Low-Temperature Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinkers: A Passive Smart Material with Potential as Viscoelastic Coupling. Part I: Synthesis and Phase Behavior

 , , ,
, , ,
Abstract
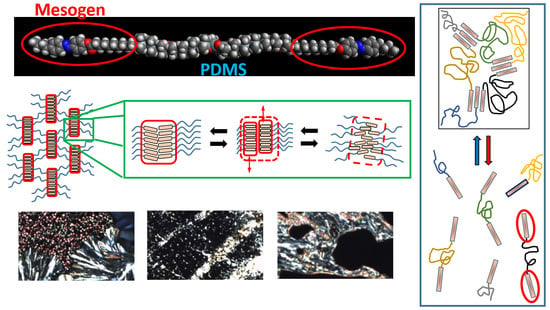
:
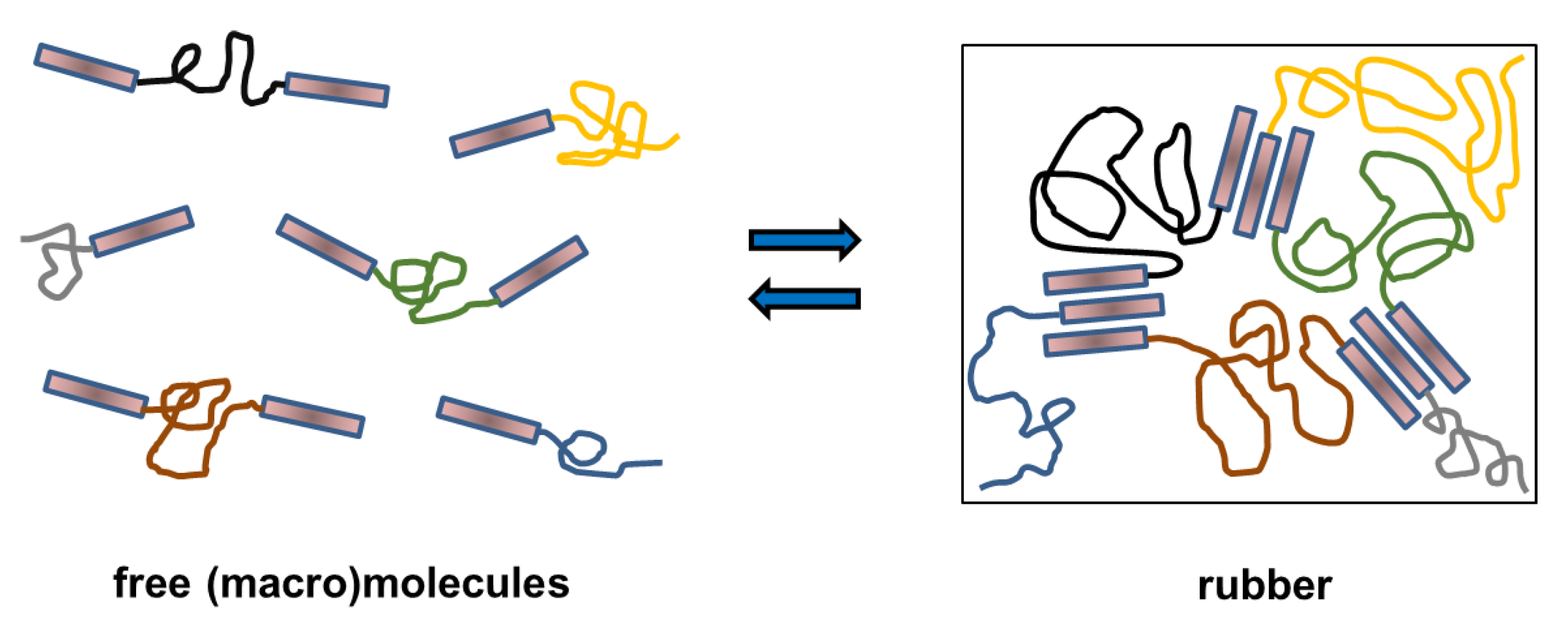
1. Introduction
2. Experimental Section
2.1. Materials
2.1.1. Commercial Chemicals
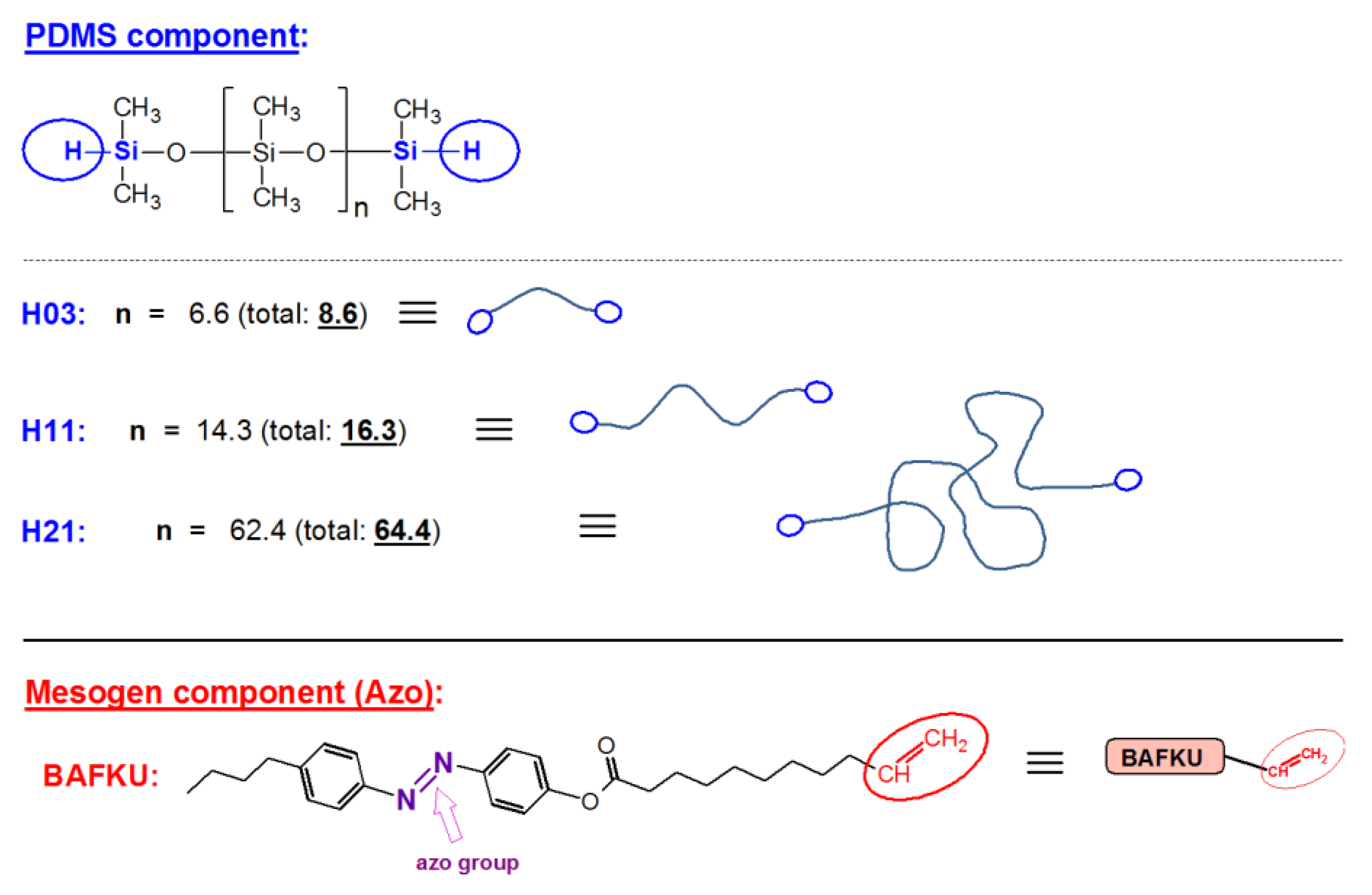
2.1.2. Synthesis of the Liquid Crystalline Mesogen named “BAFKU”
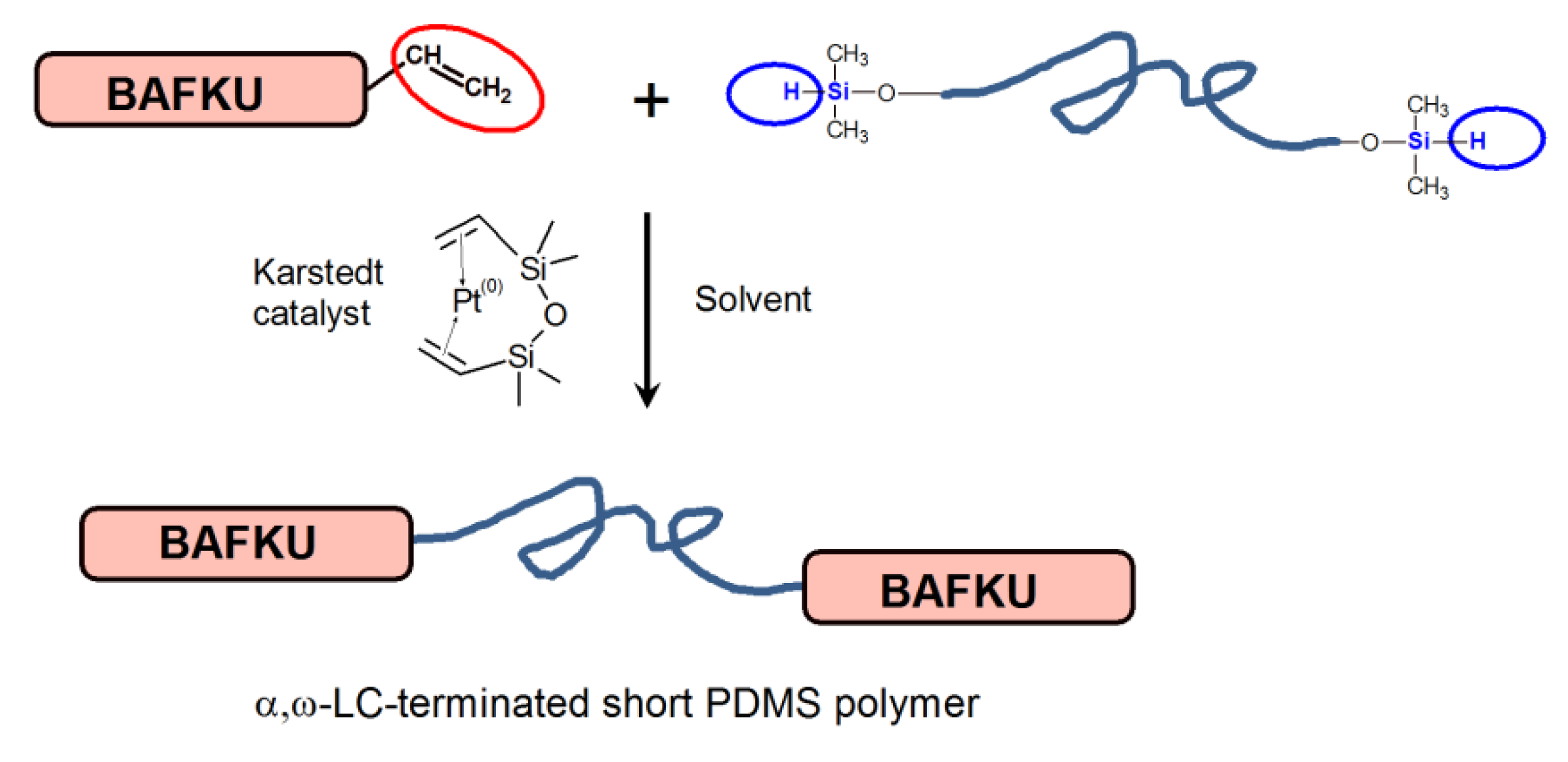
2.1.3. Preparation of the Main Chain Liquid Crystalline Copolymers Based on PDMS (DMS H03, DMS H11, or DMS H21) and LC Mesogen BAFKU
2.2. Characterization of the PDMS–BAFKU2 Copolymers
2.2.1. Chemical Microstructure
2.2.2. Thermo-Mechanical and Thermal Properties
2.2.3. Long-Range-Structure and Crystallinity Characterization
2.2.4. Polarized Light Microscopy (PLM) Observation of Phase Transitions
3. Results and Discussion
3.1. Synthesis
3.2. Thermo-Mechanical Properties
3.3. Phase Transitions in the Copolymers
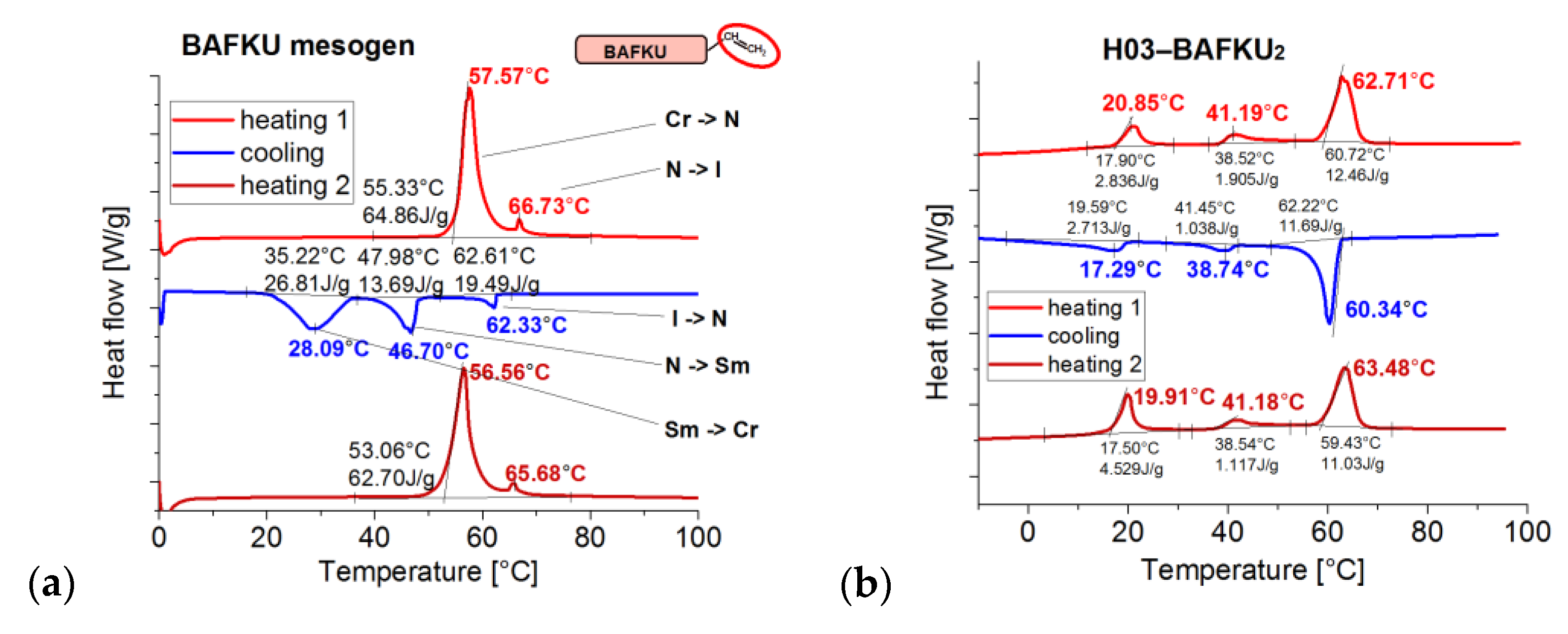
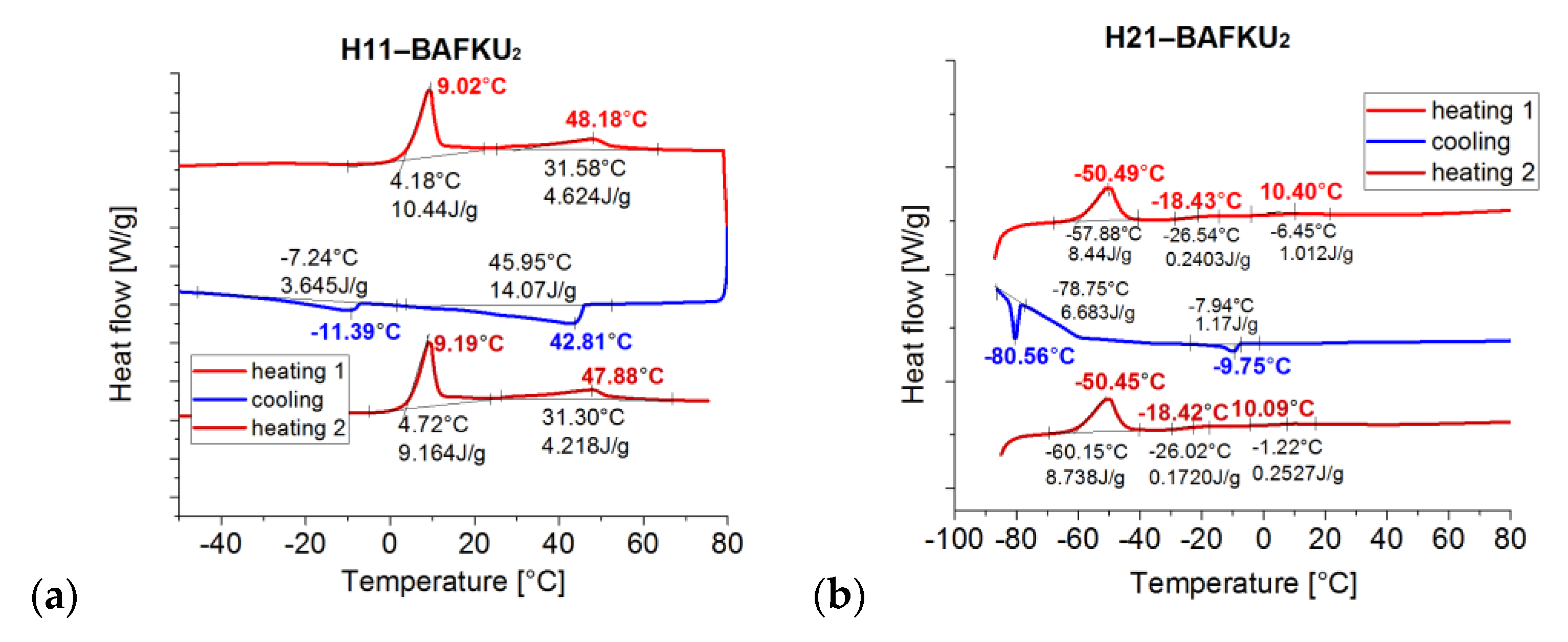
3.3.1. DSC: Specific Heat of Phase Transitions in the Copolymers
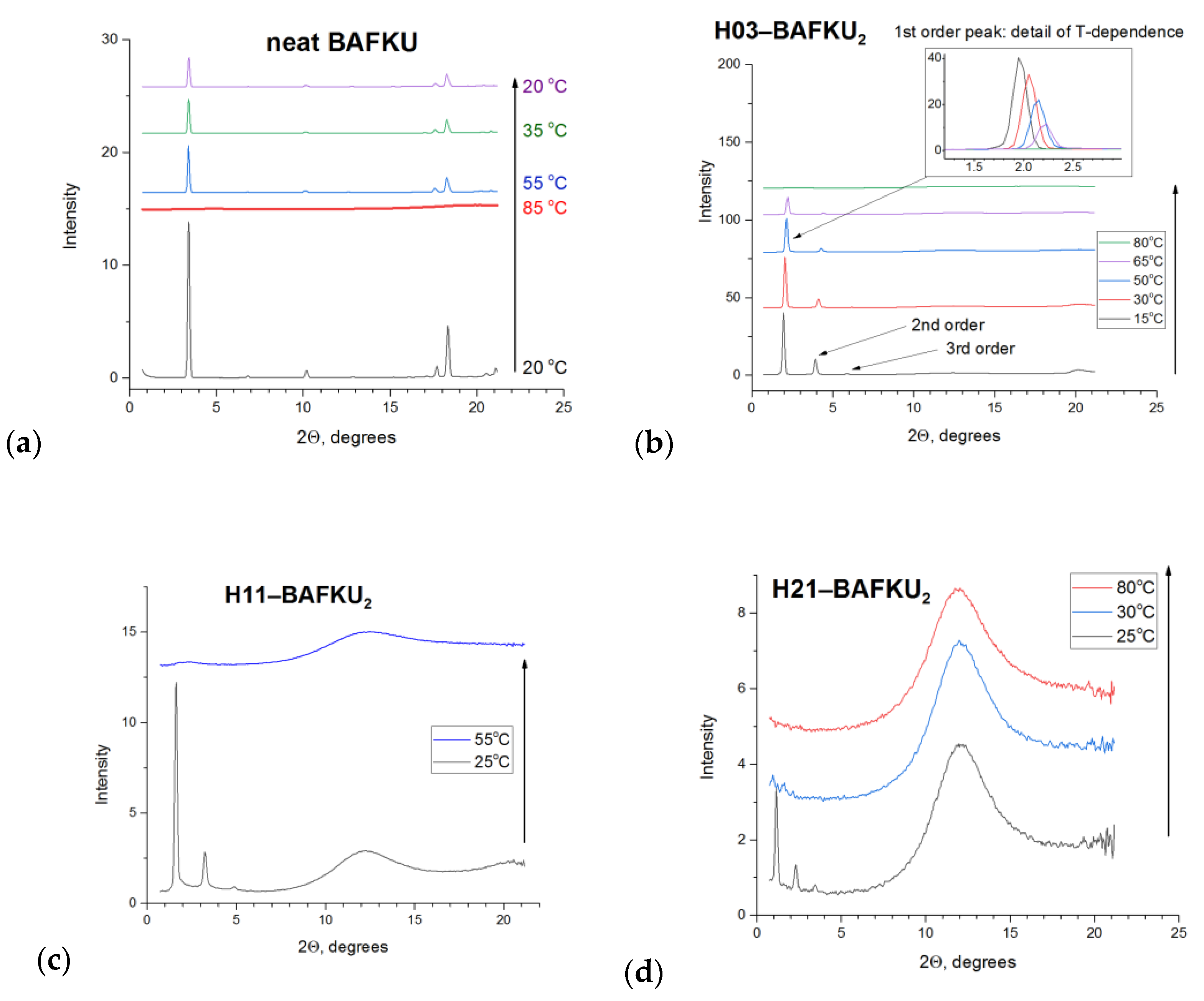
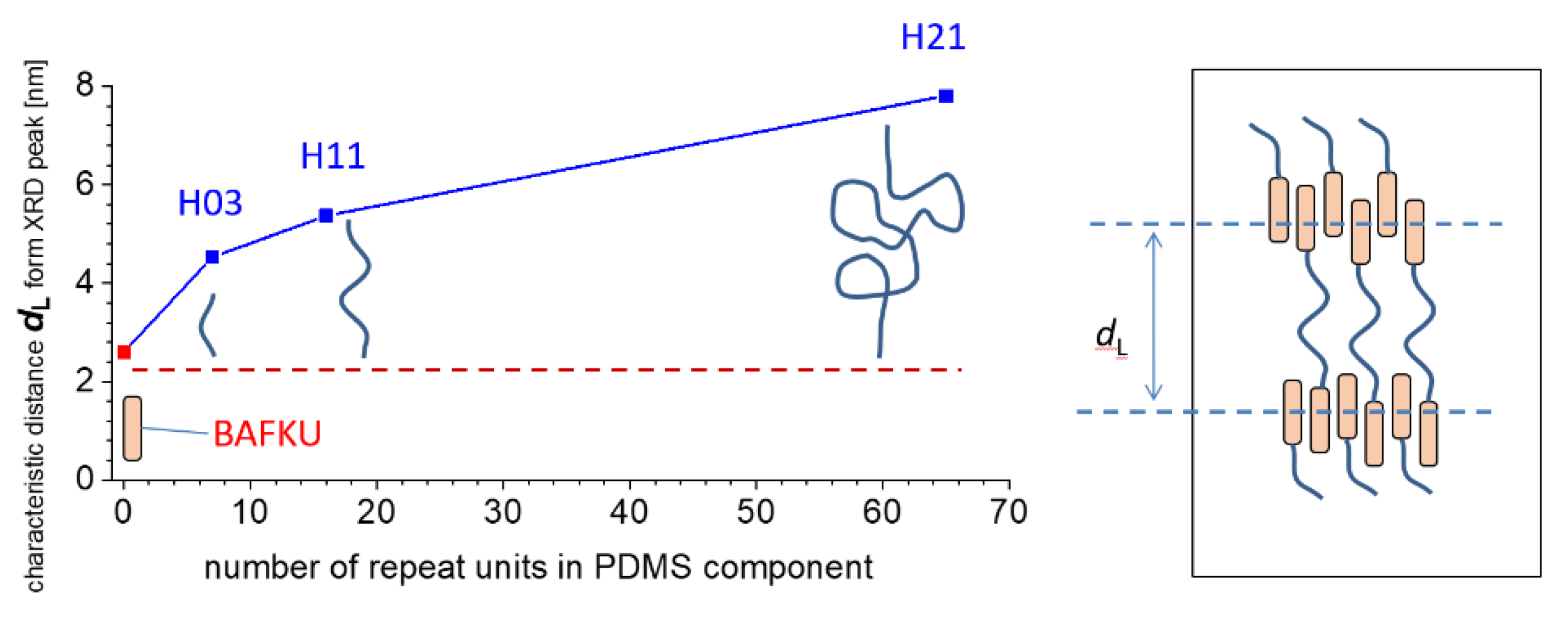
3.3.2. X-ray Diffraction (XRD) Analysis of the Phase Transitions
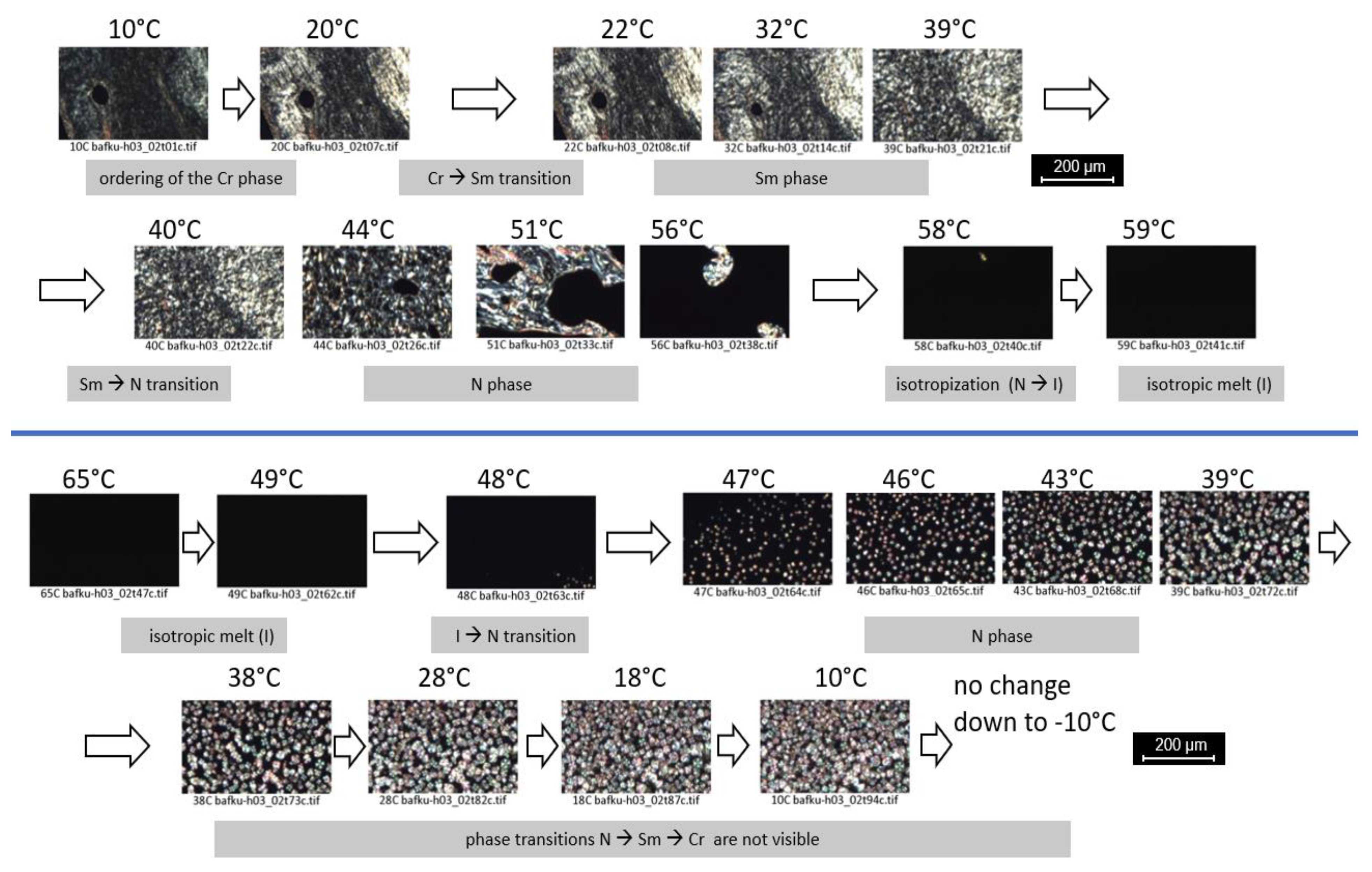
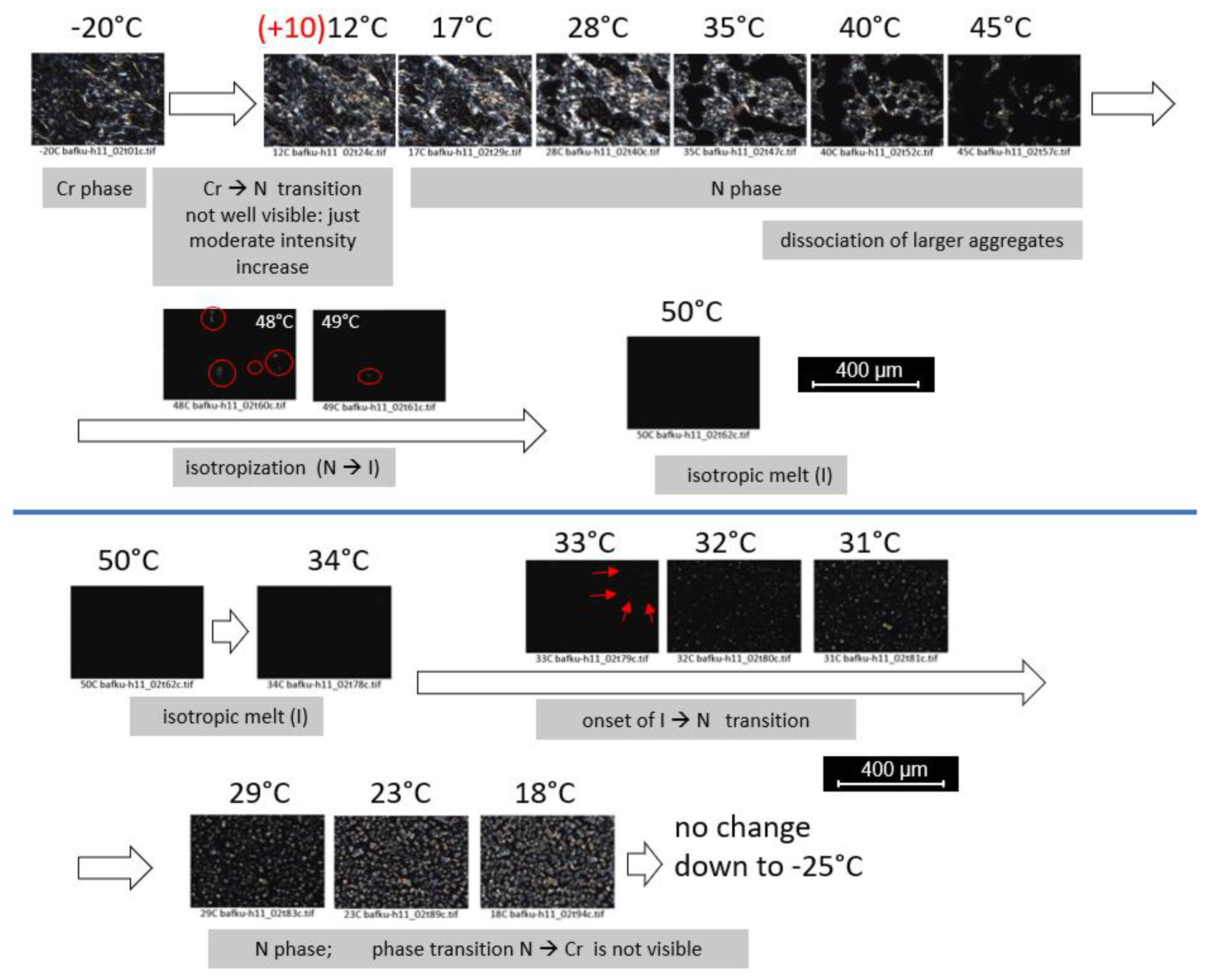
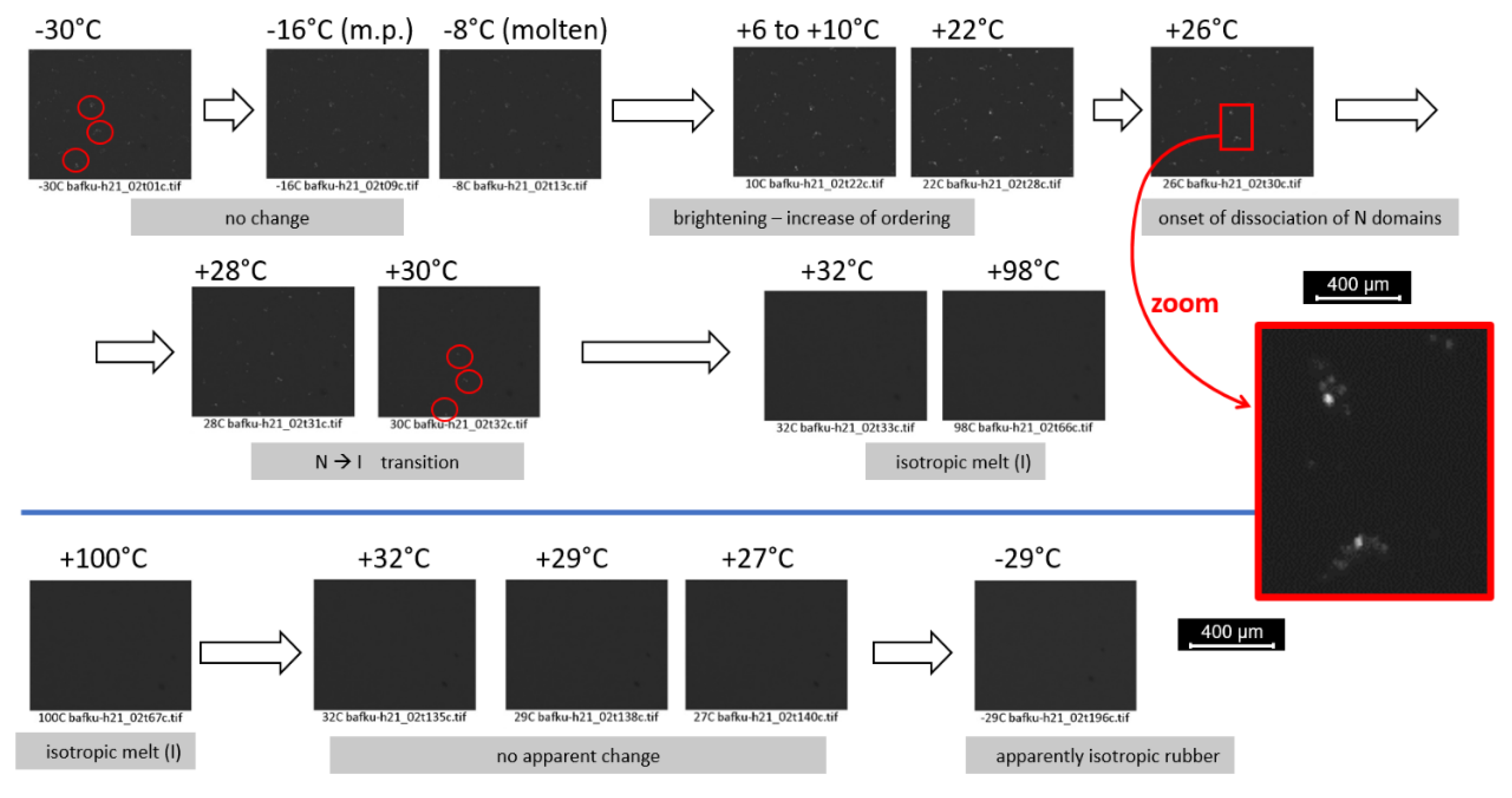
3.3.3. PLM Observation of Anisotropy and of Phase Transitions
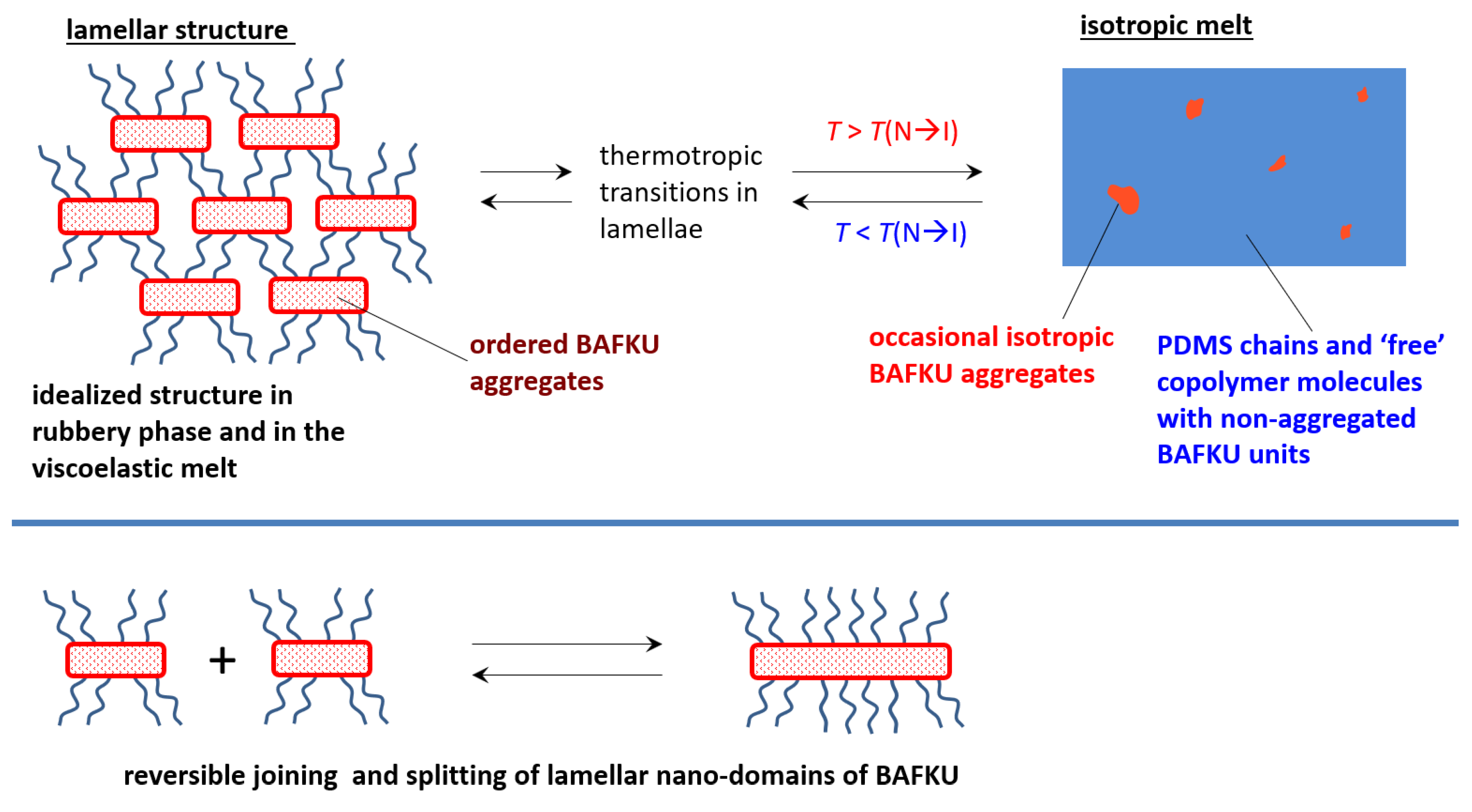
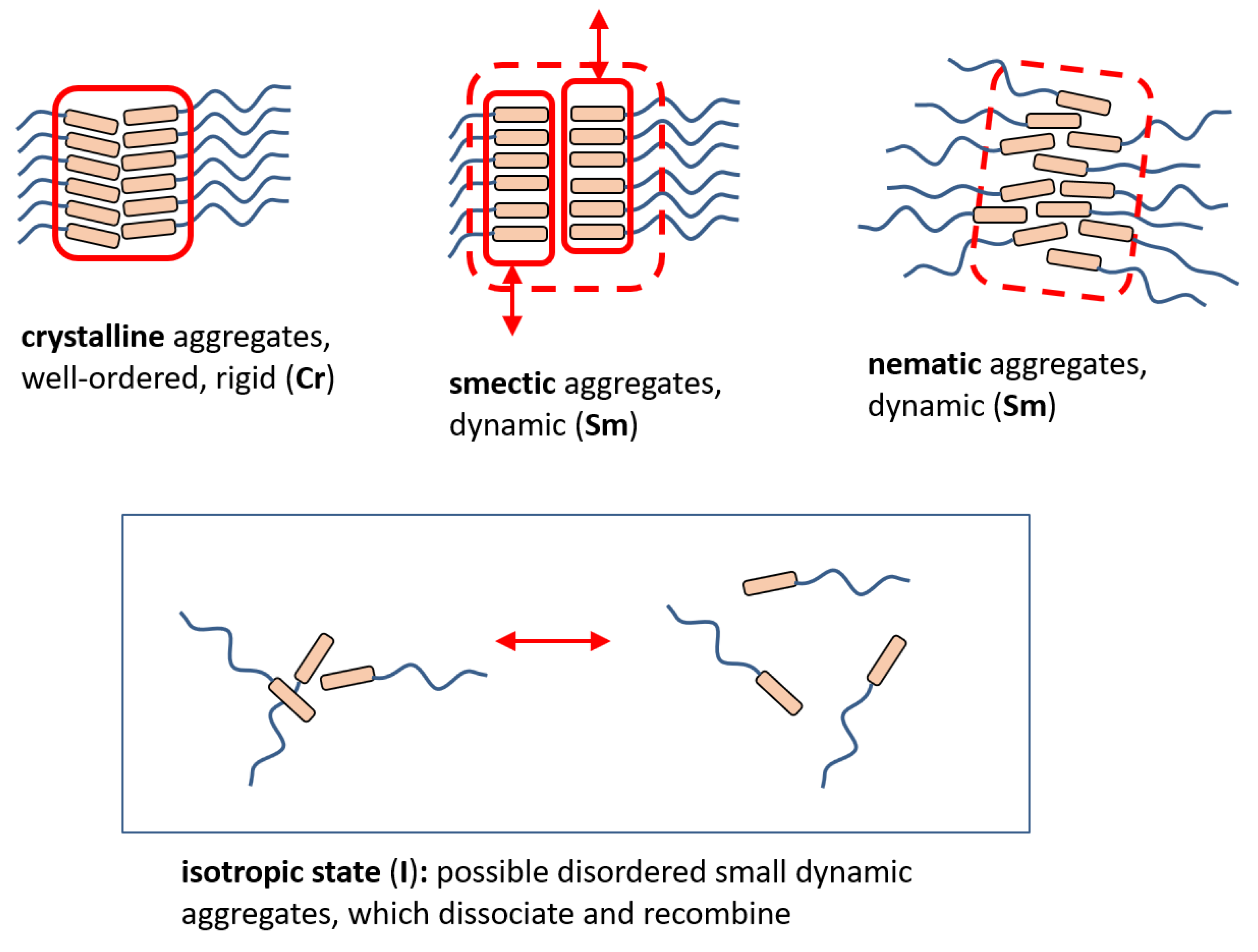
3.3.4. Structure of the BAFKU Aggregates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Dubois, J.C.; LeBarny, P.; Mauzac, M.; Noel, C.; Demus, D.; Goodby, J.W.; Gray, G.W.; Spiess, H.W.; Vill, V. Handbook of Liquid Crystals; Wiley-VCH: Weinheim, Germany, 1998; Print ISBN 9783527292707; Online ISBN 9783527620760. [Google Scholar] [CrossRef]
