Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer

 , , , , , , , , , , and add
Show full author list
, , , , , , , , , , and add
Show full author list
Abstract
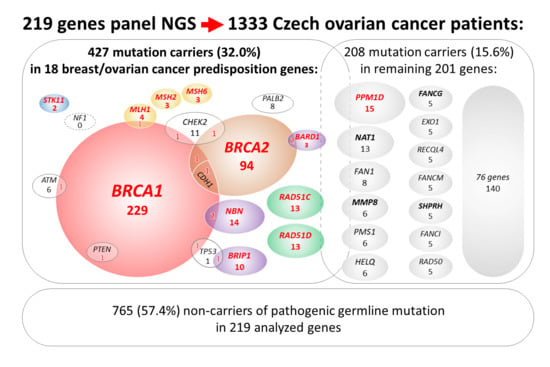
:
1. Introduction
2. Results
2.1. Description of Study Population
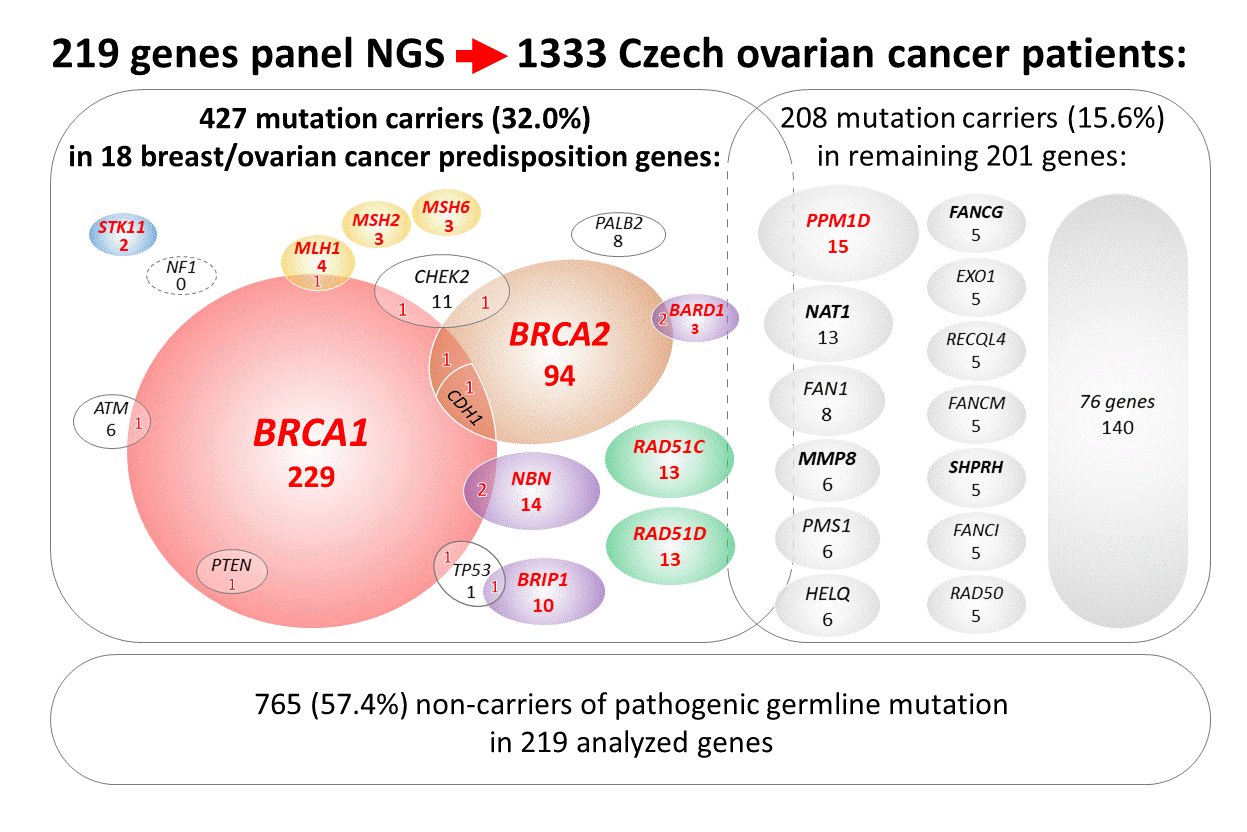
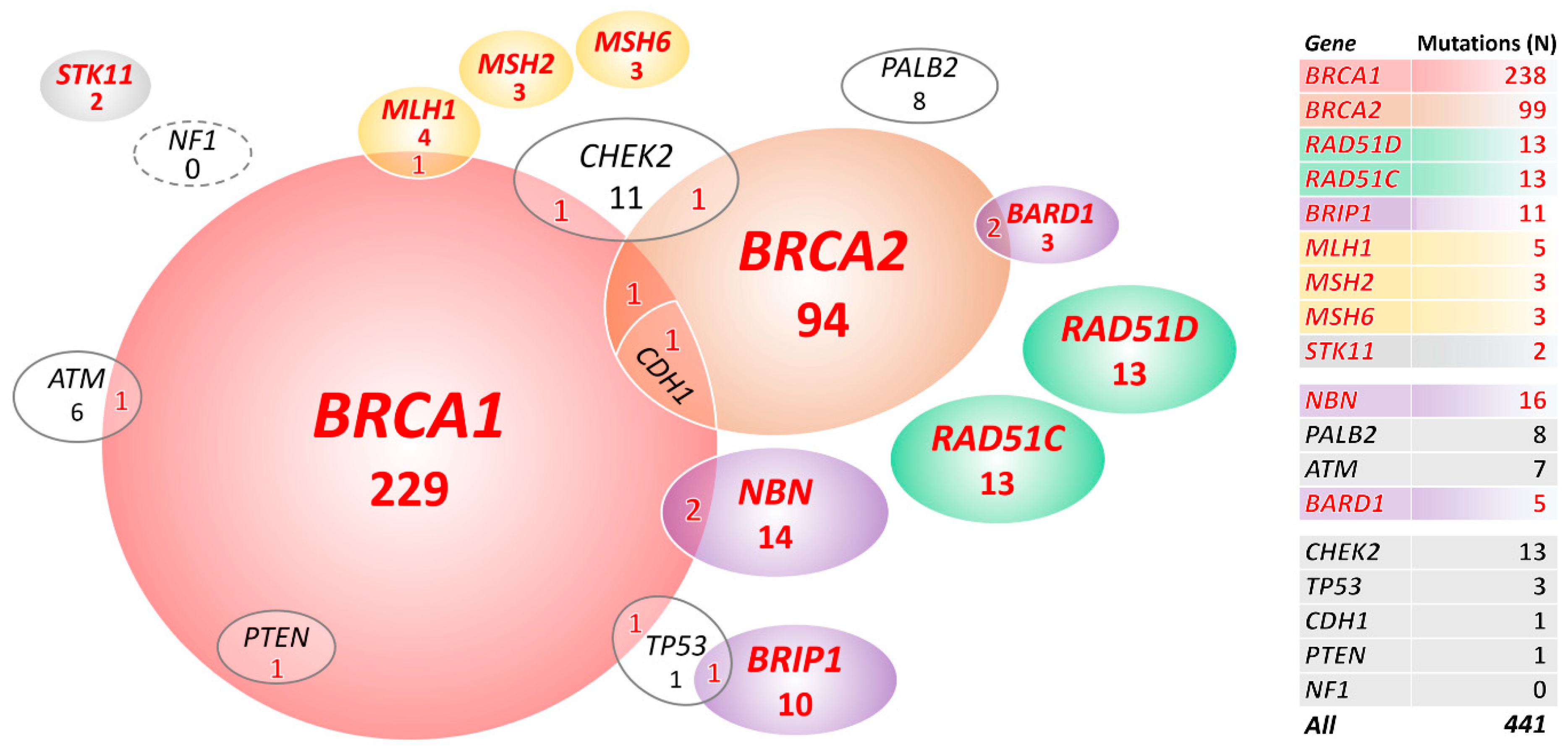
2.2. Mutations in 18 Known/Anticipated Hereditary BC/OC Genes
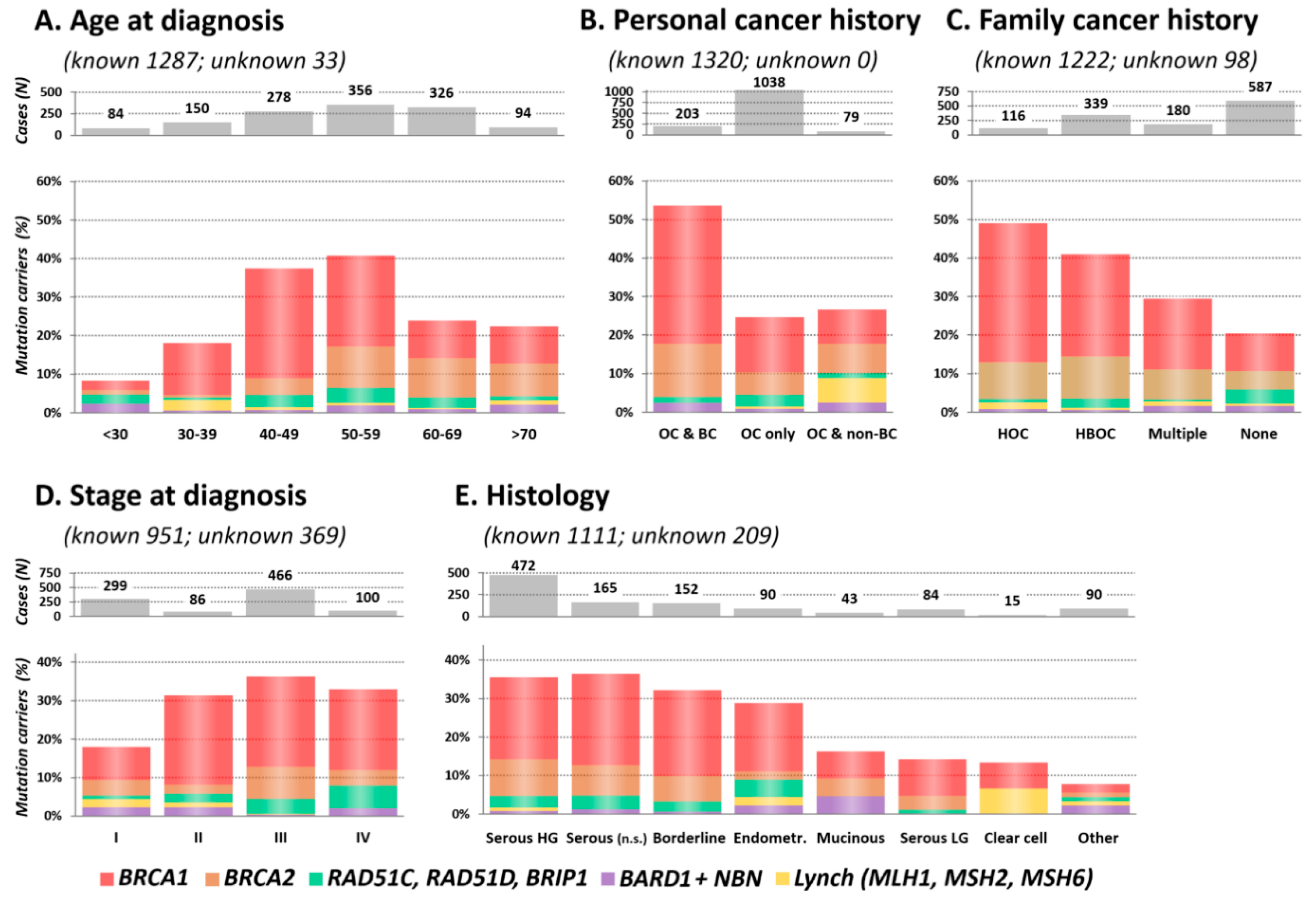
2.3. Clinical and Histopathological Characteristics of Mutation Carriers
2.3.1. Age at OC Diagnosis
2.3.2. Personal and Family Cancer History
2.3.3. Stage and Histology
2.4. Mutations in Additional 201 Analyzed Genes
3. Discussion
4. Materials and Methods
4.1. Next-Generation Sequencing
4.2. Variant Classification
4.3. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levanon, K.; Crum, C.; Drapkin, R. New insights into the pathogenesis of serous ovarian cancer and its clinical impact. J. Clin. Oncol. 2008, 26, 5284–5293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Park, E.Y.; Kim, O.; Schilder, J.M.; Coffey, D.M.; Cho, C.H.; Bast, R.C., Jr. Cell Origins of High-Grade Serous Ovarian Cancer. Cancers (Basel) 2018, 10, 433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. CA A Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, T.; Casadei, S.; Lee, M.K.; Pennil, C.C.; Nord, A.S.; Thornton, A.M.; Roeb, W.; Agnew, K.J.; Stray, S.M.; Wickramanayake, A.; et al. Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc. Natl. Acad. Sci. USA 2011, 108, 18032–18037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczkowska, M.; Krawczynska, N.; Stukan, M.; Kuzniacka, A.; Brozek, I.; Sniadecki, M.; Debniak, J.; Wydra, D.; Biernat, W.; Kozlowski, P.; et al. Spectrum and Prevalence of Pathogenic Variants in Ovarian Cancer Susceptibility Genes in a Group of 333 Patients. Cancers (Basel) 2018, 10, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krivokuca, A.; Boljevic, I.; Jovandic, S.; Magic, Z.; Mandic, A.; Tomasevic, Z.; Brankovic-Magic, M. Germline mutations in cancer susceptibility genes in high grade serous ovarian cancer in Serbia. J. Hum. Genet. 2019. [Google Scholar] [CrossRef]
