
The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction

Abstract
:Simple Summary
Abstract
1. Introduction
1.1. Genomic and Chromosomal Instabilities as Prerequisites of Solid Cancers
1.2. DNA Damage Response and Its Constituents
1.3. TP53 Function and Its Alterations in Solid Cancers
1.3.1. The Functional Aspects of TP53
1.3.2. DNA Damage, DNA Repair, and TP53
1.3.3. The Function of p53 in Interaction with Telomere Homeostasis
2. The Impact of Interactions between Altered p53 and DNA Repair on Cancer Risk
2.1. Most Frequent Mutations in Tumorigenesis
2.2. Studies on the Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Cancer Predisposition
2.3. Studies on the Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Cancer Risk
3. Studies on the Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Cancer Progression and Prognosis
4. Studies on the Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Modulating Cancer Therapy
4.1. Interactions of DNA Repair, Telomere Homeostasis, and p53 and Their Mechanistic Impact (Underlying Mechanisms In) on Cancer Therapy
4.2. The Interactions of DNA Repair, Telomere Homeostasis, and p53, and Their Underlying Mechanisms in the Resistance towards Cancer Therapy
4.3. The Interactions of DNA Repair, Telomere Homeostasis, and p53 Utilized in Cancer Therapy
5. Open Issues and Perspectives
5.1. The Interactions of DNA Repair, Telomere Homeostasis, and p53 in the Context of Epigenetic Regulations
5.2. The Interactions of DNA Repair, Telomere Homeostasis, and p53 in the Context of the Immune System
5.3. The Interactions of DNA Repair, Telomere Homeostasis, and p53 and the New Concept in Cancer Therapy
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Vodicka, P.; Vodenkova, S.; Opattova, A.; Vodickova, L. DNA damage and repair measured by comet assay in cancer patients. Mutat. Res. 2019, 843, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Vodicka, P.; Urbanova, M.; Makovicky, P.; Tomasova, K.; Kroupa, M.; Stetina, R.; Opattova, A.; Kostovcikova, K.; Siskova, A.; Schneiderova, M.; et al. Oxidative Damage in Sporadic Colorectal Cancer: Molecular Mapping of Base Excision Repair Glycosylases in Colorectal Cancer Patients. Int. J. Mol. Sci. 2020, 21, 2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasova, K.; Cumova, A.; Seborova, K.; Horak, J.; Koucka, K.; Vodickova, L.; Vaclavikova, R.; Vodicka, P. DNA Repair and Ovarian Carcinogenesis: Impact on Risk, Prognosis and Therapy Outcome. Cancers 2020, 12, 1713. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niazi, Y.; Thomsen, H.; Smolkova, B.; Vodickova, L.; Vodenkova, S.; Kroupa, M.; Vymetalkova, V.; Kazimirova, A.; Barancokova, M.; Volkovova, K.; et al. Impact of genetic polymorphisms in kinetochore and spindle assembly genes on chromosomal aberration frequency in healthy humans. Mutat. Res. 2020, 858–860, 503253. [Google Scholar] [CrossRef]
- Vodicka, P.; Musak, L.; Vodickova, L.; Vodenkova, S.; Catalano, C.; Kroupa, M.; Naccarati, A.; Polivkova, Z.; Vymetalkova, V.; Forsti, A.; et al. Genetic variation of acquired structural chromosomal aberrations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 836, 13–21. [Google Scholar] [CrossRef]
- Goldstein, M.; Kastan, M.B. The DNA damage response: Implications for tumor responses to radiation and chemotherapy. Annu. Rev. Med. 2015, 66, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Giglia-Mari, G.; Zotter, A.; Vermeulen, W. DNA damage response. Cold Spring Harb. Perspect. Biol. 2011, 3, a000745. [Google Scholar] [CrossRef]
