
Stabilization of Haloalkane Dehalogenase Structure by Interfacial Interaction with Ionic Liquids

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Construction of the Mutant, Protein Expression, and Purification
2.2. Crystallization
2.3. Data collection and Processing
2.4. Structure Solution and Refinement
2.5. Molecular Dynamics (MD) Simulations
3. Results and Discussion
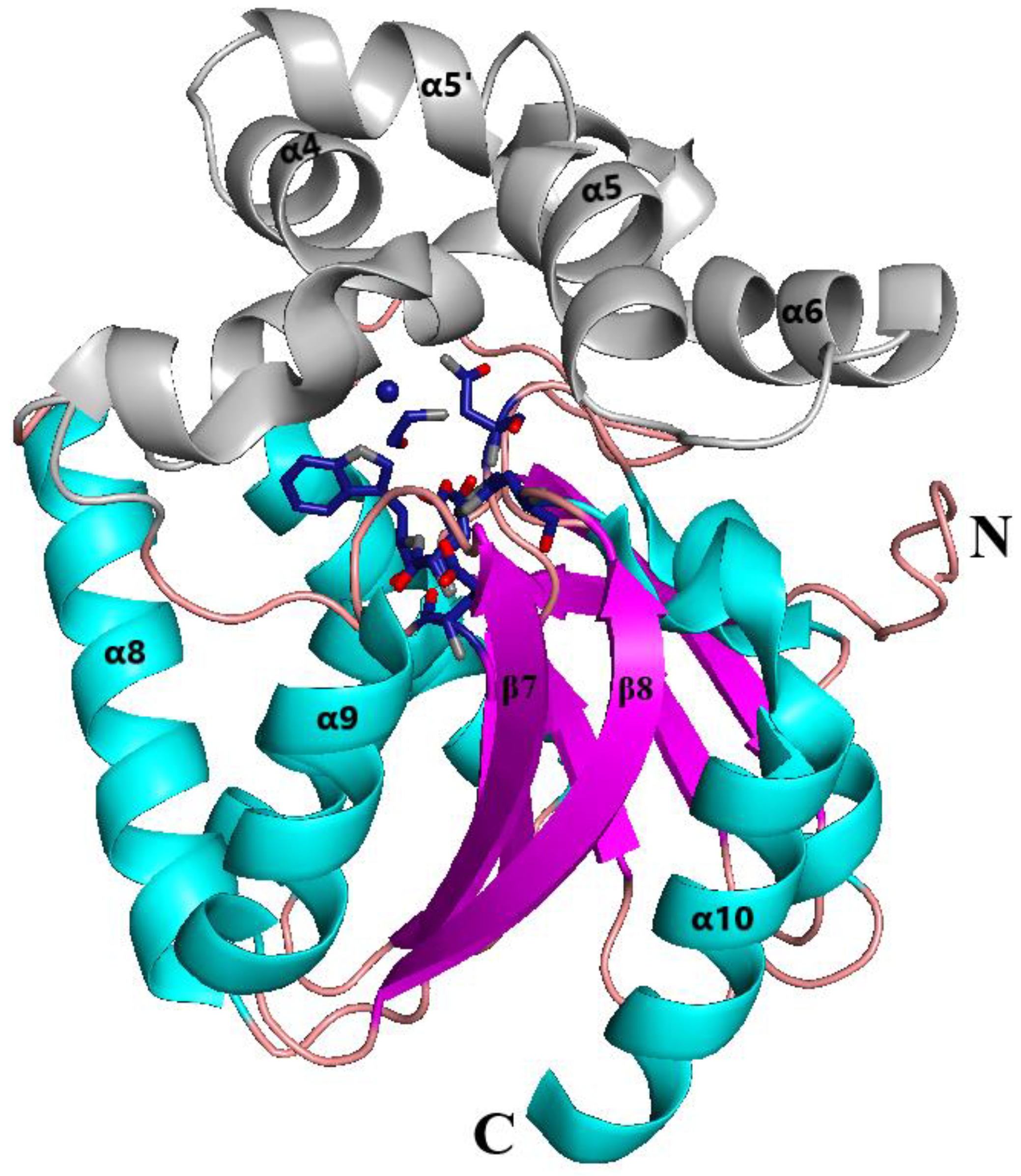
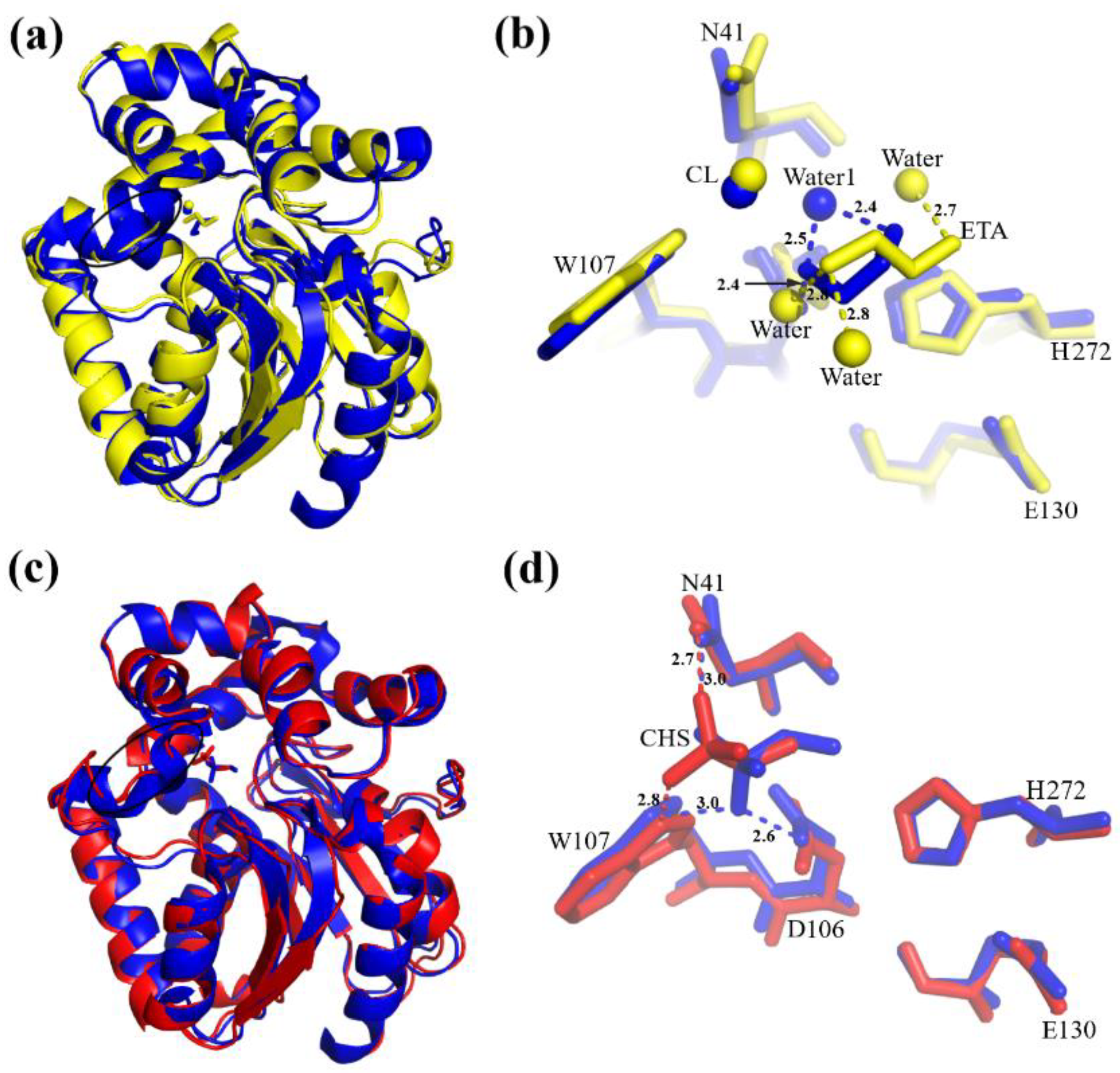
3.1. Overall Structure of the DhaA80 Variant
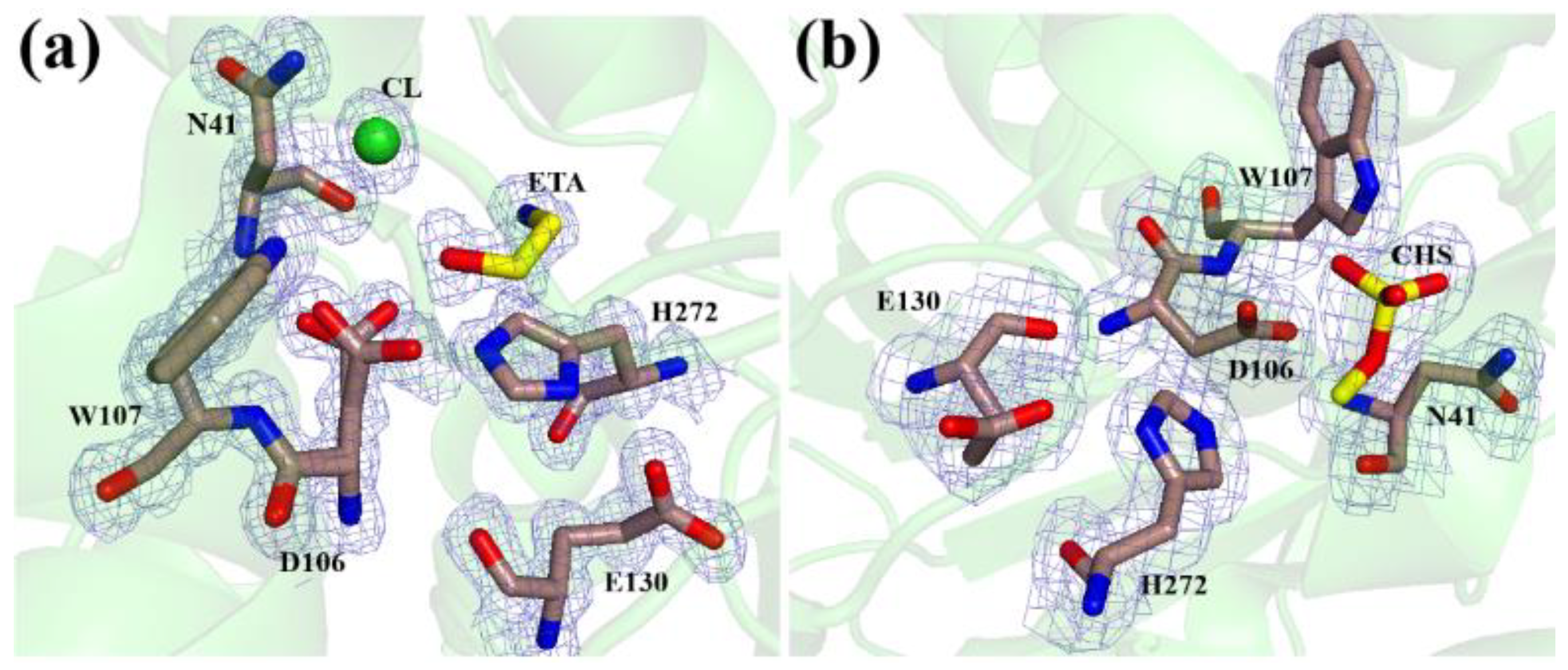
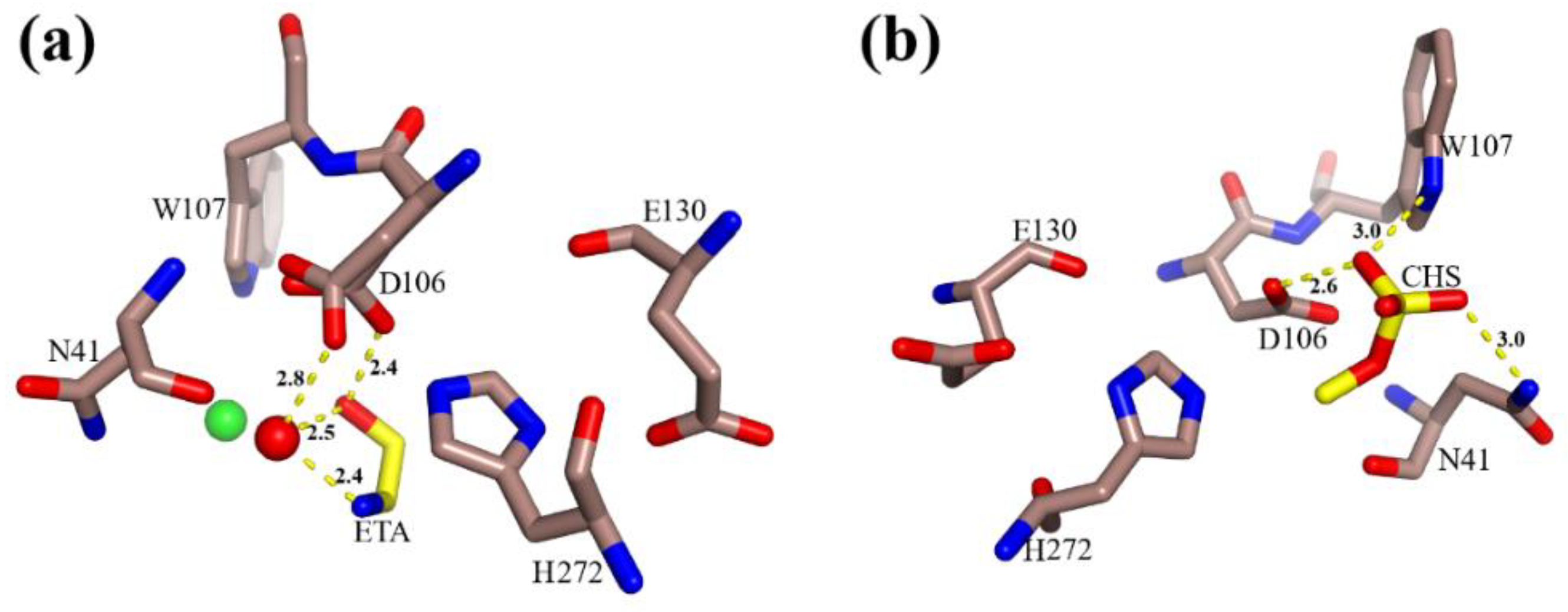
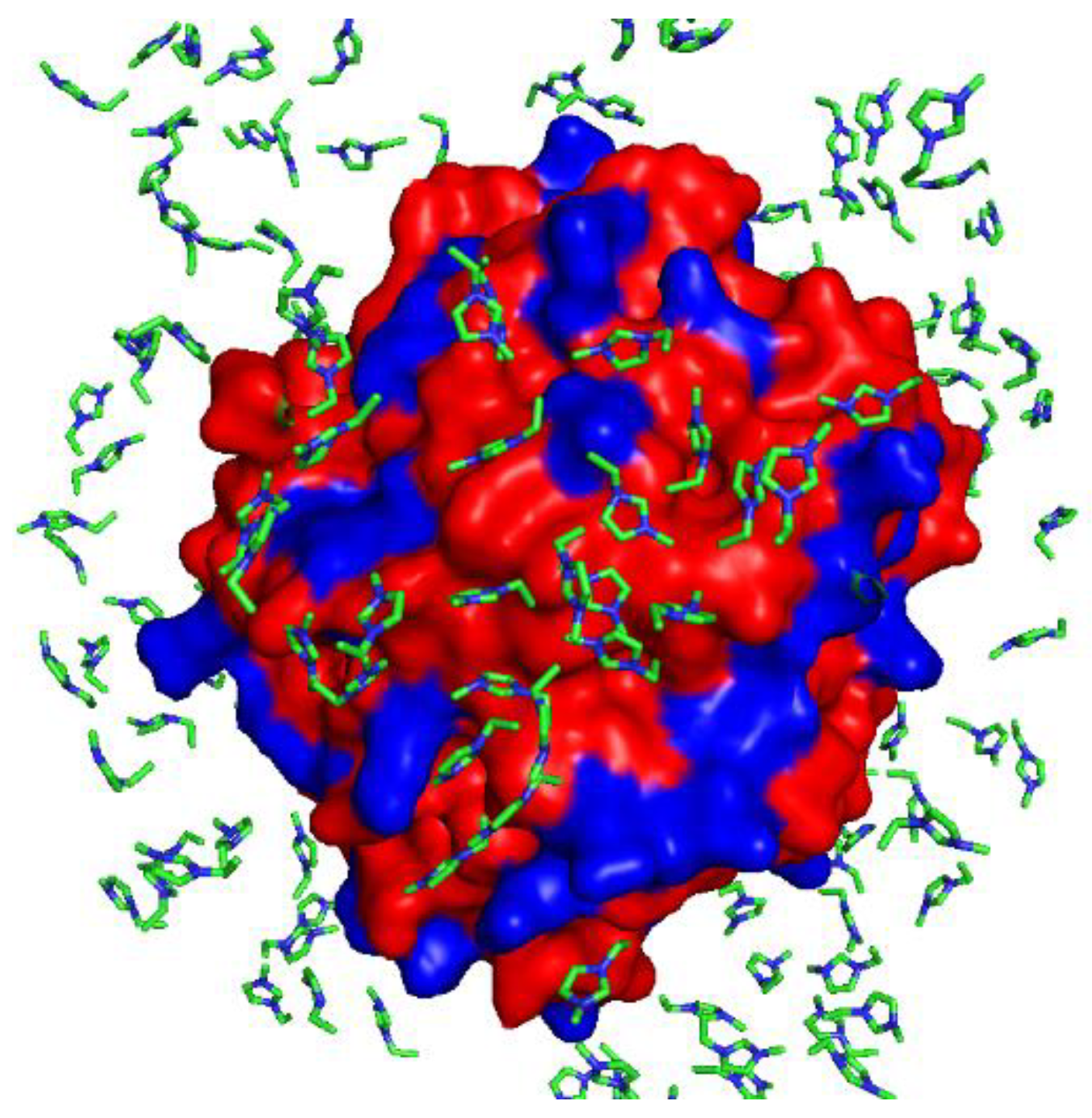
3.2. The Active Site
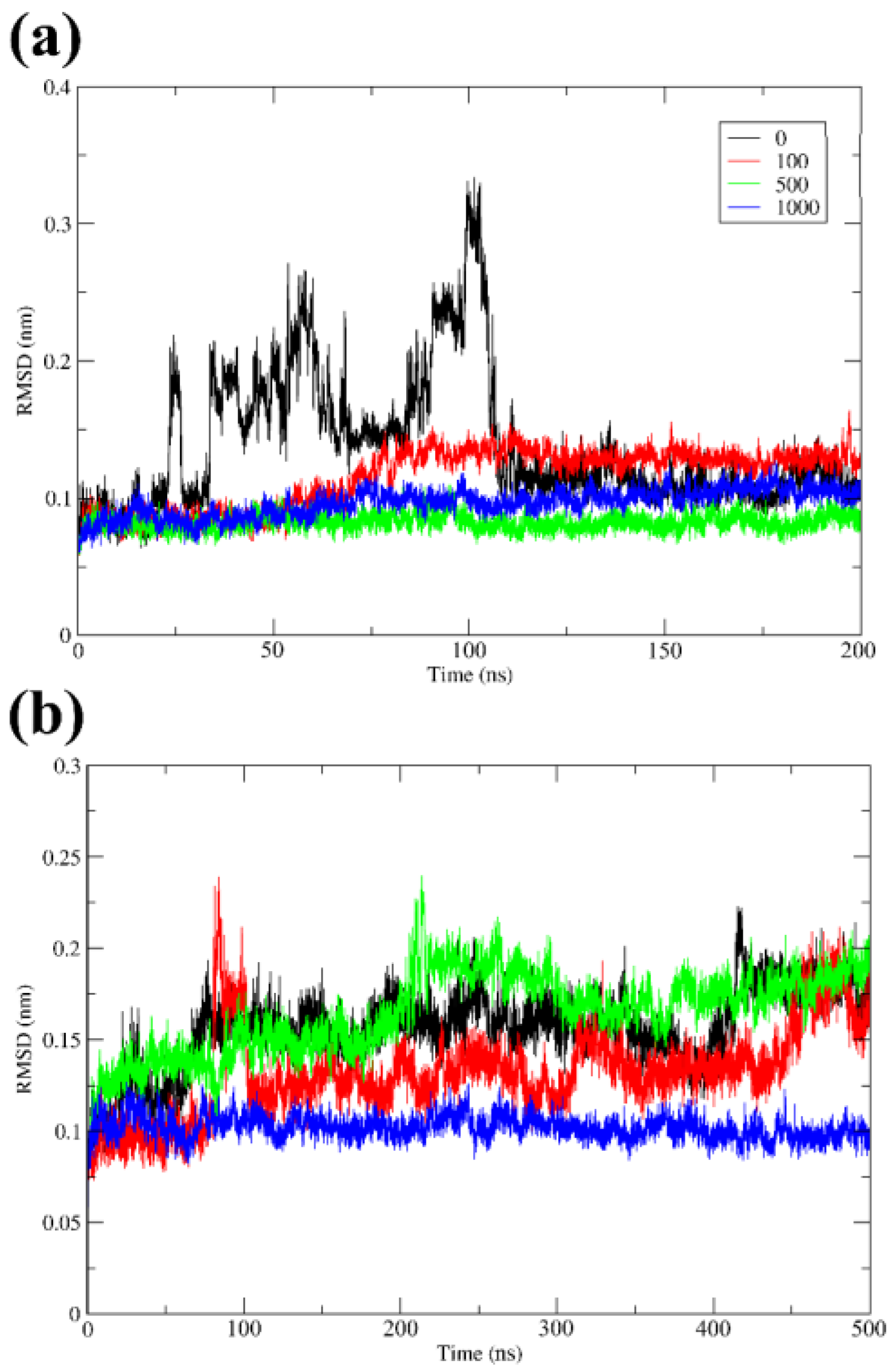
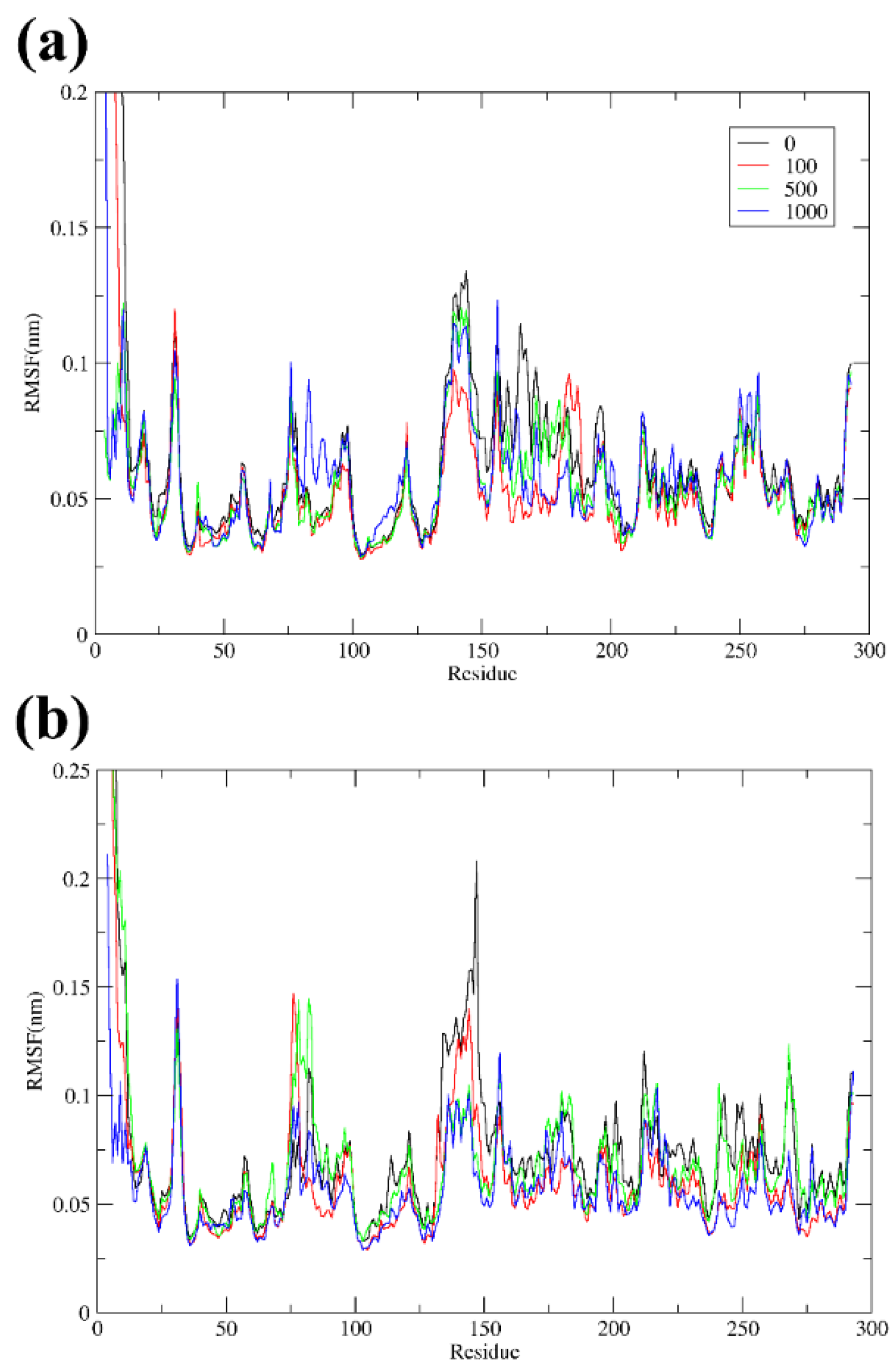
3.3. Overall Stability and Flexibility of the Structures
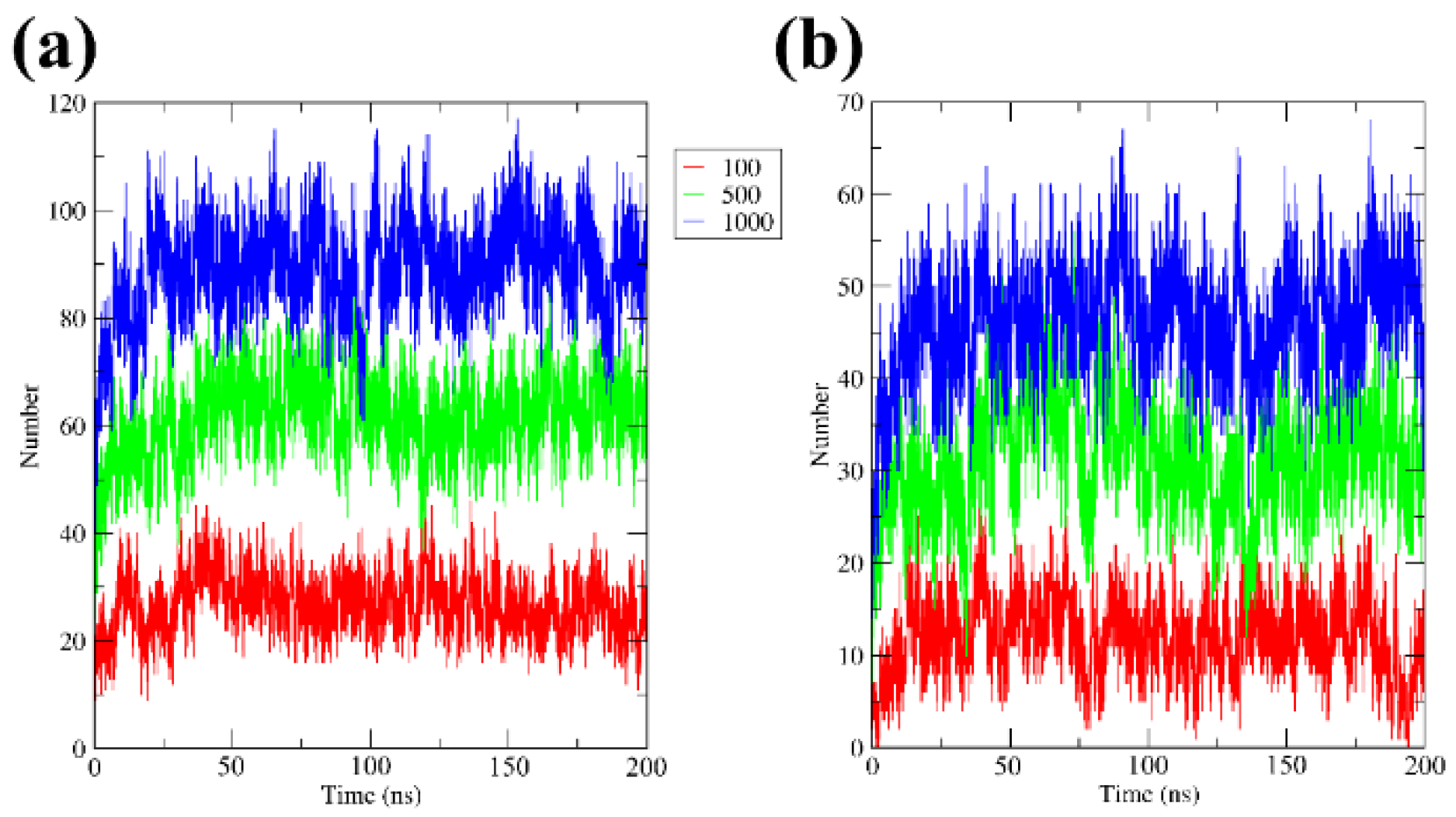
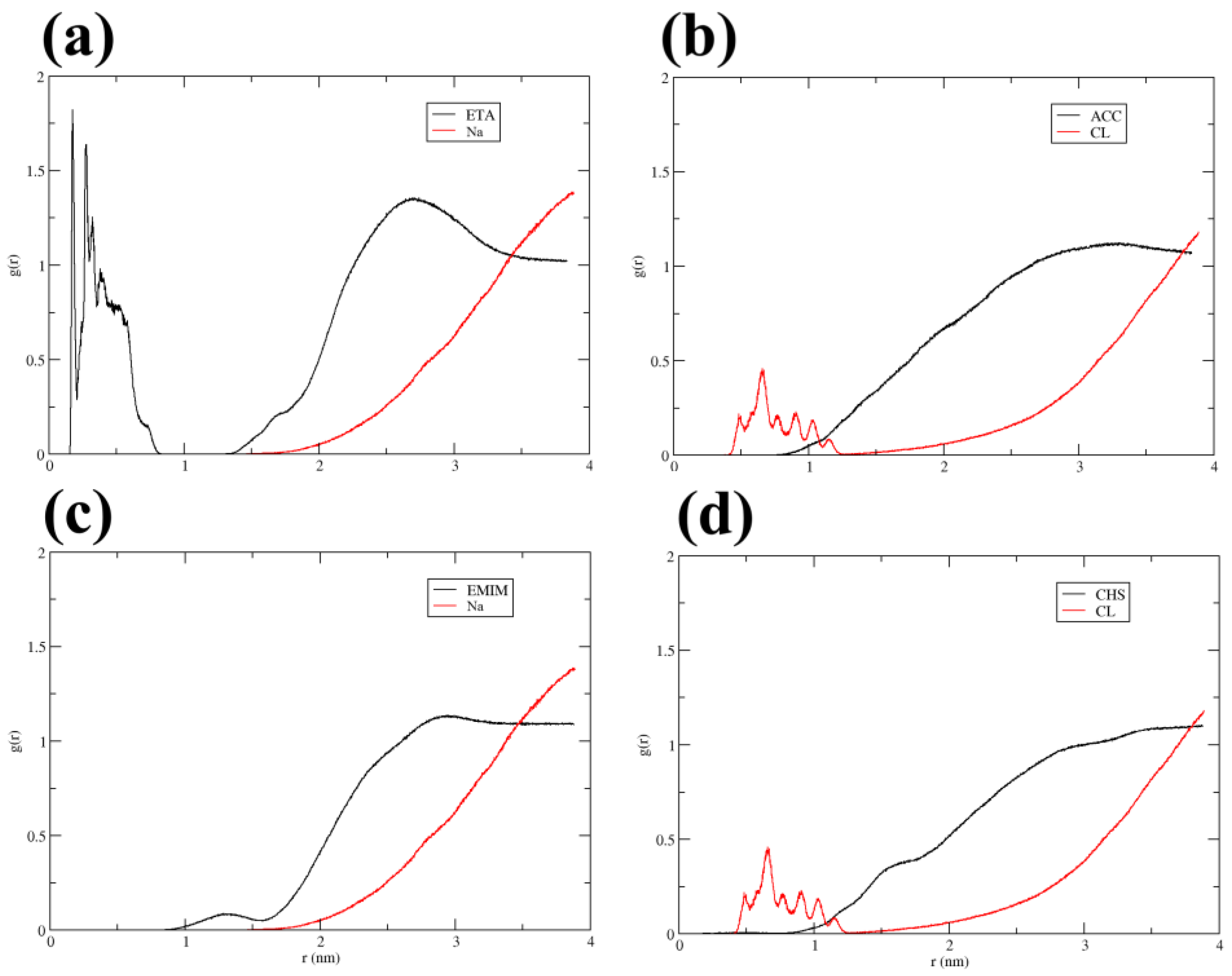
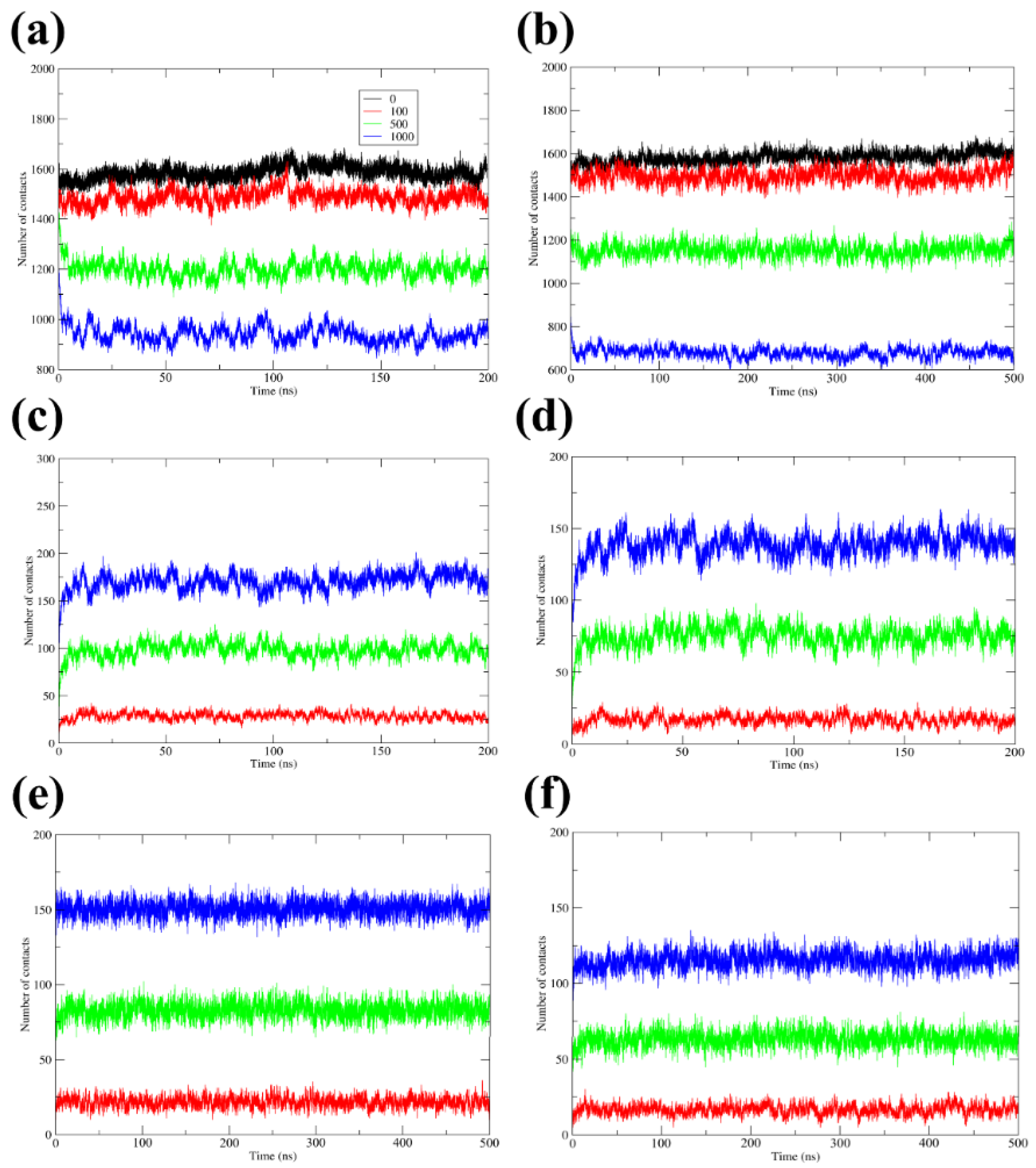
3.4. H-Bonds, Radial Distribution Functions, and Contacts
3.5. Comparison of Crystal Structure and Simulated System Data