- Hsu, C.S. The application of side-chain liquid-crystalline polymers. Prog. Polym. Sci. 1997, 22, 829–871. [Google Scholar] [CrossRef]
- Finkelmann, H.; Rehage, G. Investigations on liquid crystalline polysiloxanes, 1. Synthesis and characterization of linear polymers. Macromol. Rapid Commun. 1980, 1, 31–34. [Google Scholar] [CrossRef]
- Finkelmann, H.; Rehage, G. Investigations on liquid crystalline polysiloxanes, 2. Optical properties of cholesteric phases and influence of the flexible spacer on the mobility of the mesogenic groups. Macromol. Rapid Commun. 1980, 1, 733–740. [Google Scholar] [CrossRef]
- Finkelmann, H.; Kock, H.J.; Rehage, G. Investigations on liquid crystalline polysiloxanes 3. Liquid crystalline elastomers—a new type of liquid crystalline material. Macromol. Rapid Commun. 1981, 2, 317–322. [Google Scholar] [CrossRef]
- Finkelmann, H.; Rehage, G. Investigations on liquid crystalline polysiloxanes, 4. Cholesteric homopolymers—synthesis and optical characterization. Macromol. Rapid Commun. 1982, 3, 859–864. [Google Scholar] [CrossRef]
- Finkelmann, H.; Kock, H.J.; Gleim, W.; Rehage, G. Investigations on liquid crystalline polysiloxanes 5. Orientation of LC-elastomers by mechanical forces. Macromol. Rapid Commun. 1984, 5, 287–293. [Google Scholar] [CrossRef]
- Küpfer, J.; Finkelmann, H. Nematic liquid single crystal elastomers. Macromol. Rapid Commun. 1991, 12, 717–726. [Google Scholar] [CrossRef]
- Wang, M.; Guo, L.X.; Lin, B.P.; Zhang, X.Q.; Sun, Y.; Yang, H. Photo-responsive polysiloxane-based azobenzene liquid crystalline polymers prepared by thiol-ene click chemistry. Liq. Cryst. 2016, 43, 1626–1635. [Google Scholar] [CrossRef]
- Wang, G.F.; Xiong, Y.; Tang, H.D. Synthesis and characterization of a graft side-chain liquid crystalline polysiloxane. J. Organomet. Chem. 2015, 77, 50–54. [Google Scholar] [CrossRef]
- Zhao, W.; Lin, B.P.; Zhang, X.Q.; Sun, Y.; Yang, H. Polysiloxane Side-chain Liquid Crystalline Polymers Prerpared by Alkyne Hydrosilylation. Chin. J. Polym. Sci. 2015, 33, 1431–1441. [Google Scholar] [CrossRef]
