1. Introduction
The durability and reliability of power generation equipment are important parameters affecting the stability of the energy market. In the case of components of combustion plants, such as boiler firewalls, heat exchangers, or pressure vessels of coal-fired power plants, the service life is often limited by the corrosion attack of an aggressive environment, where hot corrosion is especially a major issue determining the service life of said components [
1,
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14]. Hot corrosion has been identified as a serious problem for many high-temperature aggressive environment applications, such as boilers [
1,
2,
4,
5,
6,
7,
8,
11,
12,
14], internal combustion engines, gas turbines [
15,
16], fluidized bed combustion boilers [
17], industrial waste incinerators [
18,
19], etc.
The most dominant substance known to promote hot corrosion processes is Na
2SO
4, mainly owing to its very good temperature stability in a wide range of oxygen partial pressures [
7]. Na
2SO
4 originates in oxidation reactions during the combustion of fuel containing both sulphur and sodium and possibly other alkali metals, such as potassium, which also form another corrosively aggressive sulphate attributed to the hot corrosion process—K
2SO
4 [
1,
7,
8,
10,
20]. Some other major aggressive substances known for their negative attribution to hot corrosion process are chlorides and vanadates [
7,
8]. A major role in the formation of highly corrosive sulphates is played by sulphur; papers [
21,
22] discuss some aspects of the role of sulphur and its contribution to corrosive processes in high-temperature environments.
Volatile alkalic sulphates (primarily Na
2SO
4) and chlorides (such as NaCl and KCl) in the gaseous state react readily with other elements of fly ash in a high-temperature oxidizing environment during the combustion process. These low-melting-point compounds condense and form deposits on the surface of boiler components. This leads to the oxidation, sulfidation, and chlorination of the material and to high-temperature corrosion. The corrosion rate increases with rising temperature [
1,
2,
3,
7,
8].
Ideally, a passivation oxide layer is formed on the surface of the material, acting as a diffusion barrier, and preventing further oxidation. Therefore, further growth of the oxide layer would require elements of the metal or oxidizing agent to be transported through the layer by solid state diffusion. In practice, the oxidation layer is often porous and cracked, which enables easier transport. In addition, ion diffusion is much faster through sulphates than through oxides. The molten salts act as a transport medium for the oxidizing agent into the material and for molten metal ions out of the material. In addition, chemical reactions are faster in the liquid state compared to the solid state. As far as the salt layer is concerned, the formation of the liquid state defines type I hot corrosion. In type I salt-induced hot corrosion, the temperature is higher than the melting temperature of the salt and therefore corrosion processes are significantly accelerated. A chemical reaction then takes place, which initially attacks the protective oxide film and continuously reduces the chromium (Cr) content of the substrate materials. As a result of the reduction in chromium content, oxidation of the substrate increases rapidly and porous deposits are formed [
5,
7,
8]. The reaction producing metal chlorides or sulphides near the oxide/metal consumes metallic elements, especially chromium. The metal elements are determined to build or repair the passivating oxide layer. In an oxidizing atmosphere, the oxide scale protecting the base iron-based high-temperature resistant alloy is usually mostly composed of chromium oxides and iron oxides. If alloy contains aluminum, alumina (Al
2O
3) is also found in the oxide layer [
8]. Solid salt deposits are deposited on the oxide scale and when the temperature rises above the melting point of the salt deposits, the salt mixture becomes liquid or partially liquid [
1,
2,
3,
4,
5,
6,
7]. This is when the temperature threshold at hot corrosion type I begins to occur. The actual melting temperature depends on the salt mixture composition, but the melting temperature of the salt mixture is usually below melting temperature of its component due to eutectic character of the mixture [
4,
5,
6,
7,
8,
9].
During the initial stage of the corrosion process, when the salt deposit is melted, a molten sulphate salt film is formed on the oxide layer of the base material and the dissolution of oxides occurs (Equation (1) [
2] for Cr
2O
3) and is promoted by an oxidizing atmosphere.
Sodium chloride reacts with the oxides, releasing chlorine, which reacts further with the oxides to form volatile chlorides (Equation (2) shows a representative reaction with chromia, but basically the same reaction is for all presented metal oxides). [
2]
Chlorine in the gaseous state, Cl
2, can rapidly penetrate through the oxide layer along cracks and reacts with material elements, such as chromium, forming volatile chlorides (Equation (3)) [
3].
In the case of the thermal spray coating layer, volatile chlorides tend to diffuse along the splat boundaries to the interface of the coating and the substrate where they can oxidize again (Equation (4)). This restores chlorine and repeats the corrosion reaction. Numerous pits at the splat boundaries formed by volatile chlorides create another path for corrosive elements and facilitate the penetration of the corrosive environment into the material [
2,
7,
8].
Other material elements such as nickel or titanium are also subject to these reactions [
1,
2,
3,
4,
5,
7,
8].
If the oxide layer on the surface of the material does not have protective properties, chlorides can substantially accelerate oxidation. Dispersed gaseous chlorine can penetrate the corrosion layer and the coating surface and can cause a high number of cracks and pores to form (the exemplar reaction with the iron in the base material is displayed in Equation (5)). The reaction of ferrous chloride with oxygen (Equation (6)) produces ferric oxide, which reacts again with sodium chloride (Equation (7)). A similar process also takes place for chromia (Equation (9)) [
2,
6].
Moreover, if there is no protective oxide layer on the surface, or when the protective layer has been damaged, such as in the case described above, molten sulphates readily penetrate towards the base material. The reaction of the sulphates with the metal oxides leads to the formation of low-melting-point alkali metal oxides (Equations (7) and (8)).