- Offit, K. BRCA mutation frequency and penetrance: New data, old debate. J. Natl. Cancer Inst. 2006, 98, 1675–1677. [Google Scholar] [CrossRef]
- Norquist, B.M.; Harrell, M.I.; Brady, M.F.; Walsh, T.; Lee, M.K.; Gulsuner, S.; Bernards, S.S.; Casadei, S.; Yi, Q.; Burger, R.A.; et al. Inherited Mutations in Women With Ovarian Carcinoma. JAMA Oncol. 2016, 2, 482–490. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ruark, E.; Xicola, R.M.; Ramsay, E.; Hughes, D.; Warren-Perry, M.; Snape, K.; Breast Cancer Susceptibility, C.; Eccles, D.; et al. Germline RAD51C mutations confer susceptibility to ovarian cancer. Nat. Genet. 2012, 44, 475–476, author reply 476. [Google Scholar] [CrossRef]
- Loveday, C.; Turnbull, C.; Ramsay, E.; Hughes, D.; Ruark, E.; Frankum, J.R.; Bowden, G.; Kalmyrzaev, B.; Warren-Perry, M.; Snape, K.; et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat. Genet. 2011, 43, 879–882. [Google Scholar] [CrossRef]
- Watson, P.; Butzow, R.; Lynch, H.T.; Mecklin, J.P.; Jarvinen, H.J.; Vasen, H.F.; Madlensky, L.; Fidalgo, P.; Bernstein, I.; International Collaborative Group on, H. The clinical features of ovarian cancer in hereditary nonpolyposis colorectal cancer. Gynecol. Oncol. 2001, 82, 223–228. [Google Scholar] [CrossRef]
- Ramus, S.J.; Song, H.; Dicks, E.; Tyrer, J.P.; Rosenthal, A.N.; Intermaggio, M.P.; Fraser, L.; Gentry-Maharaj, A.; Hayward, J.; Philpott, S.; et al. Germline Mutations in the BRIP1, BARD1, PALB2, and NBN Genes in Women With Ovarian Cancer. J. Natl. Cancer Inst. 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Banno, K.; Kisu, I.; Yanokura, M.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Hirasawa, A.; Aoki, D. Hereditary gynecological tumors associated with Peutz-Jeghers syndrome (Review). Oncol. Lett. 2013, 6, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Lilyquist, J.; LaDuca, H.; Polley, E.; Davis, B.T.; Shimelis, H.; Hu, C.; Hart, S.N.; Dolinsky, J.S.; Couch, F.J.; Goldgar, D.E. Frequency of mutations in a large series of clinically ascertained ovarian cancer cases tested on multi-gene panels compared to reference controls. Gynecol. Oncol. 2017, 147, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Carter, N.J.; Marshall, M.L.; Susswein, L.R.; Zorn, K.K.; Hiraki, S.; Arvai, K.J.; Torene, R.I.; McGill, A.K.; Yackowski, L.; Murphy, P.D.; et al. Germline pathogenic variants identified in women with ovarian tumors. Gynecol. Oncol. 2018, 151, 481–488. [Google Scholar] [CrossRef]