- Aguilera, C.; Bartulin, J.; Hisgen, B.; Ringsdorf, H. Liquid crystalline main chain polymers with highly flexible siloxane spacers. Macromol. Chem. Phys. 1983, 184, 253–262. [Google Scholar] [CrossRef]
- Braun, F.; Willner, L.; Hess, M.; Kosfeld, R. Synthesis and thermal properties of liquid-crystalline polyesters with mesogenic units and siloxane spacers in the main chain. Die Makromol. Chem. 1990, 191, 1775–1785. [Google Scholar] [CrossRef]
- Donnio, B.; Wermter, H.; Finkelmann, H.A. Simple and Versatile Synthetic Route for the Preparation of Main-Chain, Liquid-Crystalline Elastomers. Macromolecules 2000, 33, 7724–7729. [Google Scholar] [CrossRef]
- Patil, H.P.; Liao, J.; Hedden, R.C. Smectic Ordering in Main-Chain Siloxane Polymers and Elastomers Containing p-Phenylene Terephthalate Mesogens. Macromolecules 2007, 40, 6206–6216. [Google Scholar] [CrossRef]
- Burke, K.A.; Rousseau, I.A.; Mather, P.T. Reversible actuation in main-chain liquid crystalline elastomers with varying crosslink densities. Polymer 2014, 55, 5897–5907. [Google Scholar] [CrossRef]
- Samui, A.B.; Pandey, S.; Mishra, S.P. Main chain photoresponsive liquid crystalline polymer synthesized through hydrosilylation. RSC Adv. 2015, 5, 68351–68355. [Google Scholar] [CrossRef]
- Saed, M.O.; Volpe, R.H.; Traugutt, N.A.; Visvanathan, R.; Clark, N.A.; Yakacki, C.M. High strain actuation liquid crystal elastomers via modulation of mesophase structure. Soft Matter 2017, 13, 7537–7547. [Google Scholar] [CrossRef] [PubMed]
- Pandey, S.; Kolli, B.; Mishra, S.P.; Samui, A.B. Siloxane polymers containing azo moieties synthesized by click chemistry for photo responsive and liquid crystalline applications. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 1205–1215. [Google Scholar] [CrossRef]
- Shenouda, I.G.; Chien, L.C. New ferroelectric liquid-crystalline polysiloxanes containing cyanohydrin chiral mesogens: L-norleucine series. Macromolecules 1993, 26, 5020–5023. [Google Scholar] [CrossRef]