- Burrell, R.A.; McClelland, S.E.; Endesfelder, D.; Groth, P.; Weller, M.C.; Shaikh, N.; Domingo, E.; Kanu, N.; Dewhurst, S.M.; Gronroos, E.; et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 2013, 494, 492–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagel, Z.D.; Chaim, I.A.; Samson, L.D. Inter-individual variation in DNA repair capacity: A need for multi-pathway functional assays to promote translational DNA repair research. DNA Repair 2014, 19, 199–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Brenes, I.A.; Komarova, N.L.; Wodarz, D. The role of telomere shortening in carcinogenesis: A hybrid stochastic-deterministic approach. J. Theor. Biol. 2019, 460, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Tomasova, K.; Kroupa, M.; Forsti, A.; Vodicka, P.; Vodickova, L. Telomere maintenance in interplay with DNA repair in pathogenesis and treatment of colorectal cancer. Mutagenesis 2020, 35, 261–271. [Google Scholar] [CrossRef]
- Fearon, E.R.; Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 1990, 61, 759–767. [Google Scholar] [CrossRef]
- Naccarati, A.; Polakova, V.; Pardini, B.; Vodickova, L.; Hemminki, K.; Kumar, R.; Vodicka, P. Mutations and polymorphisms in TP53 gene—An overview on the role in colorectal cancer. Mutagenesis 2012, 27, 211–218. [Google Scholar] [CrossRef]
- Bouaoun, L.; Sonkin, D.; Ardin, M.; Hollstein, M.; Byrnes, G.; Zavadil, J.; Olivier, M. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum. Mutat. 2016, 37, 865–876. [Google Scholar] [CrossRef]
- Kaiser, A.M.; Attardi, L.D. Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death Differ. 2018, 25, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Stein, Y.; Rotter, V.; Aloni-Grinstein, R. Gain-of-Function Mutant p53: All the Roads Lead to Tumorigenesis. Int. J. Mol. Sci. 2019, 20, 6197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Liu, J.; Xu, D.; Zhang, T.; Hu, W.; Feng, Z. Gain-of-function mutant p53 in cancer progression and therapy. J. Mol. Cell Biol. 2020, 12, 674–687. [Google Scholar] [CrossRef] [PubMed]
- Loh, S.N. Follow the Mutations: Toward Class-Specific, Small-Molecule Reactivation of p53. Biomolecules 2020, 10, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, N.; Moreno, V.; Hughes, D.J.; Vodicka, L.; Vodicka, P.; Aglago, E.K.; Gunter, M.J.; Jenab, M. Lifestyle and dietary environmental factors in colorectal cancer susceptibility. Mol. Aspects Med. 2019, 69, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, T.; Nakagawara, A. Role of p53 in Cell Death and Human Cancers. Cancers 2011, 3, 994–1013. [Google Scholar] [CrossRef] [PubMed]
- Pitolli, C.; Wang, Y.; Candi, E.; Shi, Y.; Melino, G.; Amelio, I. p53-Mediated Tumor Suppression: DNA-Damage Response and Alternative Mechanisms. Cancers 2019, 11, 1983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantovani, F.; Collavin, L.; Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 2019, 26, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; He, X.; Lim, L.P.; de Stanchina, E.; Xuan, Z.; Liang, Y.; Xue, W.; Zender, L.; Magnus, J.; Ridzon, D.; et al. A microRNA component of the p53 tumour suppressor network. Nature 2007, 447, 1130–1134. [Google Scholar] [CrossRef] [Green Version]
- Chipuk, J.E.; Kuwana, T.; Bouchier-Hayes, L.; Droin, N.M.; Newmeyer, D.D.; Schuler, M.; Green, D.R. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science 2004, 303, 1010–1014. [Google Scholar] [CrossRef] [Green Version]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seillier, M.; Peuget, S.; Gayet, O.; Gauthier, C.; N’Guessan, P.; Monte, M.; Carrier, A.; Iovanna, J.L.; Dusetti, N.J. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012, 19, 1525–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crighton, D.; Wilkinson, S.; O’Prey, J.; Syed, N.; Smith, P.; Harrison, P.R.; Gasco, M.; Garrone, O.; Crook, T.; Ryan, K.M. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 2006, 126, 121–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mijit, M.; Caracciolo, V.; Melillo, A.; Amicarelli, F.; Giordano, A. Role of p53 in the Regulation of Cellular Senescence. Biomolecules 2020, 10, 420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar] [CrossRef]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Ahn, J.; Wilson, S.H.; Prives, C. A role for p53 in base excision repair. EMBO J. 2001, 20, 914–923. [Google Scholar] [CrossRef] [Green Version]