- Schubert, S.; van Luttikhuizen, J.L.; Auber, B.; Schmidt, G.; Hofmann, W.; Penkert, J.; Davenport, C.F.; Hille-Betz, U.; Wendeburg, L.; Bublitz, J.; et al. The identification of pathogenic variants in BRCA1/2 negative, high risk, hereditary breast and/or ovarian cancer patients: High frequency of FANCM pathogenic variants. Int. J. Cancer 2019, 144, 2683–2694. [Google Scholar] [CrossRef]
- Daly, M.B.; Pilarski, R.; Berry, M.; Buys, S.S.; Farmer, M.; Friedman, S.; Garber, J.E.; Kauff, N.D.; Khan, S.; Klein, C.; et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast and Ovarian, Version 2.2017. J. Natl. Compr. Cancer Netw. JNCCN 2017, 15, 9–20. [Google Scholar] [CrossRef]
- Domchek, S.M.; Friebel, T.M.; Neuhausen, S.L.; Wagner, T.; Evans, G.; Isaacs, C.; Garber, J.E.; Daly, M.B.; Eeles, R.; Matloff, E.; et al. Mortality after bilateral salpingo-oophorectomy in BRCA1 and BRCA2 mutation carriers: A prospective cohort study. Lancet. Oncol. 2006, 7, 223–229. [Google Scholar] [CrossRef]
- Soukupova, J.; Zemankova, P.; Lhotova, K.; Janatova, M.; Borecka, M.; Stolarova, L.; Lhota, F.; Foretova, L.; Machackova, E.; Stranecky, V.; et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 2018, 13, e0195761. [Google Scholar] [CrossRef] [Green Version]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study, G. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef]
- Rafnar, T.; Gudbjartsson, D.F.; Sulem, P.; Jonasdottir, A.; Sigurdsson, A.; Jonasdottir, A.; Besenbacher, S.; Lundin, P.; Stacey, S.N.; Gudmundsson, J.; et al. Mutations in BRIP1 confer high risk of ovarian cancer. Nat. Genet. 2011, 43, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Bonache, S.; Esteban, I.; Moles-Fernandez, A.; Tenes, A.; Duran-Lozano, L.; Montalban, G.; Bach, V.; Carrasco, E.; Gadea, N.; Lopez-Fernandez, A.; et al. Multigene panel testing beyond BRCA1/2 in breast/ovarian cancer Spanish families and clinical actionability of findings. J. Cancer Res. Clin. Oncol. 2018, 144, 2495–2513. [Google Scholar] [CrossRef]