- Zhou, Q.L.; Zhang, J.T.; Ren, Z.J.; Yan, S.K.; Xie, P.; Zhang, R.B. A Stable and High-Efficiency Blue-Light Emitting Terphenyl-Bridged Ladder Polysiloxane. Macromol. Rapid Commun. 2008, 29, 1259–1263. [Google Scholar] [CrossRef]
- Kawakami, H.; Mori, Y.; Abe, H.; Nagaoka, S. Gas transport properties of liquid crystalline polysiloxane with laterally attached side chain. J. Membr. Sci. 1997, 133, 245–253. [Google Scholar] [CrossRef]
- Rao, H.X.; Zhang, Z.Y. Preparation, Characterization, and Permeation Property of a Liquid Crystal/PDMS Membrane Material. J. Appl. Polym. Sci. 2012, 123, 191–199. [Google Scholar] [CrossRef]
- Ganicz, T.; Stanczyk, W.A.; Chmielecka, J.; Kowalski, J. Liquid crystalline polycarbosilanes and poly(di-n-butylsiloxane) as stationary phases in gas chromatography. Polym. Int. 2009, 58, 248–254. [Google Scholar] [CrossRef]
- Warner, M.; Terentjev, E. Liquid Crystal Elastomers; Oxford University Press: Oxford, UK, 2003; ISBN 9780198527671. [Google Scholar]
- Ikeda, T.; Mamiya, J.; Yu, Y. Photomechanics of Liquid-Crystalline Elastomers and Other Polymers. Angew. Chem. Int. Ed. 2007, 46, 506–528. [Google Scholar] [CrossRef]
- Ohm, C.; Brehmer, M.; Zentel, R. Liquid Crystalline Elastomers as Actuators and Sensors. Adv. Mater. 2010, 22, 3366–3387. [Google Scholar] [CrossRef]
- Strachota, A.; Kroutilová, I.; Kovářová, J.; Matějka, L. Epoxy Networks Reinforced with Polyhedral Oligomeric Silsesquioxanes (POSS). Thermomechanical Properties. Macromolecules 2004, 37, 9457–9464. [Google Scholar] [CrossRef]
- Strachota, A.; Whelan, P.; Kříž, J.; Brus, J.; Urbanová, M.; Šlouf, M.; Matějka, L. Formation of nanostructured epoxy networks containing polyhedral oligomeric silsesquioxane (POSS) blocks. Polymer 2007, 48, 3041–3058. [Google Scholar] [CrossRef]