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, H.; Yu, W.-L.; Guo, X.; Zhao, Y.-Z.; Cui, Y.; Hu, T.; Zhong, J.-Y. An effective immobilized haloalkane dehalogenase DhaA from Rhodococcus rhodochrous by adsorption, crosslink and PEGylation on meso-cellular foam. Int. J. Biol. Macromol. 2019, 125, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Fung, H.K.H.; Gadd, M.S.; Drury, T.A.; Cheung, S.; Guss, J.M.; Coleman, N.V.; Matthews, J.M. Biochemical and biophysical characterisation of haloalkane dehalogenases DmrA and DmrB inMycobacteriumstrain JS60 and their role in growth on haloalkanes. Mol. Microbiol. 2015, 97, 439–453. [Google Scholar] [CrossRef]
- Prudnikova, T.; Chaloupkova, R.; Sato, Y.; Nagata, Y.; Degtjarik, O.; Kuty, M.; Rezacova, P.; Damborsky, J.; Kuta Smatanova, I. Development of a Crystallization Protocol for the DbeA1 Variant of Novel Haloalkane Dehalogenase from Bradyrhizobium elkani USDA94. Cryst. Growth Des. 2011, 11, 516–519. [Google Scholar] [CrossRef]
- Prudnikova, T.; Kascakova, B.; Mesters, J.R.; Grinkevich, P.; Havlickova, P.; Mazur, A.; Shaposhnikova, A.; Chaloupkova, R.; Damborsky, J.; Kuty, M.; et al. Crystallization and Crystallographic Analysis of a Bradyrhizobium Elkanii USDA94 Haloalkane Dehalogenase Variant with an Eliminated Halide-Binding Site. Crystals 2019, 9, 375. [Google Scholar] [CrossRef] [Green Version]