- Lynch, H.T.; de la Chapelle, A. Hereditary colorectal cancer. N. Engl. J. Med. 2003, 348, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.M.; Li, S.; Black, M.H.; Lee, S.; Hoiness, R.; Wu, S.; Mu, W.; Huether, R.; Chen, J.; Sridhar, S.; et al. Association of Breast and Ovarian Cancers With Predisposition Genes Identified by Large-Scale Sequencing. JAMA Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kurian, A.W.; Ward, K.C.; Howlader, N.; Deapen, D.; Hamilton, A.S.; Mariotto, A.; Miller, D.; Penberthy, L.S.; Katz, S.J. Genetic Testing and Results in a Population-Based Cohort of Breast Cancer Patients and Ovarian Cancer Patients. J. Clin. Oncol. 2019, JCO1801854. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Leslie, G.; Doroszuk, A.; Schneider, S.; Allen, J.; Decker, B.; Dunning, A.M.; Redman, J.; Scarth, J.; Plaskocinska, I.; et al. Cancer Risks Associated With Germline PALB2 Pathogenic Variants: An International Study of 524 Families. J. Clin. Oncol. 2020, 38, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 2019. [Google Scholar] [CrossRef]
- Harter, P.; Hauke, J.; Heitz, F.; Reuss, A.; Kommoss, S.; Marme, F.; Heimbach, A.; Prieske, K.; Richters, L.; Burges, A.; et al. Prevalence of deleterious germline variants in risk genes including BRCA1/2 in consecutive ovarian cancer patients (AGO-TR-1). PLoS ONE 2017, 12, e0186043. [Google Scholar] [CrossRef] [PubMed]
- Whitworth, J.; Skytte, A.B.; Sunde, L.; Lim, D.H.; Arends, M.J.; Happerfield, L.; Frayling, I.M.; van Minkelen, R.; Woodward, E.R.; Tischkowitz, M.D.; et al. Multilocus Inherited Neoplasia Alleles Syndrome: A Case Series and Review. JAMA Oncol. 2016, 2, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Stradella, A.; Del Valle, J.; Rofes, P.; Feliubadalo, L.; Grau Garces, E.; Velasco, A.; Gonzalez, S.; Vargas, G.; Izquierdo, A.; Campos, O.; et al. Does multilocus inherited neoplasia alleles syndrome have severe clinical expression? J. Med. Genet. 2018. [Google Scholar] [CrossRef] [Green Version]