- Strachota, A.; Rodzeń, K.; Ribot, F.; Trchová, M.; Steinhart, M.; Starovoytova, L.; Pavlova, E. Behavior of Tin-Based “Super-POSS” Incorporated in Different Bonding Situations in Hybrid Epoxy Resins. Macromolecules 2014, 47, 4266–4287. [Google Scholar] [CrossRef]
- Rodzeń, K.; Strachota, A.; Ribot, F.; Matějka, L.; Kovářová, J.; Trchová, M.; Šlouf, M. Reactivity of the tin homolog of POSS, butylstannoxane dodecamer, in oxygen-induced crosslinking reactions with an organic polymer matrix: Study of long-time behaviour. Polym. Degrad. Stab. 2015, 118, 147–166. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Strachota, B.; Strachota, A.; Steinhart, M.; Šlouf, M. The orientation-enhancing effect of diphenyl aluminium phosphate nanorods in a liquid-crystalline epoxy matrix ordered by magnetic field. Eur. Polym. J. 2015, 72, 238–255. [Google Scholar] [CrossRef]
- Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Šlouf, M.; Zhigunov, A.; Vyroubalová, M.; Kaňková, D.; Netopilík, M. Meltable copolymeric elastomers based on polydimethylsiloxane with multiplets of pendant liquid-crystalline groups as physical crosslinker: A self-healing structural material with a potential for smart applications. Eur. Polym. J. 2020, 137. [Google Scholar] [CrossRef]
- Colombani, O.; Barioz, C.; Bouteiller, L.; Chaneac, C.; Fomperie, L.; Lortie, F.; Montes, H. Attempt toward 1D Cross-Linked Thermoplastic Elastomers: Structure and Mechanical Properties of a New System. Macromolecules 2005, 38, 1752–1759. [Google Scholar] [CrossRef]
- Botterhuis, N.E.; van Beek, D.J.M.; van Gemert, G.M.L.; Bosman, A.W.; Sijbesma, R.P. Self-Assembly and Morphology of Polydimethylsiloxane Supramolecular Thermoplastic Elastomers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3877–3885. [Google Scholar] [CrossRef]