- Hasan, K.; Gora, A.; Brezovsky, J.; Chaloupkova, R.; Moskalikova, H.; Fortova, A.; Nagata, Y.; Damborsky, J.; Prokop, Z. The effect of a unique halide-stabilizing residue on the catalytic properties of haloalkane dehalogenase DatA from Agrobacterium tumefaciens C58. FEBS J. 2013, 280, 3149–3159. [Google Scholar] [CrossRef] [PubMed]
- Ang, T.-F.; Maiangwa, J.; Salleh, A.; Normi, Y.; Leow, T. Dehalogenases: From Improved Performance to Potential Microbial Dehalogenation Applications. Molecules 2018, 23, 1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chovancova, E.; Kosinski, J.; Bujnicki, J.M.; Damborsky, J. Phylogenetic analysis of haloalkane dehalogenases. Proteins 2007, 67, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Chrast, L.; Tratsiak, K.; Planas-Iglesias, J.; Daniel, L.; Prudnikova, T.; Brezovsky, J.; Bednar, D.; Kuta Smatanova, I.; Chaloupkova, R.; Damborsky, J. Deciphering the Structural Basis of High Thermostability of Dehalogenase from Psychrophilic Bacterium Marinobacter sp. ELB17. Microorganisms 2019, 7, 498. [Google Scholar] [CrossRef] [Green Version]