- Whitworth, J.; Smith, P.S.; Martin, J.E.; West, H.; Luchetti, A.; Rodger, F.; Clark, G.; Carss, K.; Stephens, J.; Stirrups, K.; et al. Comprehensive Cancer-Predisposition Gene Testing in an Adult Multiple Primary Tumor Series Shows a Broad Range of Deleterious Variants and Atypical Tumor Phenotypes. Am. J. Hum. Genet. 2018, 103, 3–18. [Google Scholar] [CrossRef] [Green Version]
- Castera, L.; Harter, V.; Muller, E.; Krieger, S.; Goardon, N.; Ricou, A.; Rousselin, A.; Paimparay, G.; Legros, A.; Bruet, O.; et al. Landscape of pathogenic variations in a panel of 34 genes and cancer risk estimation from 5131 HBOC families. Genet. Med. 2018, 20, 1677–1686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, R.D.; Burghel, G.J.; Flaum, N.; Bulman, M.; Clamp, A.R.; Hasan, J.; Mitchell, C.L.; Schlecht, H.; Woodward, E.R.; Lallo, F.I.; et al. Prevalence of germline pathogenic BRCA1/2 variants in sequential epithelial ovarian cancer cases. J. Med. Genet. 2019, 56, 301–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaskocinska, I.; Shipman, H.; Drummond, J.; Thompson, E.; Buchanan, V.; Newcombe, B.; Hodgkin, C.; Barter, E.; Ridley, P.; Ng, R.; et al. New paradigms for BRCA1/BRCA2 testing in women with ovarian cancer: Results of the Genetic Testing in Epithelial Ovarian Cancer (GTEOC) study. J. Med. Genet. 2016, 53, 655–661. [Google Scholar] [CrossRef] [Green Version]
- Rust, K.; Spiliopoulou, P.; Tang, C.Y.; Bell, C.; Stirling, D.; Phang, T.; Davidson, R.; Mackean, M.; Nussey, F.; Glasspool, R.M.; et al. Routine germline BRCA1 and BRCA2 testing in patients with ovarian carcinoma: Analysis of the Scottish real-life experience. BJOG 2018, 125, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Ruark, E.; Snape, K.; Humburg, P.; Loveday, C.; Bajrami, I.; Brough, R.; Rodrigues, D.N.; Renwick, A.; Seal, S.; Ramsay, E.; et al. Mosaic PPM1D mutations are associated with predisposition to breast and ovarian cancer. Nature 2013, 493, 406–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbari, M.R.; Lepage, P.; Rosen, B.; McLaughlin, J.; Risch, H.; Minden, M.; Narod, S.A. PPM1D mutations in circulating white blood cells and the risk for ovarian cancer. J. Natl. Cancer Inst. 2014, 106, djt323. [Google Scholar] [CrossRef] [Green Version]