- Ślęczkowski, M.L.; Meijer, E.W.; Palmans, A.R.A. Cooperative Folding of Linear Poly(dimethyl siloxane)s via Supramolecular Interactions. Macromol. Rapid Commun. 2017, 38, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Rambarran, T.; Bertrand, A.; Gonzaga, F.; Boisson, F.; Bernard, J.; Fleury, E.; Ganachaud, F.; Brook, M.A. Sweet supramolecular elastomers from a,x-(b-cyclodextrin terminated) PDMS. Chem. Commun. 2016, 52, 6681–6684. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, A.S.; Brook, M.A. Thermoplastic Silicone Elastomers through Self-Association of Pendant Coumarin Groups. Macromolecules 2014, 47, 1656–1663. [Google Scholar] [CrossRef]
- Lamers, B.A.G.; Graf, R.; de Waal, B.F.M.; Vantomme, G.; Palmans, A.R.A.; Meijer, E.W. Polymorphism in the Assembly of Phase-Segregated Block Molecules: Pathway Control to 1D and 2D Nanostructures. J. Am. Chem. Soc. 2019, 141, 15456–15463. [Google Scholar] [CrossRef] [Green Version]
- Lamers, B.A.G.; Ślęczkowski, M.L.; Wouters, F.; Engels, T.A.P.; Meijer, E.W.; Palmans, A.R.A. Tuning polymer properties of non-covalent crosslinked PDMS by varying supramolecular interaction strength. Polym. Chem. 2020, 11, 2847–2854. [Google Scholar] [CrossRef] [Green Version]
- Dodge, L.; Chen, Y.; Brook, M.A. Silicone Boronates Reversibly Crosslink Using Lewis Acid–Lewis Base Amine Complexes. Chem. A Eur. J. 2014, 20, 9349–9356. [Google Scholar] [CrossRef]
- Li, C.H.; Wang, C.; Keplinger, C.; Zuo, J.L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C.; et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef]
- Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Strachota, B.; Šlouf, M. Low-temperature-meltable elastomers based on linear polydimethylsiloxane chains alpha,omega-terminated with mesogenic groups as physical crosslinker: A passive smart material with potential as viscoelastic coupling. Part II: Viscoelastic and rheological properties. Polymers 2020. Submitted. [Google Scholar]
- Miniewicz, A.; Girones, J.; Karpinski, P.; Mossety-Leszczak, B.; Galina, H.; Dutkiewicz, M. Photochromic and nonlinear optical properties of azo-functionalized POSS nanoparticles dispersed in nematic liquid crystals. J. Mater. Chem. C 2014, 2, 432–440. [Google Scholar] [CrossRef]