In the case of the corrosive environment containing only sulphates, sulphur oxide reacts with ferric oxide and forms ferric sulphate (Equation (9)). However, if the corrosive environment also contains chlorides, sulphur dioxide may also react with sodium oxide to form sodium sulphate and chlorine. Sulphates cause corrosion at splat boundaries compared to chlorides that cause internal corrosion. The presence of sulphur in the form of sulphates can cause internal alloy sulfidation under the protective oxide layer. Only a small amount of sulphides is formed in the corrosion layer [
1,
3,
4,
7,
8].
To prolong the service life of components, a surface treatment can be applied to protect the steel parts from corrosion in an aggressive environment. Among other deposition technologies, the thermal spray technology offers a feasible and economically acceptable way to coat the surface with a thick layer of corrosion resistant materials such as MCrAlY and MCrAlYX-type alloys. In MCrAlY(X) alloys, M stands either for Ni, Co, or both, where X is a minor alloying element, such as Re, Ta, Si, Hf, or often their combination. In recent decades, many scientific papers have been written on the topic of MCrAlY(X) hot corrosion resistance in many applications, e.g., coal-fired boilers [
7,
8], waste-to-energy boilers [
18,
19,
22], biomass boilers [
1,
11,
20], gas turbines [
15,
16], etc. In the experiment presented in this paper, several commercial MCrAlY(X) coatings deposited by HVOF technology are compared. In addition to these coatings, two conventional base materials that are commonly used for heat exchange tubes and firewalls of power plants are also tested—steel P91 and alloy 800 H. The aim of this study is to present a specific comparison of the potential benefits of increased corrosion resistance when using a coating in critically corroded areas of combustion plant heat exchanger surfaces compared to the corrosion resistance of the aforementioned conventional base materials of said components. The corrosive environment for the test is a salt mixture of 75% Na
2SO
4 and 25% NaCl, which is a simplified corrosive environment typical for the combustion chamber and flue gas path in energy producing boilers [
1,
6,
7,
8,
11,
19,
21,
22,
23].
2. Materials and Methods
Four commercially available powders were used to coat the specimens. These powders were Amperit 410.001 (NiCoCrAlY) with particle size suitable for HVOF (−45 + 22) mm, Amperit 421.001 (NiCoCrAlTaReY) particle size (−45 + 22) mm, Amperit 405.001 (NiCoCrAlYHfSi) particle size (−45 + 22) mm, and Amperit 469.001 (CoCrAlYTaCSi) particle size (−53 + 20) mm. All coatings were deposited using HP/HVOF (High Pressure/High Velocity Oxygen Fuel) technology with JP-5000 torch from the TAFA Incorporated. Previously optimized spraying parameters were used for the coating application. Stainless high-temperature-resistant steel 1.4923 was used as a base material for coatings. Further, uncoated P91 steel and alloy 800 H samples were used as a reference specimen representing an uncoated firewall or heat exchange pipe in the same environment.
The substrate surfaces were degreased and grit-blasted prior to spraying to achieve proper adhesion between the coatings and the base materials. Brown corundum F22 with grain size (0.8 to 1.0 mm) was used as abrasive medium. The applied coatings thickness ranged from 250 to 320 μm.
Hot corrosion test was based on exposure to a corrosive salt mixture at 800 °C. The test procedure was as follows: the specimens were first polished to the surface roughness of Ra
max. = 1 µm. The next step included cleaning with alcohol and heating in an oven at 250 °C. Heating is necessary for proper adhesion of the salt layer. For the simulation of hot corrosion conditions and result comparison, a mixture of salts of 75 wt.% Na
2SO
4 and 25 wt.% NaCl was chosen, which is approximately 55 mol% of Na
2SO
4 and 45 mol% of NaCl. This composition is close to the eutectic composition of NaCl/Na
2SO
4 that is about a 59:41 molar ratio. The melting temperature of NaCl/Na
2SO
4 eutectic mixture is 626 °C [
9], and the mixture used in test presented in this paper is close enough to the eutectic point of this mixture to become sufficiently melted when exposed to 800 °C. Thus, the test was performed in more aggressive type I salt-induced hot corrosion [
7,
8,
9]. This mixture of salts was mixed with alcohol and applied on the surface of evaluated specimens in the amount of (3–5) g/cm
2. This step was followed by drying of the applied mixture for 3 h in an oven at 100 °C. Before testing in furnace, each specimen was weighed and the weight was subsequently measured after each test cycle. To compare the results easily, the number of cycles was set at 50. Each cycle consisted of one hour in silicon carbide furnace and subsequent cooling for 20 min at room temperature.