- Zaky, A.; Busso, C.; Izumi, T.; Chattopadhyay, R.; Bassiouny, A.; Mitra, S.; Bhakat, K.K. Regulation of the human AP-endonuclease (APE1/Ref-1) expression by the tumor suppressor p53 in response to DNA damage. Nucleic Acids Res. 2008, 36, 1555–1566. [Google Scholar] [CrossRef]
- Offer, H.; Wolkowicz, R.; Matas, D.; Blumenstein, S.; Livneh, Z.; Rotter, V. Direct involvement of p53 in the base excision repair pathway of the DNA repair machinery. FEBS Lett. 1999, 450, 197–204. [Google Scholar] [CrossRef]
- Jung, H.J.; Kim, H.L.; Kim, Y.J.; Weon, J.I.; Seo, Y.R. A novel chemopreventive mechanism of selenomethionine: Enhancement of APE1 enzyme activity via a Gadd45a, PCNA and APE1 protein complex that regulates p53-mediated base excision repair. Oncol. Rep. 2013, 30, 1581–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, Y.R.; Fishel, M.L.; Amundson, S.; Kelley, M.R.; Smith, M.L. Implication of p53 in base excision DNA repair: In vivo evidence. Oncogene 2002, 21, 731–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Offer, H.; Zurer, I.; Banfalvi, G.; Reha’k, M.; Falcovitz, A.; Milyavsky, M.; Goldfinger, N.; Rotter, V. p53 modulates base excision repair activity in a cell cycle-specific manner after genotoxic stress. Cancer Res. 2001, 61, 88–96. [Google Scholar]
- Smith, M.L.; Seo, Y.R. p53 regulation of DNA excision repair pathways. Mutagenesis 2002, 17, 149–156. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.W.; Yeh, H.; Schaeffer, L.; Roy, R.; Moncollin, V.; Egly, J.M.; Wang, Z.; Freidberg, E.C.; Evans, M.K.; Taffe, B.G.; et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat. Genet. 1995, 10, 188–195. [Google Scholar] [CrossRef]
- Schwartz, D.; Goldfinger, N.; Kam, Z.; Rotter, V. p53 controls low DNA damage-dependent premeiotic checkpoint and facilitates DNA repair during spermatogenesis. Cell Growth Differ. 1999, 10, 665–675. [Google Scholar]
- Zurer, I.; Hofseth, L.J.; Cohen, Y.; Xu-Welliver, M.; Hussain, S.P.; Harris, C.C.; Rotter, V. The role of p53 in base excision repair following genotoxic stress. Carcinogenesis 2004, 25, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Hollstein, M.; Xu, Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat. Cell Biol. 2007, 9, 573–580. [Google Scholar] [CrossRef]
- Menon, V.; Povirk, L. Involvement of p53 in the repair of DNA double strand breaks: Multifaceted Roles of p53 in homologous recombination repair (HRR) and non-homologous end joining (NHEJ). Subcell. Biochem. 2014, 85, 321–336. [Google Scholar] [CrossRef] [Green Version]
- Yoon, D.; Wang, Y.; Stapleford, K.; Wiesmuller, L.; Chen, J. P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J. Mol. Biol. 2004, 336, 639–654. [Google Scholar] [CrossRef]
- Zhang, Y.X.; Pan, W.Y.; Chen, J. p53 and its isoforms in DNA double-stranded break repair. J. Zhejiang Univ. Sci. B 2019, 20, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Mehigan, B.J.; Ashman, J.N.; Baker, R.P.; Macdonald, A.; Greenman, J.; Monson, J.R.; Cawkwell, L. Mismatch repair, p53 and chromosomal aberrations in primary colorectal carcinomas. Acta Oncol. 2006, 45, 61–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.S.; Huang, Y.Y.; Pan, S.C.; Cheng, C.T.; Liu, C.C.; Shih, C.H.; Ho, H.L.; Yeh, Y.C.; Chou, T.Y.; Lee, M.Y.; et al. Involvement of increased p53 expression in the decrease of mitochondrial DNA copy number and increase of SUVmax of FDG-PET scan in esophageal squamous cell carcinoma. Mitochondrion 2019, 47, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Wright, W.E. Senescence and immortalization: Role of telomeres and telomerase. Carcinogenesis 2005, 26, 867–874. [Google Scholar] [CrossRef]