- Kieffer, J.; Karkoulis, D. PyFAI, a versatile library for azimuthal regrouping. J. Phys. Conf. Ser. 2013, 425, 1–6. [Google Scholar] [CrossRef]
- Slouf, M.; Krejcikova, S.; Vackova, T.; Kratochvil, J.; Novak, L. In situ observation of nucleated polymer crystallization in polyoxymethylene sandwich composites. Front. Mater. 2015, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Vackova, T.; Kratochvil, J.; Ostafinska, A.; Krejcikova, S.; Nevoralova, M.; Slouf, M. Impact of particle morphology on structure, crystallization kinetics, and properties of PCL composites with TiO2-based particles. Polym. Bull. 2017, 74, 445–464. [Google Scholar] [CrossRef]
- Klonos, P.A. Crystallization, glass transition, and molecular dynamics in PDMS of low molecular weights: A calorimetric and dielectric study. Polymer 2018, 159, 169–180. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | BAFKU Mesogen | Precursor DMS H03 | Precursor DMS H11 | Precursor DMS H21 | Chloroform | Karstedt’s Catalyst 2 wt.% Solution | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sample Name | |||||||||||||||||||||
| [g] | [mmol] of C=C Groups | wt% | Vol% | [g] | [mmol] of SiH Groups | wt% | Vol% | [g] | [mmol] of SiH Groups | wt% | Vol% | [g] | [mmol] of SiH Groups | wt% | Vol% | [g] | [mL] | [g] as 2% Solution | [mmol] | ||
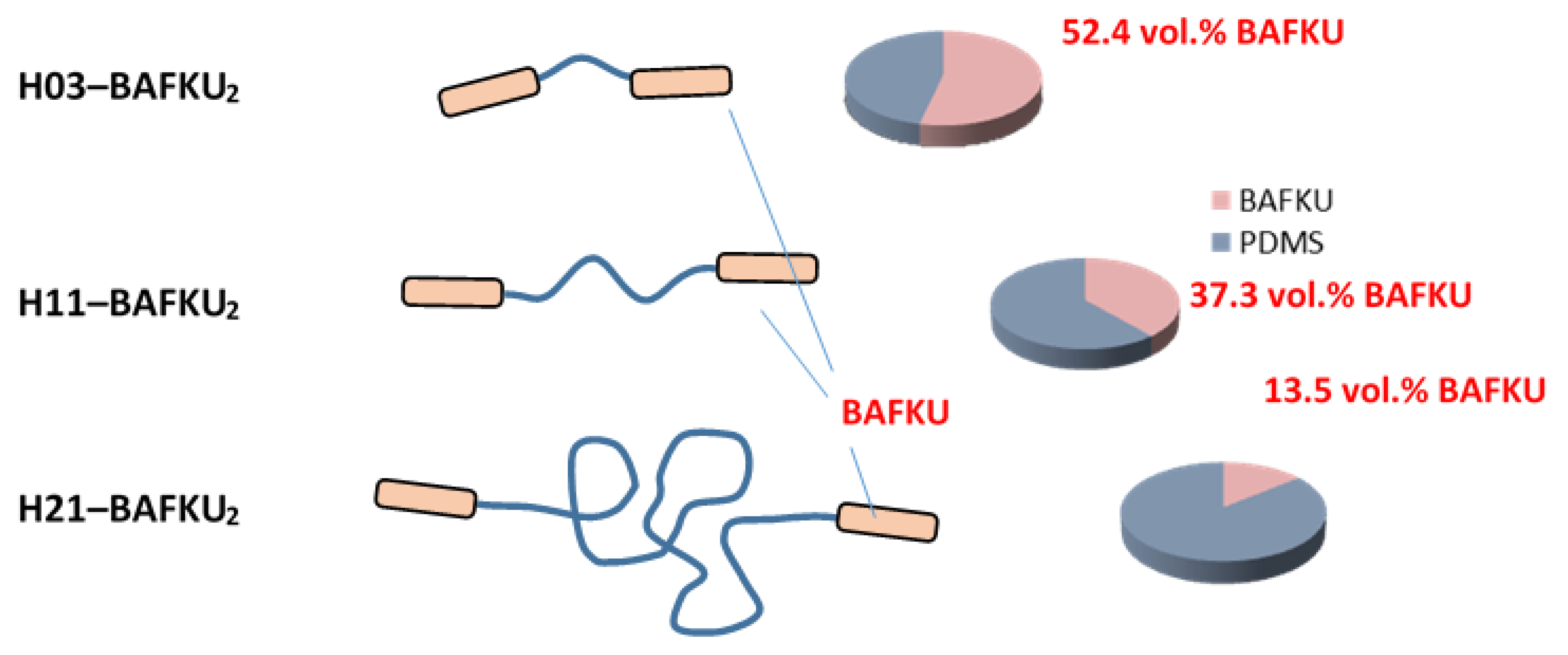
| H03–BAFKU2 | 0.5 | 1.19 | 57.4 | 52.44 | 0.371 | 1.19 | 42.6 | 47.56 | 2.956 | 24.76 | 0.0087 | 0.0228 | |||||||||
| H11–BAFKU2 | 0.5 | 1.19 | 41.43 | 37.29 | 0.711 | 1.19 | 58.57 | 62.71 | 2.956 | 24.76 | 0.0087 | 0.0228 | |||||||||
| H21–BAFKU2 | 0.5 | 1.19 | 15.1 | 13.47 | 2.832 | 1.19 | 84.9 | 86.53 | 2.956 | 24.76 | 0.0087 | 0.0228 | |||||||||
| BAFKU | |||
|---|---|---|---|
| 2θ [°] | q [Å−1] | d [nm] | comment |
| 3.40 | 0.242 | 2.60 | BAFKU length |
| 6.80 | 0.484 | 1.30 | -“- (second order of 3.40°) |
| 10.18 | 0.724 | 0.87 | BAFKU width |
| 17.70 | 1.255 | 0.50 | standard Van der Waals distance in crystal |