- Koudelakova, T.; Chaloupkova, R.; Brezovsky, J.; Prokop, Z.; Sebestova, E.; Hesseler, M.; Khabiri, M.; Plevaka, M.; Kulik, D.; Kuta Smatanova, I.; et al. Engineering Enzyme Stability and Resistance to an Organic Cosolvent by Modification of Residues in the Access Tunnel. Angew. Chem. Int. Ed. 2013, 52, 1959–1963. [Google Scholar] [CrossRef]
- Reslan, M.; Kayser, V. Ionic liquids as biocompatible stabilizers of proteins. Biophys. Rev. 2018, 10, 781–793. [Google Scholar] [CrossRef] [PubMed]
- Vrikkis, R.M.; Fraser, K.J.; Fujita, K.; MacFarlane, D.R.; Elliott, G.D. Biocompatible Ionic Liquids: A New Approach for Stabilizing Proteins in Liquid Formulation. J. Biomech. Eng. 2009, 131, 074514. [Google Scholar] [CrossRef]
- Stepankova, V.; Vanacek, P.; Damborsky, J.; Chaloupkova, R. Comparison of catalysis by haloalkane dehalogenases in aqueous solutions of deep eutectic and organic solvents. Green Chem. 2014, 16, 2754–2761. [Google Scholar] [CrossRef]
- Khabiri, M.; Minofar, B.; Brezovský, J.; Damborský, J.; Ettrich, R. Interaction of organic solvents with protein structures at protein-solvent interface. J. Mol. Model. 2012, 19, 4701–4711. [Google Scholar] [CrossRef]
- Micaêlo, N.M.; Soares, C.M. Protein Structure and Dynamics in Ionic Liquids. Insights from Molecular Dynamics Simulation Studies. J. Phys. Chem. B 2008, 112, 2566–2572. [Google Scholar] [CrossRef]