- Jones, K.R.; Elmore, L.W.; Jackson-Cook, C.; Demasters, G.; Povirk, L.F.; Holt, S.E.; Gewirtz, D.A. p53-Dependent accelerated senescence induced by ionizing radiation in breast tumour cells. Int. J. Radiat. Biol. 2005, 81, 445–458. [Google Scholar] [CrossRef]
- Artandi, S.E.; Attardi, L.D. Pathways connecting telomeres and p53 in senescence, apoptosis, and cancer. Biochem. Biophys. Res. Commun. 2005, 331, 881–890. [Google Scholar] [CrossRef]
- Roake, C.M.; Artandi, S.E. Control of Cellular Aging, Tissue Function, and Cancer by p53 Downstream of Telomeres. Cold Spring Harb. Perspect. Med. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Toufektchan, E.; Toledo, F. The Guardian of the Genome Revisited: p53 Downregulates Genes Required for Telomere Maintenance, DNA Repair, and Centromere Structure. Cancers 2018, 10, 135. [Google Scholar] [CrossRef] [Green Version]
- Ge, Y.; Wu, S.; Zhang, Z.; Li, X.; Li, F.; Yan, S.; Liu, H.; Huang, J.; Zhao, Y. Inhibition of p53 and/or AKT as a new therapeutic approach specifically targeting ALT cancers. Protein Cell 2019, 10, 808–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, A.; Shin, V.Y.; Ho, C.Y.S.; Au, C.H.; Slavin, T.P.; Weitzel, J.N.; Chan, T.L.; Ma, E.S.K. Mutation screening of germline TP53 mutations in high-risk Chinese breast cancer patients. BMC Cancer 2020, 20, 1053. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Sun, Q. TP53 mutations, expression and interaction networks in human cancers. Oncotarget 2017, 8, 624–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, M.; Oshima, M. Mutant p53 in colon cancer. J. Mol. Cell Biol. 2019, 11, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, H.J.; Bae, J.M.; Wen, X.; Jung, S.; Kim, Y.; Kim, K.J.; Cho, N.Y.; Kim, J.H.; Han, S.W.; Kim, T.Y.; et al. p53 expression status is associated with cancer-specific survival in stage III and high-risk stage II colorectal cancer patients treated with oxaliplatin-based adjuvant chemotherapy. Br. J. Cancer 2019, 120, 797–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zhang, J.; Tong, J.H.M.; Chan, A.W.H.; Yu, J.; Kang, W.; To, K.F. Targeting the Oncogenic p53 Mutants in Colorectal Cancer and Other Solid Tumors. Int. J. Mol. Sci. 2019, 20, 5999. [Google Scholar] [CrossRef] [Green Version]
- Donehower, L.A.; Soussi, T.; Korkut, A.; Liu, Y.; Schultz, A.; Cardenas, M.; Li, X.; Babur, O.; Hsu, T.K.; Lichtarge, O.; et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell Rep. 2019, 28, 1370–1384.e5. [Google Scholar] [CrossRef] [Green Version]
- Baker, S.J.; Preisinger, A.C.; Jessup, J.M.; Paraskeva, C.; Markowitz, S.; Willson, J.K.; Hamilton, S.; Vogelstein, B. p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res. 1990, 50, 7717–7722. [Google Scholar]
- Iacopetta, B.; Russo, A.; Bazan, V.; Dardanoni, G.; Gebbia, N.; Soussi, T.; Kerr, D.; Elsaleh, H.; Soong, R.; Kandioler, D.; et al. Functional categories of TP53 mutation in colorectal cancer: Results of an International Collaborative Study. Ann. Oncol. 2006, 17, 842–847. [Google Scholar] [CrossRef]
- Bargonetti, J.; Prives, C. Gain-of-function mutant p53: History and speculation. J. Mol. Cell Biol. 2019, 11, 605–609. [Google Scholar] [CrossRef]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grady, W.M.; Markowitz, S.D. The molecular pathogenesis of colorectal cancer and its potential application to colorectal cancer screening. Dig. Dis. Sci. 2015, 60, 762–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, X.P.; Frayling, I.M.; Sgouros, J.G.; Du, M.Q.; Willcocks, T.C.; Talbot, I.C.; Tomlinson, I.P. The spectrum of p53 mutations in colorectal adenomas differs from that in colorectal carcinomas. Gut 2002, 50, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Hainaut, P.; Pfeifer, G.P. Somatic TP53 Mutations in the Era of Genome Sequencing. Cold Spring Harb. Perspect. Med. 2016, 6, a026179. [Google Scholar] [CrossRef] [Green Version]