| 18.36 | 1.301 | 0.48 | standard Van der Waals distance in crystal |
| H03 | H11 | H21 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 2θ [°] | q [Å−1] | d [nm] | 2θ [°] | q [Å−1] | d [nm] | 2θ [°] | q [Å−1] | d [nm] | comment |
| 1.95 | 0.139 | 4.53 | 1.644 | 0.117 | 5.37 | 1.13 | 0.0804 | 7.81 | main reflection |
| 3.90 | 0.277 | 2.26 | 3.28 | 0.233 | 2.69 | 2.26 | 0.161 | 3.90 | 2nd order |
| 5.85 | 0.416 | 1.509 | 4.93 | 0.351 | 1.79 | 3.39 | 0.241 | 2.60 | 3rd order |
| 8–16 | 0.57–1.13 | 1.1–0.55 | 8–16 (12.24) | 0.57–1.13 (0.896) | 1.1–0.55 (0.723) | 8–16 (12.24) | 0.57–1.13 (0.896) | 1.1–0.55 (0.723) | PDMS |
| 20.1 | 1.42 | 0.44 | 20.6 | 1.46 | 0.43 | -- | -- | -- | org.–org. (+PDMS) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Strachota, B.; Šlouf, M.; Zhigunov, A.; Vyroubalová, M.; Kaňková, D.; Netopilík, M.; Walterová, Z. Low-Temperature Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinkers: A Passive Smart Material with Potential as Viscoelastic Coupling. Part I: Synthesis and Phase Behavior. Polymers 2020, 12, 2476. https://doi.org/10.3390/polym12112476
Horodecka S, Strachota A, Mossety-Leszczak B, Strachota B, Šlouf M, Zhigunov A, Vyroubalová M, Kaňková D, Netopilík M, Walterová Z. Low-Temperature Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinkers: A Passive Smart Material with Potential as Viscoelastic Coupling. Part I: Synthesis and Phase Behavior. Polymers. 2020; 12(11):2476. https://doi.org/10.3390/polym12112476
Chicago/Turabian StyleHorodecka, Sabina, Adam Strachota, Beata Mossety-Leszczak, Beata Strachota, Miroslav Šlouf, Alexander Zhigunov, Michaela Vyroubalová, Dana Kaňková, Miloš Netopilík, and Zuzana Walterová. 2020. "Low-Temperature Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinkers: A Passive Smart Material with Potential as Viscoelastic Coupling. Part I: Synthesis and Phase Behavior" Polymers 12, no. 11: 2476. https://doi.org/10.3390/polym12112476