- Kleiblova, P.; Shaltiel, I.A.; Benada, J.; Sevcik, J.; Pechackova, S.; Pohlreich, P.; Voest, E.E.; Dundr, P.; Bartek, J.; Kleibl, Z.; et al. Gain-of-function mutations of PPM1D/Wip1 impair the p53-dependent G1 checkpoint. J. Cell Biol. 2013, 201, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Pharoah, P.D.P.; Song, H.; Dicks, E.; Intermaggio, M.P.; Harrington, P.; Baynes, C.; Alsop, K.; Australian Ovarian Cancer Study, G.; Bogdanova, N.; Cicek, M.S.; et al. PPM1D Mosaic Truncating Variants in Ovarian Cancer Cases May Be Treatment-Related Somatic Mutations. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [Green Version]
- Hein, D.W.; Fakis, G.; Boukouvala, S. Functional expression of human arylamine N-acetyltransferase NAT1*10 and NAT1*11 alleles: A mini review. Pharm. Genom. 2018, 28, 238–244. [Google Scholar] [CrossRef]
- Butcher, N.J.; Minchin, R.F. Arylamine N-acetyltransferase 1: A novel drug target in cancer development. Pharm. Rev. 2012, 64, 147–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seelinger, M.; Otterlei, M. Helicase-Like Transcription Factor HLTF and E3 Ubiquitin Ligase SHPRH Confer DNA Damage Tolerance through Direct Interactions with Proliferating Cell Nuclear Antigen (PCNA). Int. J. Mol. Sci. 2020, 21, 693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalepa, G.; Clapp, D.W. Fanconi anaemia and cancer: An intricate relationship. Nat. Rev. Cancer 2018, 18, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Bao, Y.; Riaz, M.; Tiller, J.; Liew, D.; Zhuang, X.; Amor, D.J.; Huq, A.; Petelin, L.; Nelson, M.; et al. Population genomic screening of all young adults in a health-care system: A cost-effectiveness analysis. Genet. Med. 2019. [Google Scholar] [CrossRef]
- Best, A.F.; Tucker, M.A.; Frone, M.N.; Greene, M.H.; Peters, J.A.; Katki, H.A. A Pragmatic Testing-Eligibility Framework for Population Mutation Screening: The Example of BRCA1/2. Cancer Epidemiol Biomark. Prev. 2019, 28, 293–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turnbull, C.; Sud, A.; Houlston, R.S. Cancer genetics, precision prevention and a call to action. Nat. Genet. 2018, 50, 1212–1218. [Google Scholar] [CrossRef]