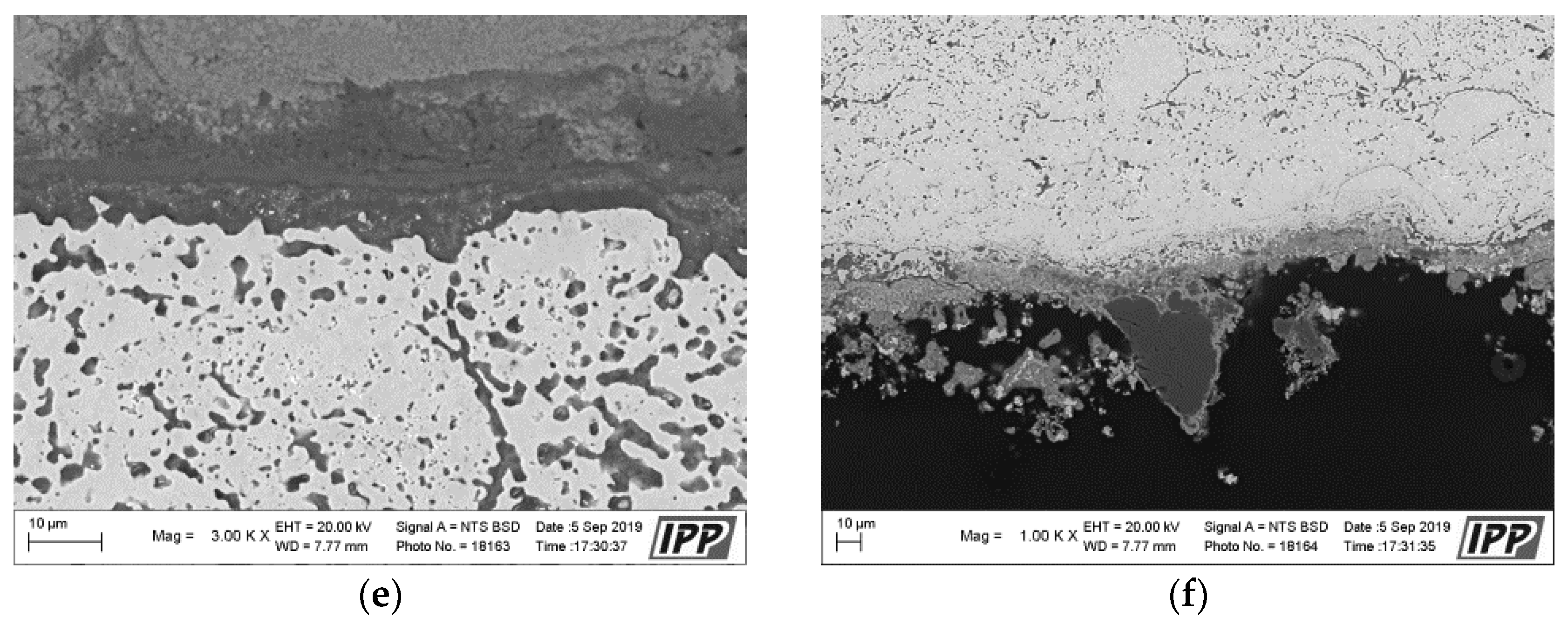
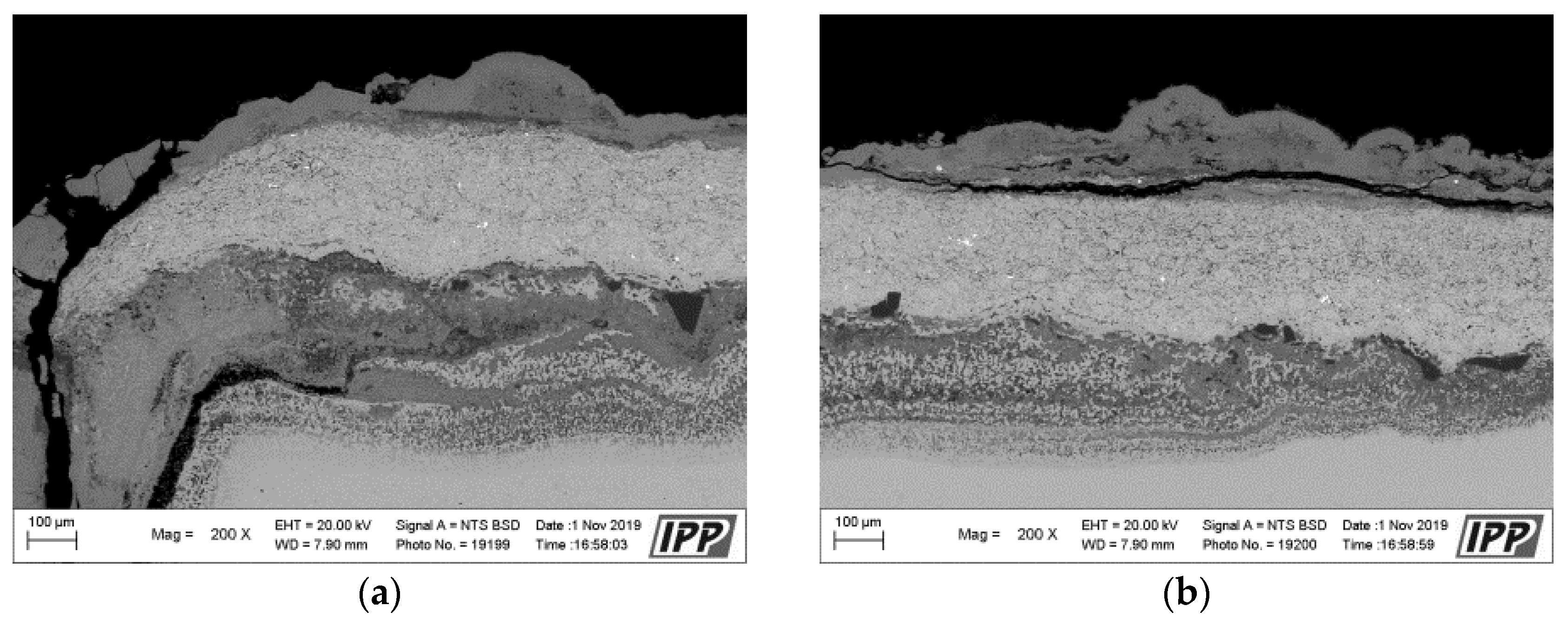
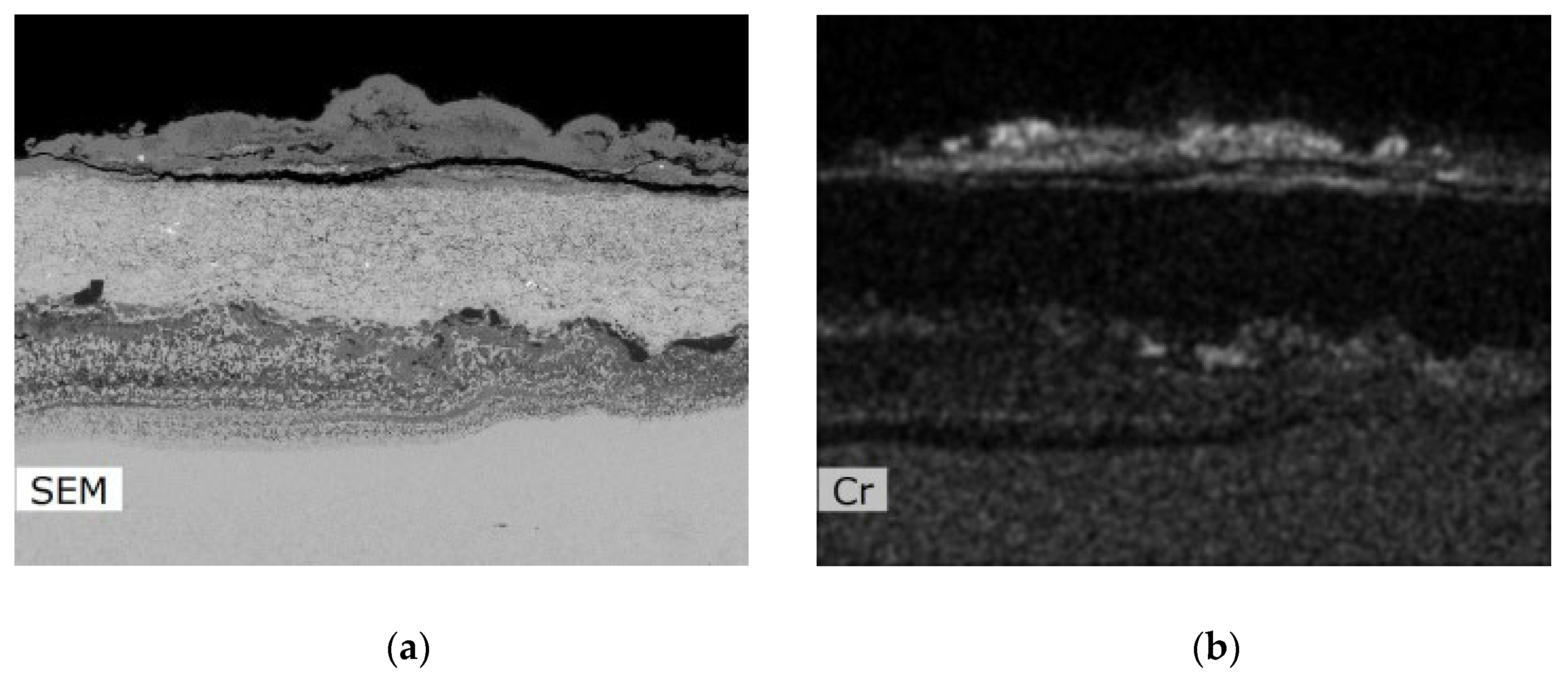
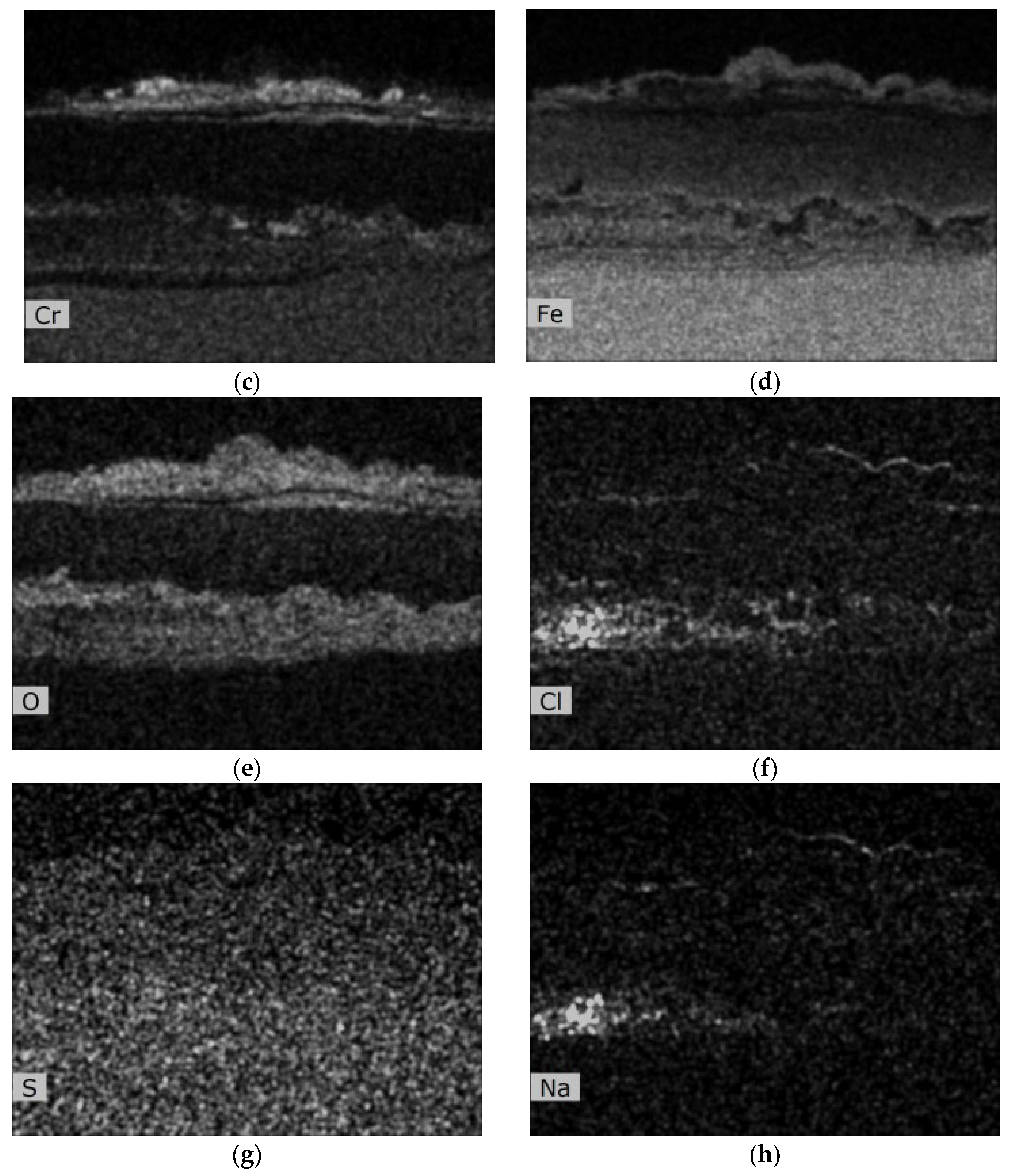
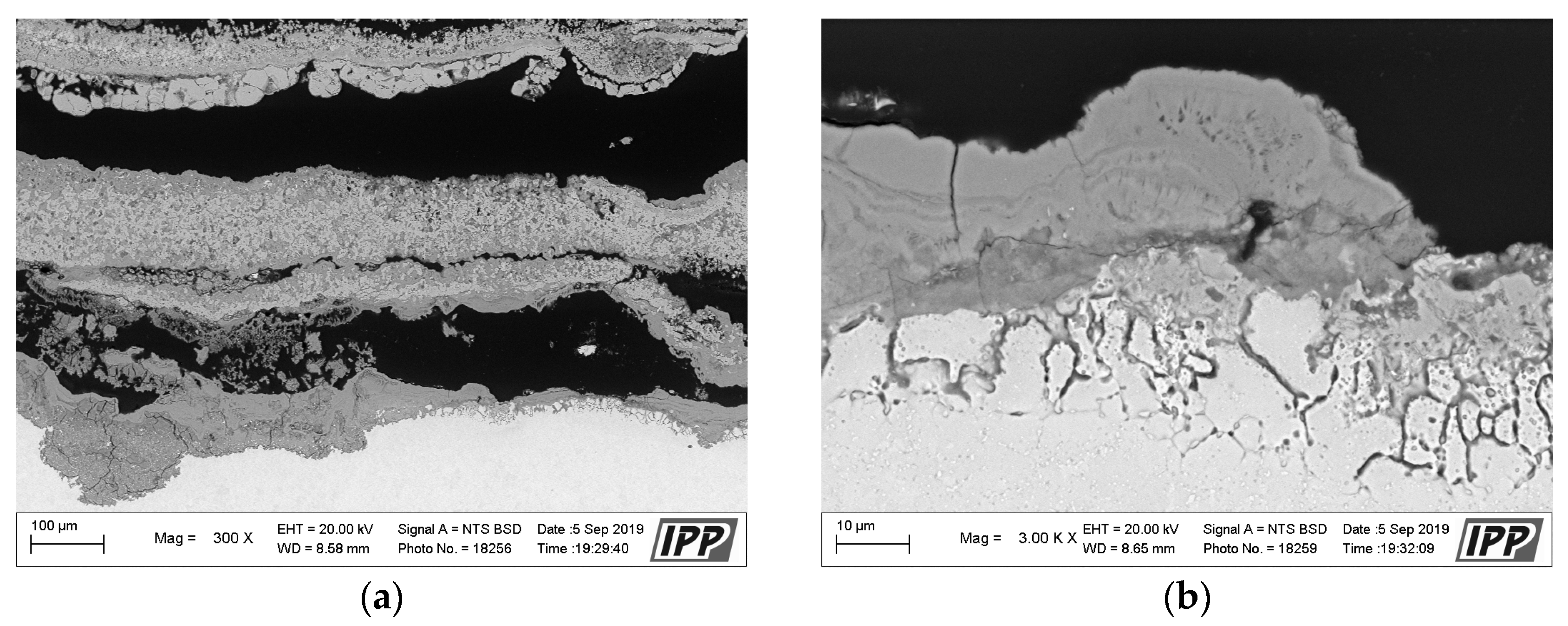
Metallographic evaluation was conducted using a scanning electron microscope EVO MA 15 (Carl Zeiss SMT, Oberkochen, Germany) equipped with Quantax EDS system XFlash® 5010 (Bruker, Billerica, MA, USA). Exposed specimens were further evaluated by EDS elemental analysis to specify the changes in the chemical composition. It is important to mention that the specimen preparation for metallographic evaluation is not a standard procedure usually used after the corrosion testing, but it is an essential part of the microstructure study of thermal sprayed coatings. Corroded thermal sprayed coatings require special care when separating apart from the original specimen. Pure alcohol was used as a cutting coolant. The separated part was further rinsed again with alcohol. Lateral specimen sides and beveled sharp edges of coating were ground. It was also necessary to grind the bottom edge of the base material and remove any impurities to prevent the possible oxidation. Each specimen was further rinsed with alcohol and dried in warm air. The sample prepared in this way was finally cold-mounted in epoxy and polished. The corroded samples prepared in this manner were subsequently subjected to the analyses described above.