- Russo, A.; Bazan, V.; Iacopetta, B.; Kerr, D.; Soussi, T.; Gebbia, N.; Group, T.C.C.S. The TP53 colorectal cancer international collaborative study on the prognostic and predictive significance of p53 mutation: Influence of tumor site, type of mutation, and adjuvant treatment. J. Clin. Oncol. 2005, 23, 7518–7528. [Google Scholar] [CrossRef] [PubMed]
- Carethers, J.M.; Jung, B.H. Genetics and Genetic Biomarkers in Sporadic Colorectal Cancer. Gastroenterology 2015, 149, 1177–1190.e3. [Google Scholar] [CrossRef] [Green Version]
- Baran, B.; Mert Ozupek, N.; Yerli Tetik, N.; Acar, E.; Bekcioglu, O.; Baskin, Y. Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterol. Res. 2018, 11, 264–273. [Google Scholar] [CrossRef] [Green Version]
- Spencer, J.M.; Morgan, M.B.; Trapp, K.M.; Moon, S.D. Topical formulation engendered alteration in p53 and cyclobutane pyrimidine dimer expression in chronic photodamaged patients. J. Drugs Dermatol. 2013, 12, 336–340. [Google Scholar]
- Cardin, R.; Piciocchi, M.; Tieppo, C.; Maddalo, G.; Zaninotto, G.; Mescoli, C.; Rugge, M.; Farinati, F. Oxidative DNA damage in Barrett mucosa: Correlation with telomeric dysfunction and p53 mutation. Ann. Surg. Oncol. 2013, 20 (Suppl. 3), S583–S589. [Google Scholar] [CrossRef]
- ElBadre, H.M.; El-Mahdy, R.I.; Mohamed, N.A.; Zakhary, M.M.; Maximous, D.W. Tissue Indices of Telomere Length and p53 in Patients with Different Gastrointestinal Tumors: Correlation with Clinicopathological Status. Appl. Biochem. Biotechnol. 2018, 186, 764–778. [Google Scholar] [CrossRef]
- Borbora, D.; Dutta, H.K.; Devi, K.R.; Mahanta, J.; Medhi, P.; Narain, K. Long telomeres cooperate with p53, MDM2, and p21 polymorphisms to raise pediatric solid tumor risk. Pediatr. Int. 2019, 61, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Scalise, J.R.; Pocas, R.C.; Caneloi, T.P.; Lopes, C.O.; Kanno, D.T.; Marques, M.G.; Valdivia, J.C.; Maximo, F.R.; Pereira, J.A.; Ribeiro, M.L.; et al. DNA Damage Is a Potential Marker for TP53 Mutation in Colorectal Carcinogenesis. J. Gastrointest. Cancer 2016, 47, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Caja, F.; Vodickova, L.; Kral, J.; Vymetalkova, V.; Naccarati, A.; Vodicka, P. DNA Mismatch Repair Gene Variants in Sporadic Solid Cancers. Int. J. Mol. Sci. 2020, 21, 5561. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhang, Y.; Chen, M.; Wang, Y.; Feng, Y.; Xu, Z.; Zhang, D.; Sun, Y.; Fu, Z. Association of genetic variants in ATR-CHEK1 and ATM-CHEK2 pathway genes with risk of colorectal cancer in a Chinese population. Oncotarget 2018, 9, 26616–26624. [Google Scholar] [CrossRef] [Green Version]
- Jordan, J.J.; Inga, A.; Conway, K.; Edmiston, S.; Carey, L.A.; Wu, L.; Resnick, M.A. Altered-function p53 missense mutations identified in breast cancers can have subtle effects on transactivation. Mol. Cancer Res. 2010, 8, 701–716. [Google Scholar] [CrossRef] [Green Version]
- Siegel, M.B.; He, X.; Hoadley, K.A.; Hoyle, A.; Pearce, J.B.; Garrett, A.L.; Kumar, S.; Moylan, V.J.; Brady, C.M.; Van Swearingen, A.E.; et al. Integrated RNA and DNA sequencing reveals early drivers of metastatic breast cancer. J. Clin. Investig. 2018, 128, 1371–1383. [Google Scholar] [CrossRef]
- Zagouri, F.; Kotoula, V.; Kouvatseas, G.; Sotiropoulou, M.; Koletsa, T.; Gavressea, T.; Valavanis, C.; Trihia, H.; Bobos, M.; Lazaridis, G.; et al. Protein expression patterns of cell cycle regulators in operable breast cancer. PLoS ONE 2017, 12, e0180489. [Google Scholar] [CrossRef] [Green Version]