- Gabai-Kapara, E.; Lahad, A.; Kaufman, B.; Friedman, E.; Segev, S.; Renbaum, P.; Beeri, R.; Gal, M.; Grinshpun-Cohen, J.; Djemal, K.; et al. Population-based screening for breast and ovarian cancer risk due to BRCA1 and BRCA2. Proc. Natl. Acad. Sci. USA 2014, 111, 14205–14210. [Google Scholar] [CrossRef] [Green Version]
- Manchanda, R.; Patel, S.; Gordeev, V.S.; Antoniou, A.C.; Smith, S.; Lee, A.; Hopper, J.L.; MacInnis, R.J.; Turnbull, C.; Ramus, S.J.; et al. Cost-effectiveness of Population-Based BRCA1, BRCA2, RAD51C, RAD51D, BRIP1, PALB2 Mutation Testing in Unselected General Population Women. J. Natl. Cancer Inst. 2018, 110, 714–725. [Google Scholar] [CrossRef]
- George, A.; Kaye, S.; Banerjee, S. Delivering widespread BRCA testing and PARP inhibition to patients with ovarian cancer. Nat. Rev. Clin. Oncol. 2017, 14, 284–296. [Google Scholar] [CrossRef]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet. Oncol. 2014, 15, 852–861, Correction in 2015, 16, e158. [Google Scholar] [CrossRef]
- Chandran, E.A.; Kennedy, I. Significant Tumor Response to the Poly (ADP-ribose) Polymerase Inhibitor Olaparib in Heavily Pretreated Patient With Ovarian Carcinosarcoma Harboring a Germline RAD51D Mutation. JCO Precis. Oncol. 2018, 1–4. [Google Scholar] [CrossRef]
- Ngoi, N.Y.L.; Tay, D.; Heong, V.; Thian, Y.L.; Ong, P.Y.; Ow, S.G.W.; Jeyasekharan, A.D.; Lim, Y.W.; Lim, S.E.; Lee, S.C.; et al. Reversal of Bowel Obstruction with Platinum-Based Chemotherapy and Olaparib in Recurrent, Short Platinum-Free Interval, RAD51C Germline Mutation–Associated Ovarian Cancer. JCO Precis. Oncol. 2018, 1–8. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | 1320 OC Patients (a) N Mutations (%) | 2278 PMC N mutations (%) | OR (95% CI); p (a) |
|---|---|---|---|
| Increased OC risk (b) | |||
| BRCA1 (c) | 229 (17.35) | 5 (0.22) | 95.2 (40.1–295.2); 1.83 × 10−97 |
| BRCA2 (c) | 94 (7.12) | 7 (0.31) | 24.9 (11.6–63.6); 1.16 × 10−33 |
| RAD51D | 13 (0.98) | 2 (0.09) | 11.3 (2.6–103.4); 9.66 × 10−5 |
| RAD51C | 13 (0.98) | 4 (0.18) | 5.7 (1.7–23.8); 0.001 |