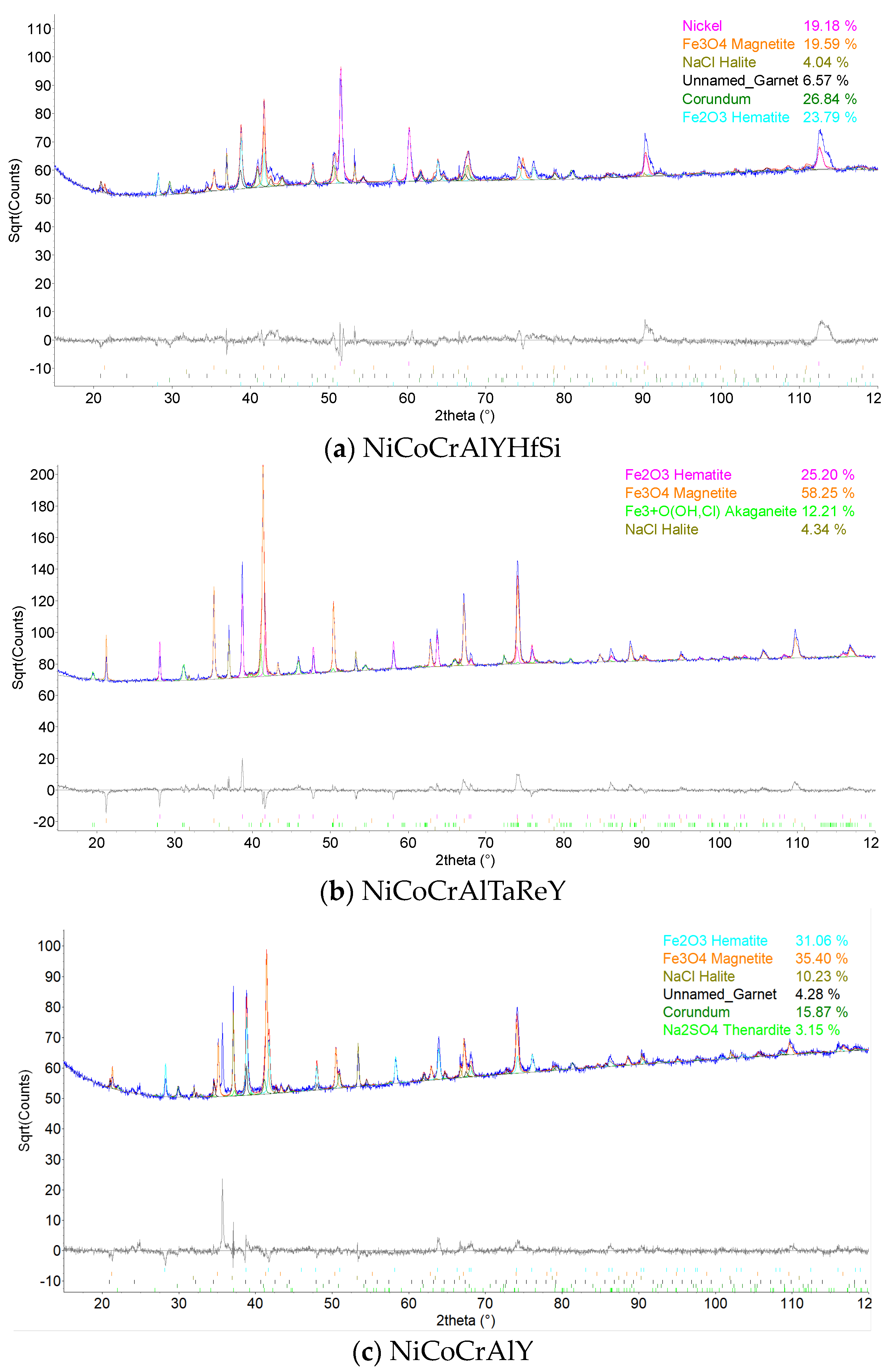
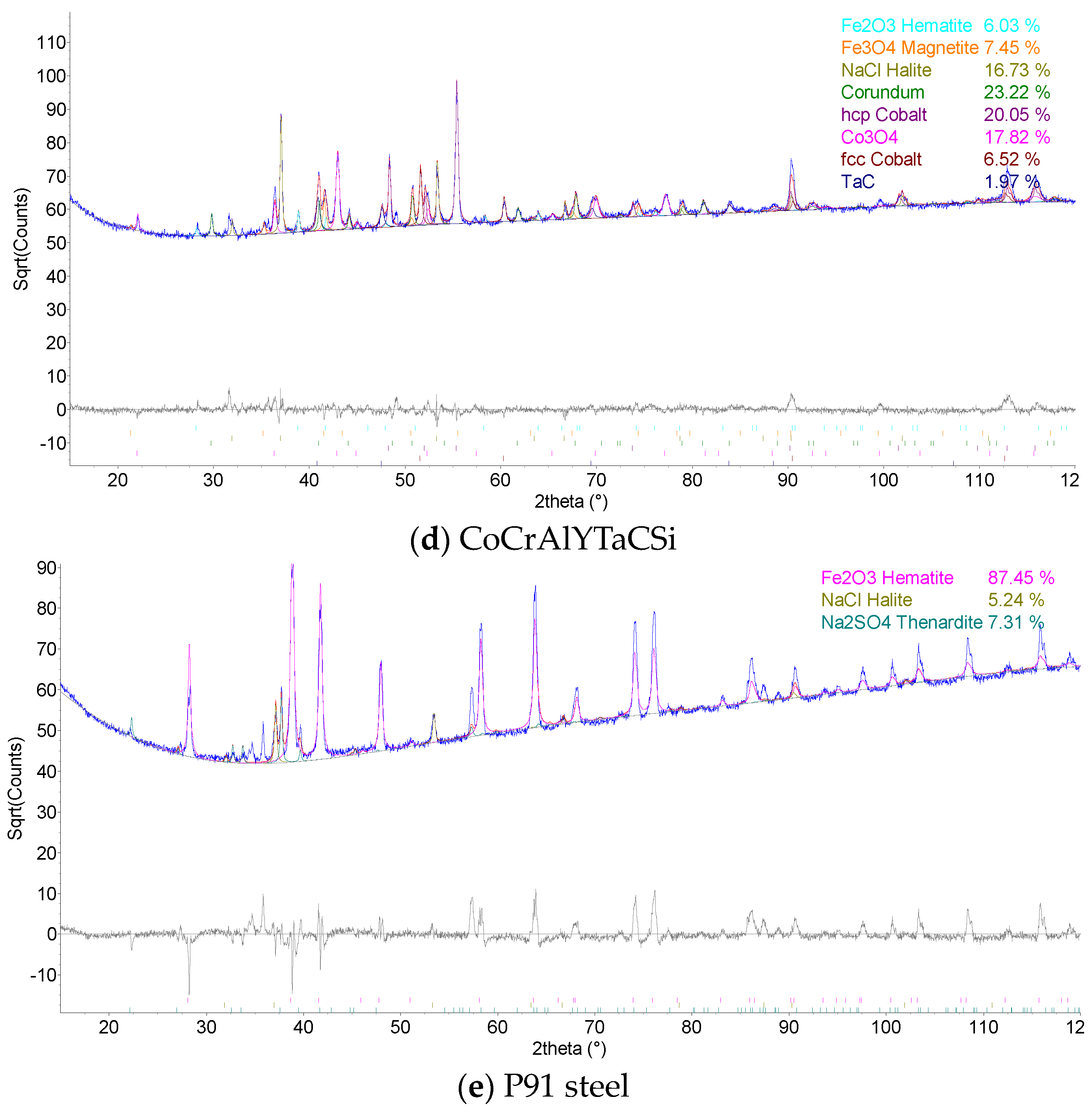
The coatings’ phase compositions were evaluated using X-ray diffraction (XRD), the D8 Discover powder diffractometer, Bruker, in Bragg–Brentano geometry with a 1D detector and CoKα radiation. The scanned region was from 15 to 100° 2θ with a 0.03° 2θ step size and with a 96 s counting time per step. The obtained diffraction patterns were subjected to quantitative Rietveld analysis performed in the TOPAS 5, which uses the so-called fundamental parameters approach. Crystalline phases in the XRD patterns were identified using the ICSD database.
