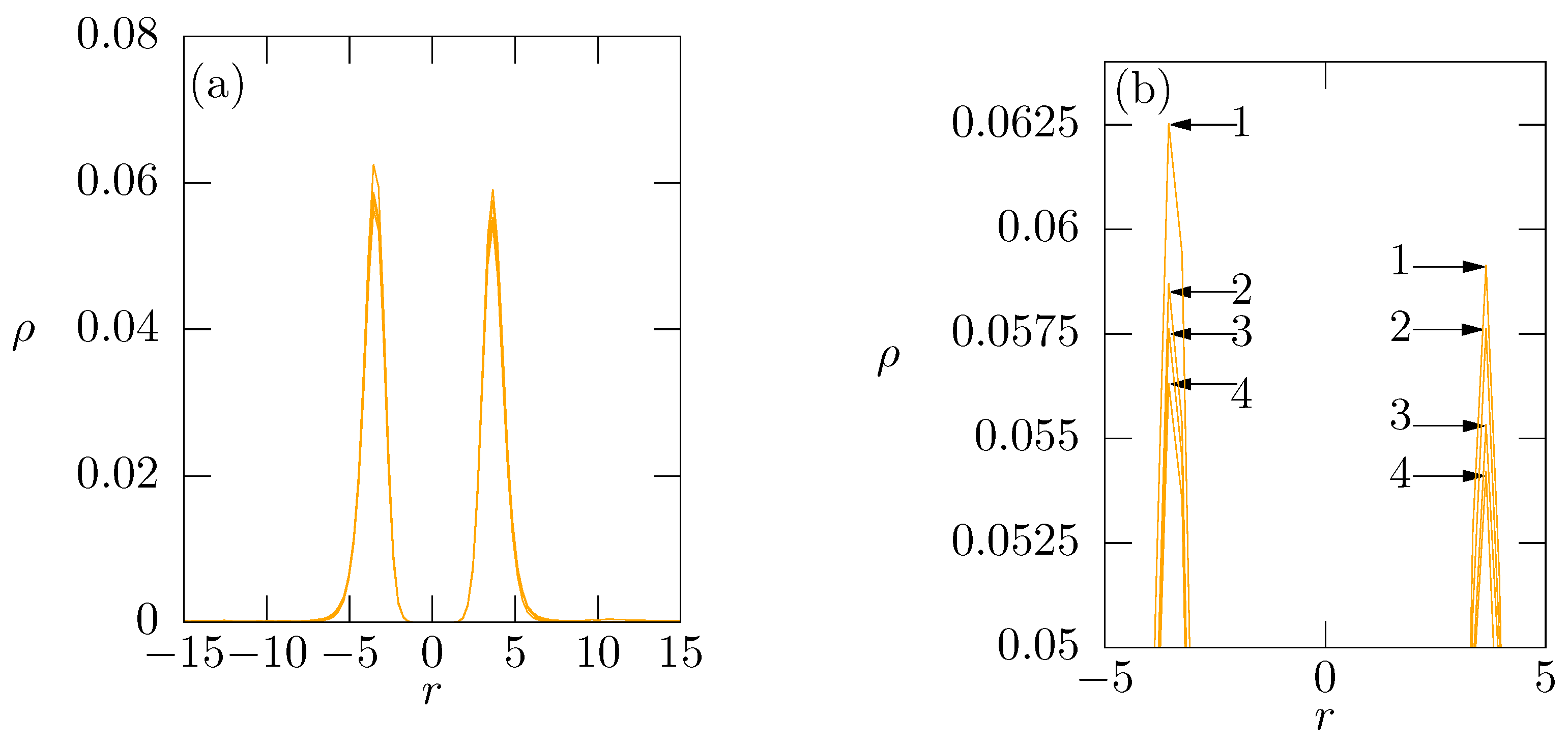{kind=link}
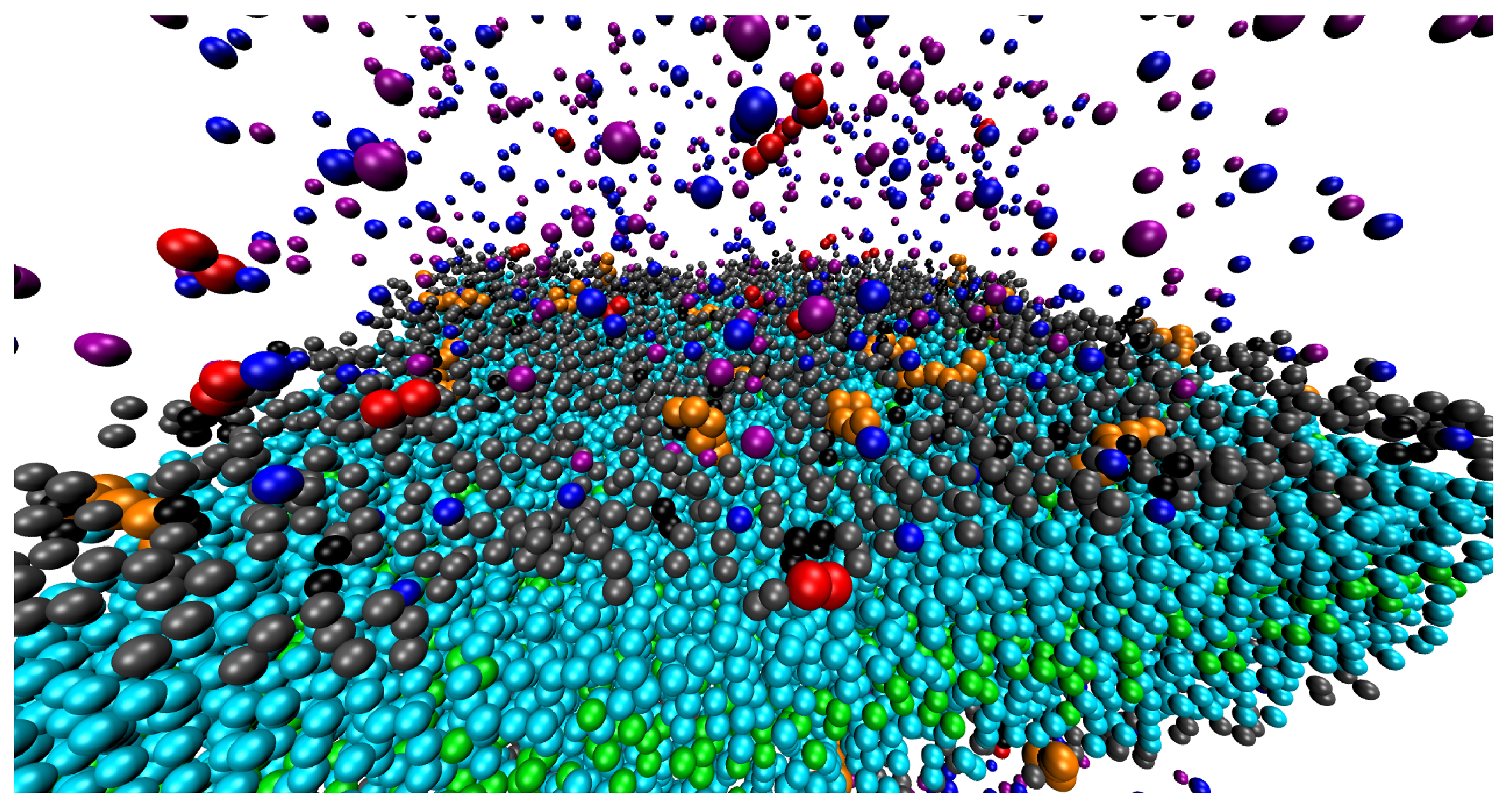{kind=link}
| / | ||||||||
|---|---|---|---|---|---|---|---|---|
| H | Hq | G | T, E | S | Na/Cl | Me | P | |
| H | 25/4.5 | 25/4.5 | 27/4.5 | 50/9.0 | 27/4.5 | 27/4.5 | 25/4.5 | 27/4.5 |
| Hq | 25/4.5 | 27/4.5 | 50/9.0 | 27/4.5 | 27/4.5 | 27/4.5 | 27.4/5 | |
| G | 25/4.5 | 50/9.0 | 28/4.5 | 28/4.5 | 28/4.5 | 28/4.5 | ||
| T, E | 25/4.5 | 75/20.0 | 75/20.0 | 75/20.0 | 27/4.5 | |||
| S | 25/4.5 | 25/4.5 | 25/4.5 | 25/4.5 | ||||
| Na/Cl | 25/4.5 | 25/4.5 | 25/4.5 | |||||
| Me | 25/4.5 | 25/4.5 | ||||||
| P | 25/4.5 | |||||||
| Membrane with | Me | P | Na/Cl | Membrane with | Me | P | Na/Cl |
|---|---|---|---|---|---|---|---|
| 1. 10% Na | 0 | 0 | 171/0 | 9. salt + 25 Me + 16 P | 25 | 16 | 584/413 |
| 2. 20% Na 10% Cl | 0 | 0 | 342/171 | 10. salt + 25 Me + 32 P | 25 | 32 | 538/367 |
| 3. salt + 25 Me | 25 | 0 | 392/221 | 11. salt + 50 Me + 16 P | 50 | 16 | 634/463 |
| 4. salt + 50 Me | 50 | 0 | 442/271 | 12. salt + 50 Me + 32 P | 50 | 32 | 638/467 |
| 5. salt + 100 Me | 100 | 0 | 542/371 | 13. salt + 100 Me + 16 P | 100 | 16 | 734/564 |