- Mollevi, D.G.; Serrano, T.; Ginesta, M.M.; Valls, J.; Torras, J.; Navarro, M.; Ramos, E.; Germa, J.R.; Jaurrieta, E.; Moreno, V.; et al. Mutations in TP53 are a prognostic factor in colorectal hepatic metastases undergoing surgical resection. Carcinogenesis 2007, 28, 1241–1246. [Google Scholar] [CrossRef]
- Huang, D.; Sun, W.; Zhou, Y.; Li, P.; Chen, F.; Chen, H.; Xia, D.; Xu, E.; Lai, M.; Wu, Y.; et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev. 2018, 37, 173–187. [Google Scholar] [CrossRef]
- Westra, J.L.; Schaapveld, M.; Hollema, H.; de Boer, J.P.; Kraak, M.M.; de Jong, D.; ter Elst, A.; Mulder, N.H.; Buys, C.H.; Hofstra, R.M.; et al. Determination of TP53 mutation is more relevant than microsatellite instability status for the prediction of disease-free survival in adjuvant-treated stage III colon cancer patients. J. Clin. Oncol. 2005, 23, 5635–5643. [Google Scholar] [CrossRef]
- Fry, E.A.; Niehans, G.E.; Kratzke, R.A.; Kai, F.; Inoue, K. Survival of Lung Cancer Patients Dependent on the LOH Status for DMP1, ARF, and p53. Int. J. Mol. Sci. 2020, 21, 7971. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, N.; Gao, G.; Deng, H.B.; Wang, Z.H.; Deng, L.L.; Yang, Y.; Lu, C. Prognostic value of TP53 co-mutation status combined with EGFR mutation in patients with lung adenocarcinoma. J. Cancer Res. Clin. Oncol. 2020, 146, 2851–2859. [Google Scholar] [CrossRef] [PubMed]
- Sirotkovic-Skerlev, M.; Plavetic, N.D.; Sedlic, F.; Kuna, S.K.; Vrbanec, D.; Belev, B.; Plestina, S.; Kovac, Z.; Kulic, A. Prognostic value of circulating Bcl-2 and anti-p53 antibodies in patients with breast cancer: A long term follow-up (17.5 years). Cancer Biomark. 2020. [Google Scholar] [CrossRef] [PubMed]
- Grosser, B.; Kohlruss, M.; Slotta-Huspenina, J.; Jesinghaus, M.; Pfarr, N.; Steiger, K.; Novotny, A.; Gaida, M.M.; Schmidt, T.; Hapfelmeier, A.; et al. Impact of Tumor Localization and Molecular Subtypes on the Prognostic and Predictive Significance of p53 Expression in Gastric Cancer. Cancers 2020, 12, 1689. [Google Scholar] [CrossRef] [PubMed]
- Vakili, S.A.; George, A.; Ayatollahi, S.A.; Martorell, M.; Ostrander, E.A.; Salehi, B.; Martins, N.; Sharifi-Rad, J. Phenolic compounds, saponins and alkaloids on cancer progression: Emphasis on p53 expression and telomere length. Cell. Mol. Biol. 2020, 66, 110–119. [Google Scholar] [CrossRef]
- Opattova, A.; Horak, J.; Vodenkova, S.; Kostovcikova, K.; Cumova, A.; Macinga, P.; Galanova, N.; Rejhova, A.; Vodickova, L.; Kozics, K.; et al. Ganoderma Lucidum induces oxidative DNA damage and enhances the effect of 5-Fluorouracil in colorectal cancer in vitro and in vivo. Mutat. Res. 2019, 845, 403065. [Google Scholar] [CrossRef]
- Li, X.L.; Zhou, J.; Chen, Z.R.; Chng, W.J. P53 mutations in colorectal cancer—Molecular pathogenesis and pharmacological reactivation. World J. Gastroenterol. 2015, 21, 84–93. [Google Scholar] [CrossRef]
- Thottassery, J.V.; Zambetti, G.P.; Arimori, K.; Schuetz, E.G.; Schuetz, J.D. p53-dependent regulation of MDR1 gene expression causes selective resistance to chemotherapeutic agents. Proc. Natl. Acad. Sci. USA 1997, 94, 11037–11042. [Google Scholar] [CrossRef] [Green Version]
- Sampath, J.; Sun, D.; Kidd, V.J.; Grenet, J.; Gandhi, A.; Shapiro, L.H.; Wang, Q.; Zambetti, G.P.; Schuetz, J.D. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J. Biol. Chem. 2001, 276, 39359–39367. [Google Scholar] [CrossRef] [Green Version]
- Pabla, N.; Huang, S.; Mi, Q.S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Pearlman, A.H.; Hsieh, P. DNA mismatch repair and the DNA damage response. DNA Repair 2016, 38, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundar, R.; Brown, J.; Ingles Russo, A.; Yap, T.A. Targeting ATR in cancer medicine. Curr. Probl. Cancer 2017, 41, 302–315. [Google Scholar] [CrossRef]