- Klähn, M.; Lim, G.S.; Seduraman, A.; Wu, P. On the different roles of anions and cations in the solvation of enzymes in ionic liquids. Phys. Chem. Chem. Phys. 2011, 13, 1649–1662. [Google Scholar] [CrossRef] [PubMed]
- Krug, M.; Weiss, M.S.; Heinemann, U.; Mueller, U. XDSAPP: A graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 2012, 45, 568–572. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.W.; McCoy, A.; et al. Overview of theCCP4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, P.W.; Beran, B.; Bi, C.; Bluhm, W.F.; Dimitropoulos, D.; Goodsell, D.S.; Prlic, A.; Quesada, M.; Quinn, G.B.; Westbrook, J.D.; et al. The RCSB Protein Data Bank: Redesigned web site and web services. Nucleic Acids Res. 2010, 39, D392–D401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagin, A.; Teplyakov, A. MOLREP: An Automated Program for Molecular Replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Longa, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development ofCoot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Lua, R.C.; Lichtarge, O. PyETV: A PyMOL evolutionary trace viewer to analyze functional site predictions in protein complexes. Bioinformatics 2010, 26, 2981–2982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins Struct. Funct. Bioinform. 2006, 65, 712–725. [Google Scholar] [CrossRef] [Green Version]