4. Conclusions
The aim of this study was to evaluate the resistance of selected thermally sprayed coatings against the high-temperature corrosion in the aggressive environment of 75% Na2SO4 and 25% NaCl salt mixture and demonstrate a comparison of the potential increase in corrosion resistance when applying these selected commercial coatings. For this reason, in addition to the four commercial MCrAlY(X) coatings, P91 steel and 800 H alloy (both in the form of bulk material) are also tested in the present corrosion test as examples of some conventional base materials used in power equipment.
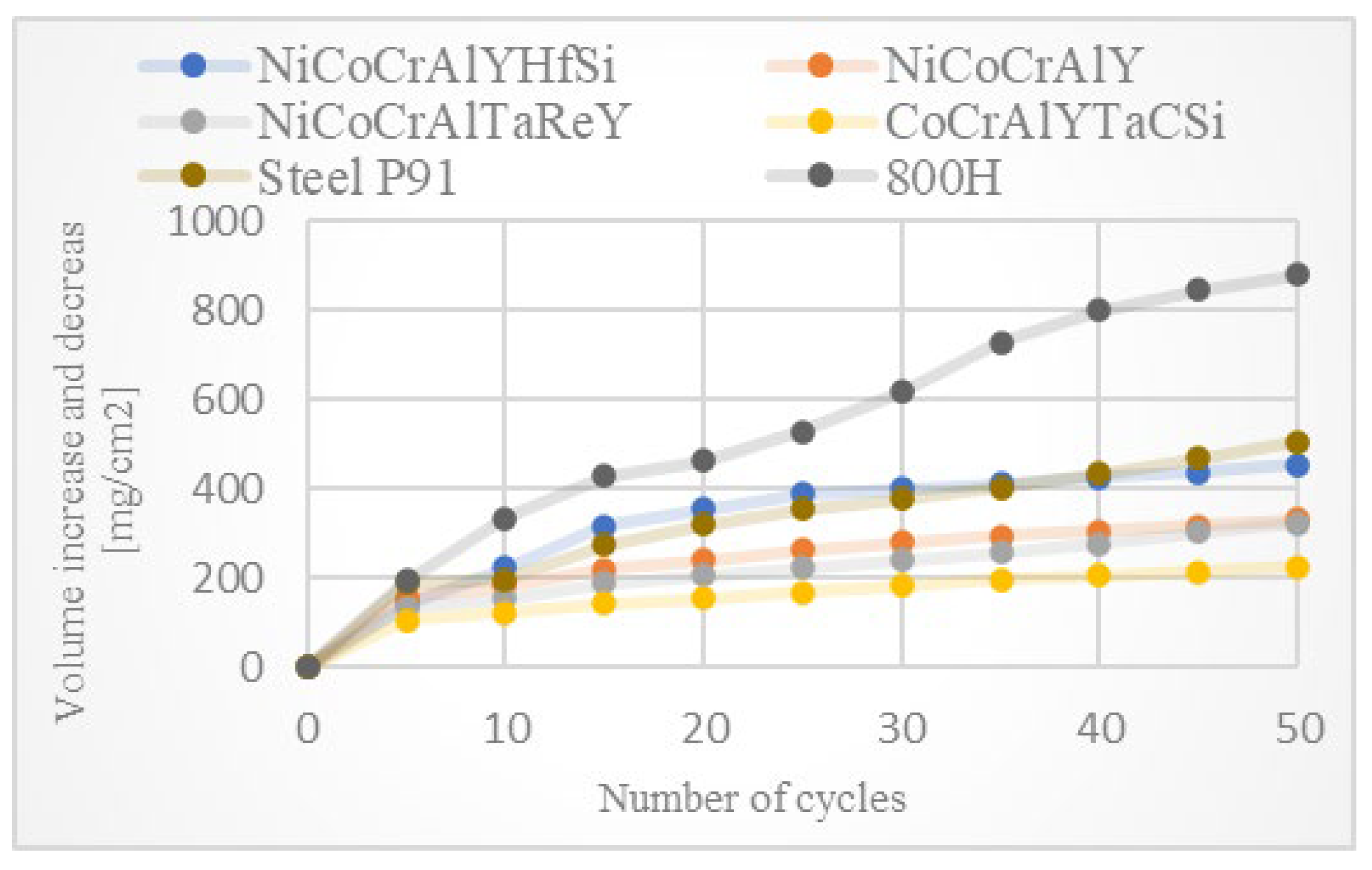
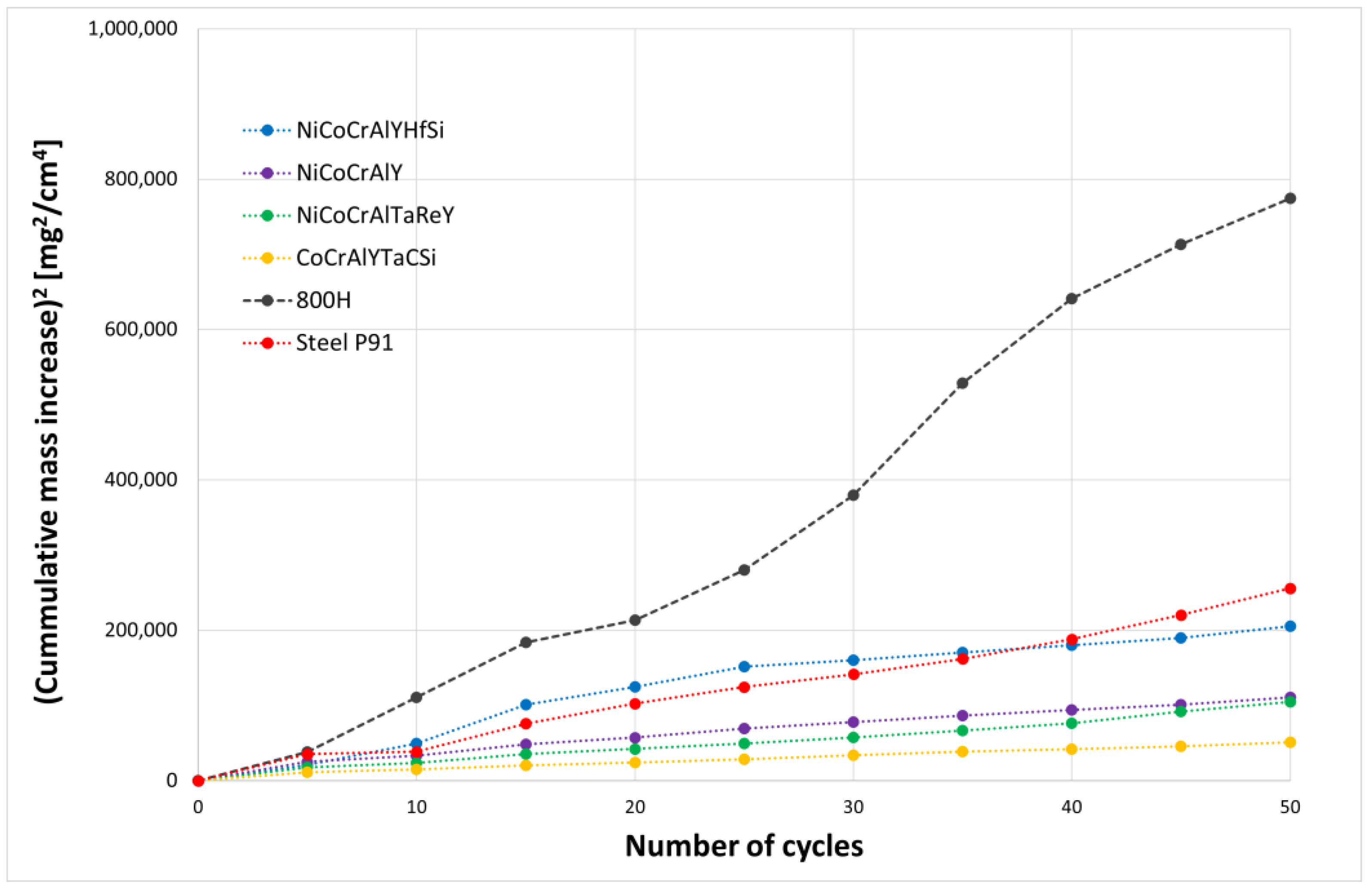
The results of thermogravimetric evaluation of corrosion products kinetics, supported by the SEM and EDS observation of tested specimens, show that the corrosive environment of chloride salts is very aggressive for many common materials. The CoCrAlYTaCSi-coated specimen exhibited the best corrosion resistance to this environment. The tested sample showed a relatively integral protective layer of alumina after fifty 1-h cycles at 800 °C, which seems to have fulfilled its protective function against penetration of the aggressive environment of alkali metal sulphates and chlorides. Based on the test performed, the CoCrAlYTaCSi coating can be recommended as a potentially suitable surface protection for power plant boiler environments dominated by alkaline sulphates and chlorides.
All specimens experienced corrosion attack on all uncoated surfaces and the coated specimens exhibited coating delamination to some extent. The observed corrosion mechanisms of the NiCoCrAlYHfSi, NiCoCrAlY, NiCoCrAlTaReY, and CoCrAlYTaCSi coatings were found to be similar. Lateral corrosion attack spread through the base material to the coating–substrate interface from the edges towards the center of the samples. The corrosion attack on splats and their interfaces was observed on all coatings. Uncoated P91 steel exhibited decent test results roughly comparable to the NiCoCrAlYHfSi-coated specimen (worst resistance among tested coatings), but since potential industrial utilization of HVOF-sprayed NiCoCrAlYHfSi coating on P91 would not probably lead to any noticeable increase in corrosion resistance and would only serve to add some minor thickness to coated firewall or tube, it cannot be recommended for a corrosion environment with the dominant influence of sodium sulphates and chlorides. The corrosion attack on the 800 H alloy specimen proved that this material is completely unsuitable for applications in aggressive environments with chloride salts.

 ,
, 























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}