- Williamson, C.T.; Miller, R.; Pemberton, H.N.; Jones, S.E.; Campbell, J.; Konde, A.; Badham, N.; Rafiq, R.; Brough, R.; Gulati, A.; et al. ATR inhibitors as a synthetic lethal therapy for tumours deficient in ARID1A. Nat. Commun. 2016, 7, 13837. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.H.; Oh, D.Y. ATM in DNA repair in cancer. Pharmacol. Ther. 2019, 203, 107391. [Google Scholar] [CrossRef] [PubMed]
- Karnitz, L.M.; Zou, L. Molecular Pathways: Targeting ATR in Cancer Therapy. Clin. Cancer Res. 2015, 21, 4780–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anuja, K.; Chowdhury, A.R.; Saha, A.; Roy, S.; Rath, A.K.; Kar, M.; Banerjee, B. Radiation-induced DNA damage response and resistance in colorectal cancer stem-like cells. Int. J. Radiat. Biol. 2019, 95, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, A.M.; Ryan, A.J. ATM and ATR as therapeutic targets in cancer. Pharmacol. Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Z.; Oleinick, N.L.; Zhang, J. ATR/CHK1 inhibitors and cancer therapy. Radiother. Oncol. 2018, 126, 450–464. [Google Scholar] [CrossRef]
- Jabbour-Leung, N.A.; Chen, X.; Bui, T.; Jiang, Y.; Yang, D.; Vijayaraghavan, S.; McArthur, M.J.; Hunt, K.K.; Keyomarsi, K. Sequential Combination Therapy of CDK Inhibition and Doxorubicin Is Synthetically Lethal in p53-Mutant Triple-Negative Breast Cancer. Mol. Cancer Ther. 2016, 15, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.W.; Dreaden, E.C.; Morandell, S.; Zhou, W.; Dhara, S.S.; Sriram, G.; Lam, F.C.; Patterson, J.C.; Quadir, M.; Dinh, A.; et al. Enhancing chemotherapy response through augmented synthetic lethality by co-targeting nucleotide excision repair and cell-cycle checkpoints. Nat. Commun. 2020, 11, 4124. [Google Scholar] [CrossRef]
- Leijen, S.; van Geel, R.M.; Sonke, G.S.; de Jong, D.; Rosenberg, E.H.; Marchetti, S.; Pluim, D.; van Werkhoven, E.; Rose, S.; Lee, M.A.; et al. Phase II Study of WEE1 Inhibitor AZD1775 Plus Carboplatin in Patients With TP53-Mutated Ovarian Cancer Refractory or Resistant to First-Line Therapy Within 3 Months. J. Clin. Oncol. 2016, 34, 4354–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, E.K.; Fader, A.N.; Santin, A.D.; Liu, J.F. Uterine serous carcinoma: Molecular features, clinical management, and new and future therapies. Gynecol. Oncol. 2021, 160, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Oza, A.M.; Estevez-Diz, M.; Grischke, E.M.; Hall, M.; Marme, F.; Provencher, D.; Uyar, D.; Weberpals, J.I.; Wenham, R.M.; Laing, N.; et al. A Biomarker-enriched, Randomized Phase II Trial of Adavosertib (AZD1775) Plus Paclitaxel and Carboplatin for Women with Platinum-sensitive TP53-mutant Ovarian Cancer. Clin. Cancer Res. 2020, 26, 4767–4776. [Google Scholar] [CrossRef] [PubMed]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilsker, D.F.; Barrett, A.M.; Dull, A.B.; Lawrence, S.M.; Hollingshead, M.G.; Chen, A.; Kummar, S.; Parchment, R.E.; Doroshow, J.H.; Kinders, R.J. Evaluation of Pharmacodynamic Responses to Cancer Therapeutic Agents Using DNA Damage Markers. Clin. Cancer Res. 2019, 25, 3084–3095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plimack, E.R.; Hoffman-Censits, J.H.; Viterbo, R.; Trabulsi, E.J.; Ross, E.A.; Greenberg, R.E.; Chen, D.Y.; Lallas, C.D.; Wong, Y.N.; Lin, J.; et al. Accelerated methotrexate, vinblastine, doxorubicin, and cisplatin is safe, effective, and efficient neoadjuvant treatment for muscle-invasive bladder cancer: Results of a multicenter phase II study with molecular correlates of response and toxicity. J. Clin. Oncol. 2014, 32, 1895–1901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef] [Green Version]
- Christmann, M.; Kaina, B. Epigenetic regulation of DNA repair genes and implications for tumor therapy. Mutat. Res. 2019, 780, 15–28. [Google Scholar] [CrossRef]