- Martínez, L.; Andrade, R.; Birgin, E.G.; Martínez, J.M. PACKMOL: A package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 2009, 30, 2157–2164. [Google Scholar] [CrossRef] [PubMed]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Konar, S.; Sinha, S.K.; Datta, S.; Ghorai, P.K. The effect of ionic liquid on the structure of active site pocket and catalytic activity of a β-glucosidase from Halothermothrix orenii. J. Mol. Liq. 2020, 306, 112879. [Google Scholar] [CrossRef]
- Harada, L.; Pereira, J.; Campos, W.; Silva, E.; Moutinho, C.; Vila, M.; Oliveira, J.M., Jr.; Teixeira, J.A.; Balcao, V.M.; Tubino, M. Insights into Protein-Ionic Liquid Interactions Aiming at Macromolecule Delivery Systems. J. Braz. Chem. Soc. 2018, 29, 1983–1998. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DhaA80 | Soaked with [ETA][ACC] | Soaked with [EMIM][CHS] |
|---|---|---|
| Data collection | ||
| Beamline | BESSY MX14.2 | BESSY MX14.2 |
| Space group | P212121 | P212121 |
| a, b, c (Å) | 52.31, 69.95, 83.82 | 50.89, 69.55, 83.92 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 44.38–1.25 (1.28–1.25) | 32.13–1.75 (1.79–1.75) |
| Total no. of reflections | 847,732 (97,232) | 23,531 (2646) |
| No. of unique reflections | 103,516 (61,553) | 7740 (1572) |
| Completeness (%) | 99.2 (97.95) | 99.2 (94.64) |
| 〈I/σ(I)〉 | 4.82 (1.25) | 1.94 (1.75) |
| Rmeas * | 5.4 (59.9) | 3.7 (48.2) |
| CC1/2 | 0.99 (0.85) | 0.98 (0.77) |
| Wilson B factor (Å2) | 9.3 | 22.9 |
| Refinement | ||
| No. of reflections used for refinement | 82,895 | 29,090 |
| Rwork ‡/Rfree § (%) | 12.50/15.30 | 20.80/22.40 |
| No. of non-H atoms | 2852 | 2502 |
| No. of protein atoms | 2483 | 2361 |
| No. of chloride ions | 1 | 0 |
| No. of IL ligands | 1 | 1 |
| No. of water molecules | 362 | 333 |
| Average B factor (Å2) | 14.0 | 29.0 |
| Ramachandran plot | ||
| Most favored (%) | 97.0 | 97.0 |
| Allowed (%) | 3.0 | 3.0 |
| Outliers (%) | 0 | 0 |
| R.M.S. deviations | ||
| Bonds (Å) | 0.017 | 0.004 |
| Angles (°) | 2.072 | 1.071 |
| PDB ID | 7O3O | 7O8B |
| System No. | Ionic Liquid | No. of Ion Pairs | Molar Concentration (M) | No. of Crystal Water Molecules | Total No. of Atoms |
|---|---|---|---|---|---|
| 1 | water 1 | 0 | 0 | 362 | 15,308 |