| 6. salt + 150 Me | 150 | 0 | 642/471 | 14. salt + 100 Me + 32 P | 100 | 32 | 738/567 |
| 7. salt + 25 Me + 16 P | 25 | 16 | 488/317 | 15. salt + 150 Me + 16 P | 150 | 16 | 738/567 |
| 8. salt + 25 Me + 32 P | 25 | 32 | 584/413 | 16. salt + 150 Me + 32 P | 150 | 32 | 834/663 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blovský, T.; Šindelka, K.; Limpouchová, Z.; Procházka, K. Changes in Ion Concentrations upon the Binding of Short Polyelectrolytes on Phospholipid Bilayers: Computer Study Addressing Interesting Physiological Consequences. Polymers 2022, 14, 3634. https://doi.org/10.3390/polym14173634
Blovský T, Šindelka K, Limpouchová Z, Procházka K. Changes in Ion Concentrations upon the Binding of Short Polyelectrolytes on Phospholipid Bilayers: Computer Study Addressing Interesting Physiological Consequences. Polymers. 2022; 14(17):3634. https://doi.org/10.3390/polym14173634
Chicago/Turabian StyleBlovský, Tomáš, Karel Šindelka, Zuzana Limpouchová, and Karel Procházka. 2022. "Changes in Ion Concentrations upon the Binding of Short Polyelectrolytes on Phospholipid Bilayers: Computer Study Addressing Interesting Physiological Consequences" Polymers 14, no. 17: 3634. https://doi.org/10.3390/polym14173634