- Slyskova, J.; Korenkova, V.; Collins, A.R.; Prochazka, P.; Vodickova, L.; Svec, J.; Lipska, L.; Levy, M.; Schneiderova, M.; Liska, V.; et al. Functional, genetic, and epigenetic aspects of base and nucleotide excision repair in colorectal carcinomas. Clin. Cancer Res. 2012, 18, 5878–5887. [Google Scholar] [CrossRef] [Green Version]
- Farkas, S.A.; Vymetalkova, V.; Vodickova, L.; Vodicka, P.; Nilsson, T.K. DNA methylation changes in genes frequently mutated in sporadic colorectal cancer and in the DNA repair and Wnt/beta-catenin signaling pathway genes. Epigenomics 2014, 6, 179–191. [Google Scholar] [CrossRef]
- Naccarati, A.; Pardini, B.; Stefano, L.; Landi, D.; Slyskova, J.; Novotny, J.; Levy, M.; Polakova, V.; Lipska, L.; Vodicka, P. Polymorphisms in miRNA-binding sites of nucleotide excision repair genes and colorectal cancer risk. Carcinogenesis 2012, 33, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Schneiderova, M.; Naccarati, A.; Pardini, B.; Rosa, F.; Gaetano, C.D.; Jiraskova, K.; Opattova, A.; Levy, M.; Veskrna, K.; Veskrnova, V.; et al. MicroRNA-binding site polymorphisms in genes involved in colorectal cancer etiopathogenesis and their impact on disease prognosis. Mutagenesis 2017, 32, 533–542. [Google Scholar] [CrossRef] [PubMed]
- Pardini, B.; Rosa, F.; Barone, E.; Di Gaetano, C.; Slyskova, J.; Novotny, J.; Levy, M.; Garritano, S.; Vodickova, L.; Buchler, T.; et al. Variation within 3′-UTRs of base excision repair genes and response to therapy in colorectal cancer patients: A potential modulation of microRNAs binding. Clin. Cancer Res. 2013, 19, 6044–6056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naccarati, A.; Rosa, F.; Vymetalkova, V.; Barone, E.; Jiraskova, K.; Di Gaetano, C.; Novotny, J.; Levy, M.; Vodickova, L.; Gemignani, F.; et al. Double-strand break repair and colorectal cancer: Gene variants within 3′ UTRs and microRNAs binding as modulators of cancer risk and clinical outcome. Oncotarget 2016, 7, 23156–23169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gronke, K.; Hernandez, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef]
- Mukherjee, S.; Abdisalaam, S.; Bhattacharya, S.; Srinivasan, K.; Sinha, D.; Asaithamby, A. Mechanistic link between DNA damage sensing, repairing and signaling factors and immune signaling. Adv. Protein Chem. Struct. Biol. 2019, 115, 297–324. [Google Scholar] [CrossRef]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [Green Version]


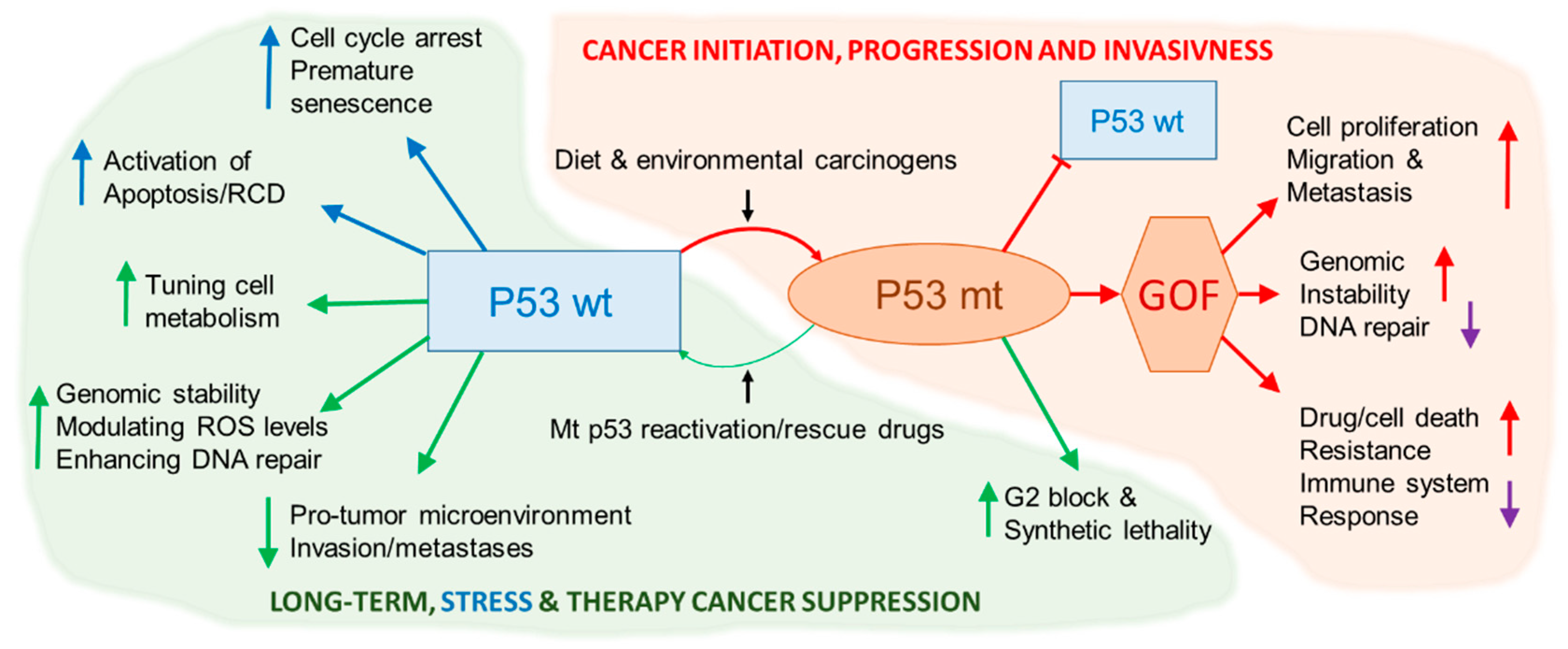{kind=link}
| TP53 Status | DNA Repair Pathway | Effect on DNA Repair | Citation |
|---|---|---|---|
| TP53 missense mutation | BER | APE1 regulation | [41] |
| Hot-spot tumor-derived TP53 mutants | BER | Does not enhance activity of repair pathway in comparison with wt p53 | [39] |
| Arg273His mutant TP53 | NER | Does not inhibit XPD (Rad3) and XPB DNA helicase activities in comparison with wt p53 | [47] |
| p53 knock out | NER | Increase in chromosomal abnormalities | [48] |
| R248W and R273H mutant TP53 | HR | Interaction with MRE11; inter-chromosomal translocations | [50] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vodicka, P.; Andera, L.; Opattova, A.; Vodickova, L. The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction. Cancers 2021, 13, 479. https://doi.org/10.3390/cancers13030479
Vodicka P, Andera L, Opattova A, Vodickova L. The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction. Cancers. 2021; 13(3):479. https://doi.org/10.3390/cancers13030479
Chicago/Turabian StyleVodicka, Pavel, Ladislav Andera, Alena Opattova, and Ludmila Vodickova. 2021. "The Interactions of DNA Repair, Telomere Homeostasis, and p53 Mutational Status in Solid Cancers: Risk, Prognosis, and Prediction" Cancers 13, no. 3: 479. https://doi.org/10.3390/cancers13030479