| 2 | [ETA][ACC] | 100 | 0.34 | 362 | 14,521 |
| 3 | [ETA][ACC] | 500 | 1.8 | 362 | 11,394 |
| 4 | [ETA][ACC] | 1000 | 3.8 | 362 | 7999 |
| 5 | water 2 | 0 | 0 | 163 | 15,454 |
| 6 | [EMIM][CHS] | 100 | 0.34 | 163 | 14,169 |
| 7 | [EMIM][CHS] | 500 | 1.8 | 163 | 9965 |
| 8 | [EMIM][CHS] | 1000 | 3.8 | 163 | 5640 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaposhnikova, A.; Kuty, M.; Chaloupkova, R.; Damborsky, J.; Kuta Smatanova, I.; Minofar, B.; Prudnikova, T. Stabilization of Haloalkane Dehalogenase Structure by Interfacial Interaction with Ionic Liquids. Crystals 2021, 11, 1052. https://doi.org/10.3390/cryst11091052
Shaposhnikova A, Kuty M, Chaloupkova R, Damborsky J, Kuta Smatanova I, Minofar B, Prudnikova T. Stabilization of Haloalkane Dehalogenase Structure by Interfacial Interaction with Ionic Liquids. Crystals. 2021; 11(9):1052. https://doi.org/10.3390/cryst11091052
Chicago/Turabian StyleShaposhnikova, Anastasiia, Michal Kuty, Radka Chaloupkova, Jiri Damborsky, Ivana Kuta Smatanova, Babak Minofar, and Tatyana Prudnikova. 2021. "Stabilization of Haloalkane Dehalogenase Structure by Interfacial Interaction with Ionic Liquids" Crystals 11, no. 9: 1052. https://doi.org/10.3390/cryst11091052