| BRIP1 (c) | 10 (0.76) | 5 (0.22) | 3.5 (1.1–13); 0.03 |
| MLH1 (c) | 4 (0.3) | 1 (0.04) | 6.9 (0.7–340.4); 0.06 (d) |
| MSH2 | 3 (0.23) | 0 | 0.049 (d) |
| MSH6 | 3 (0.23) | 0 | 0.049 (d) |
| STK11 | 2 (0.15) | 0 | 0.13 |
| Potentially increase or insufficient evidence OC risk (b) | |||
| NBN (c) | 14 (1.06) | 7 (0.31) | 3.5 (1.3–10.2); 0.006 |
| PALB2 | 8 (0.61) | 9 (0.40) | 1.5 (0.5–4.5); 0.45 |
| ATM (c) | 6 (0.45) | 8 (0.35) | 1.3 (0.4–4.3); 0.78 |
| BARD1 (c) | 3 (0.23) | 0 | 0.049 |
| No increased risk of OC (b) | |||
| CHEK2 (c) | 11 (0.83) | 8 (0.35) | 2.4 (0.9–6.8); 0.06 |
| TP53(c) | 1 (0.08) | 2 (0.09) | 0.9 (0–16.6); 1 |
| CDH1(c) | 0 | 0 | - |
| PTEN (c) | 0 | 0 | - |
| NF1 | 0 | 0 | - |
| Gene | Patients N Mutations (%) | 2278 PMC N Mutations (%) | OR (95% CI); p (Bonferroni Corrected p) |
|---|---|---|---|
| All 1333 OC patients | |||
| PPM1D | 16 (1.20) | 2 (0.09) | 13.82 (3.24–124.22); 7.4 × 10−6 (0.001) |
| NAT1 | 13 (0.98) | 5 (0.22) | 4.48 (1.49–16.07); 0.003 (n.s.) |
| SHPRH | 5 (0.38) | 1 (0.04) | 8.57 (0.96–404.83); 0.028 (n.s.) |
| 934 OC patients without mutations in 10 genes significantly associated with OC in our study | |||
| PPM1D | 12 (1.28) | 2 (0.09) | 14.80 (3.28–136.67); 1.7 × 10−5 (0.003) |
| NAT1 | 8 (0.86) | 5 (0.22) | 3.96 (1.13–15.30); 0.026 (n.s.) |
| MMP8 | 6 (0.64) | 4 (0.18) | 3.67 (0.87–17.74); 0.041 (n.s.) |
| FANCG | 5 (0.53) | 2 (0.09) | 6.12 (1.00–64.45); 0.025 (n.s.) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lhotova, K.; Stolarova, L.; Zemankova, P.; Vocka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. https://doi.org/10.3390/cancers12040956
Lhotova K, Stolarova L, Zemankova P, Vocka M, Janatova M, Borecka M, Cerna M, Jelinkova S, Kral J, Volkova Z, et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers. 2020; 12(4):956. https://doi.org/10.3390/cancers12040956
Chicago/Turabian StyleLhotova, Klara, Lenka Stolarova, Petra Zemankova, Michal Vocka, Marketa Janatova, Marianna Borecka, Marta Cerna, Sandra Jelinkova, Jan Kral, Zuzana Volkova, and et al. 2020. "Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer" Cancers 12, no. 4: 956. https://doi.org/10.3390/cancers12040956