
The Role of ARHGAP1 in Rho GTPase Inactivation during Metastasizing of Breast Cancer Cell Line MCF-7 after Treatment with Doxorubicin

Abstract
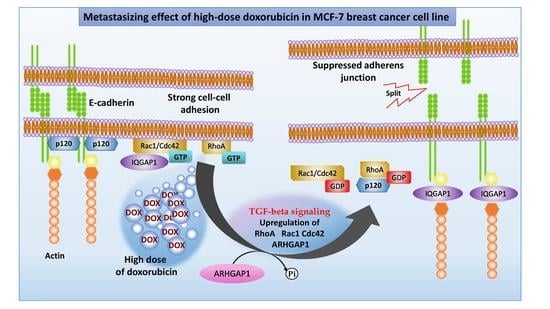
:
1. Introduction
2. Results
2.1. Qualitative and Quantitative Mass Spectrometry Analysis
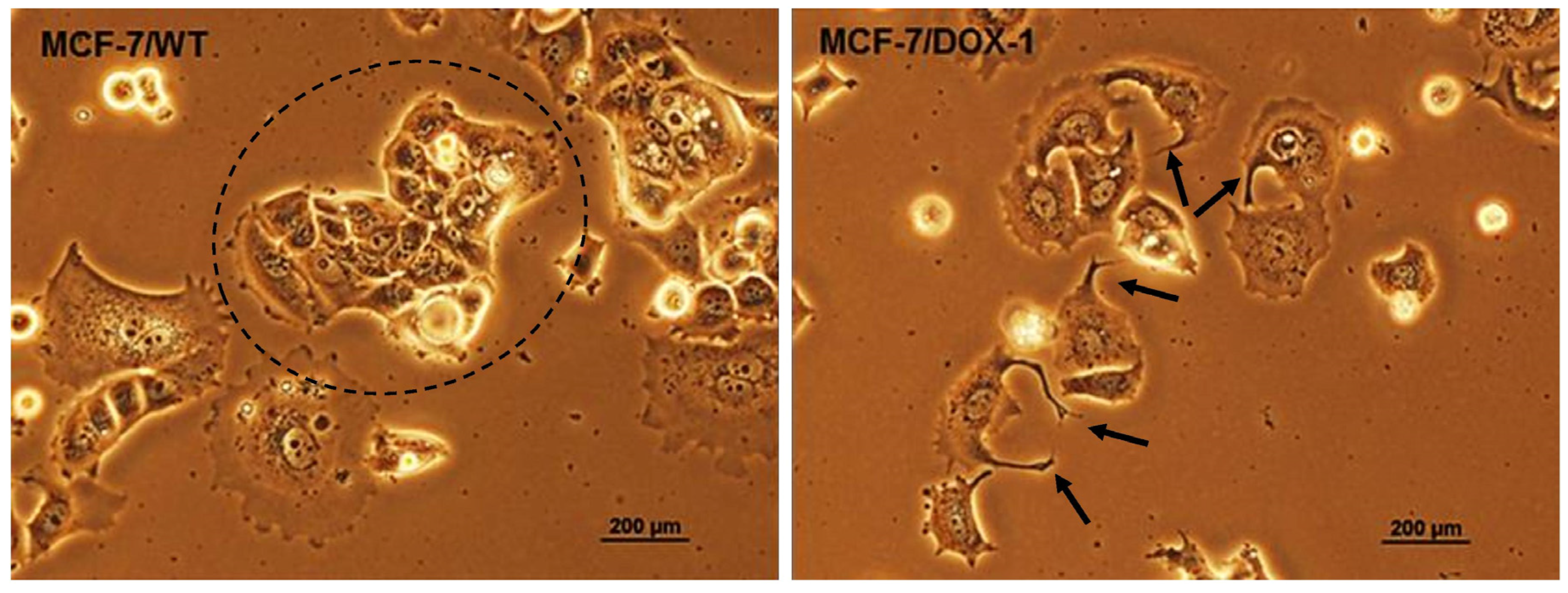
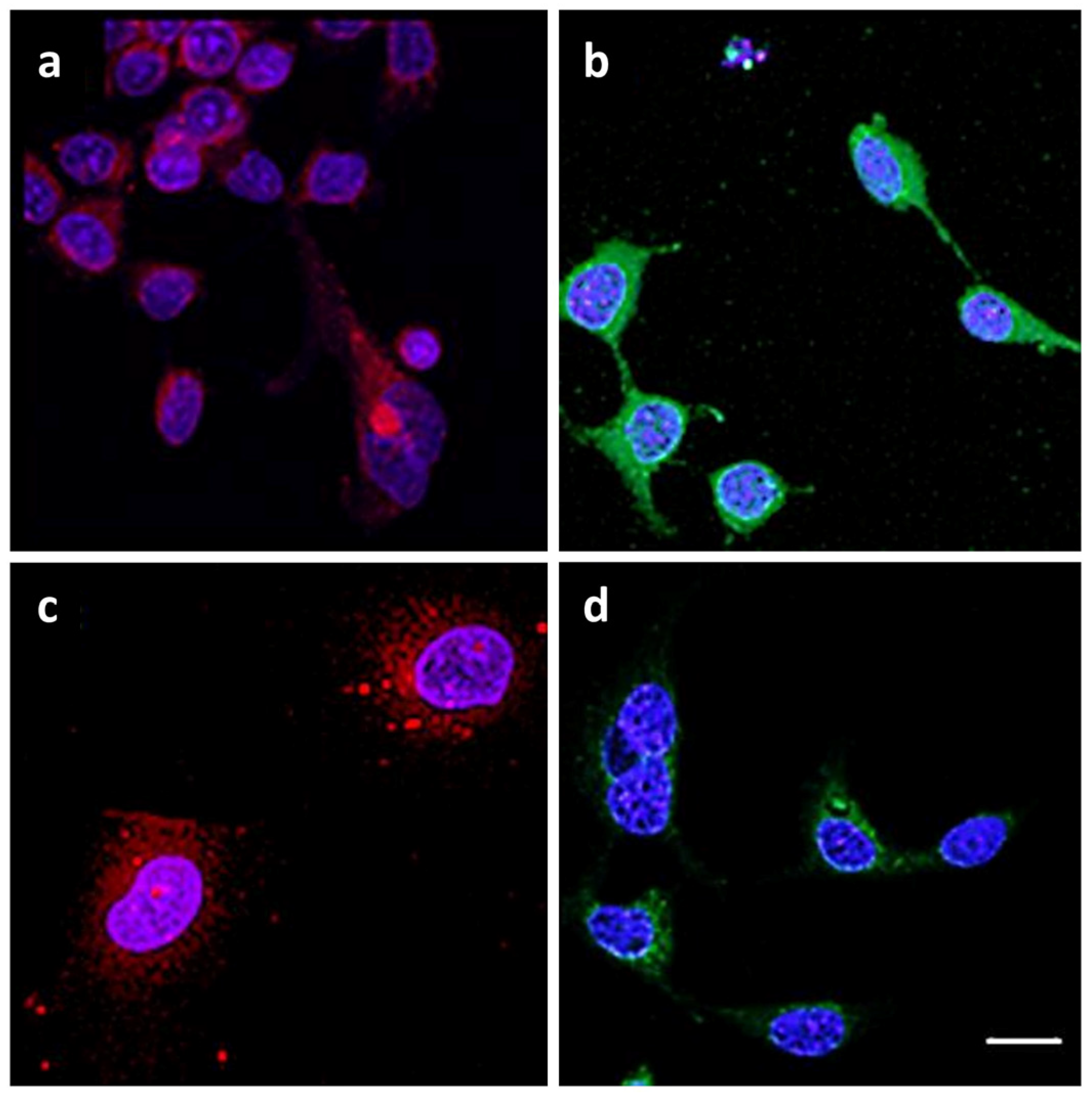
2.2. Analysis of Morphological Changes and Immunohistochemistry
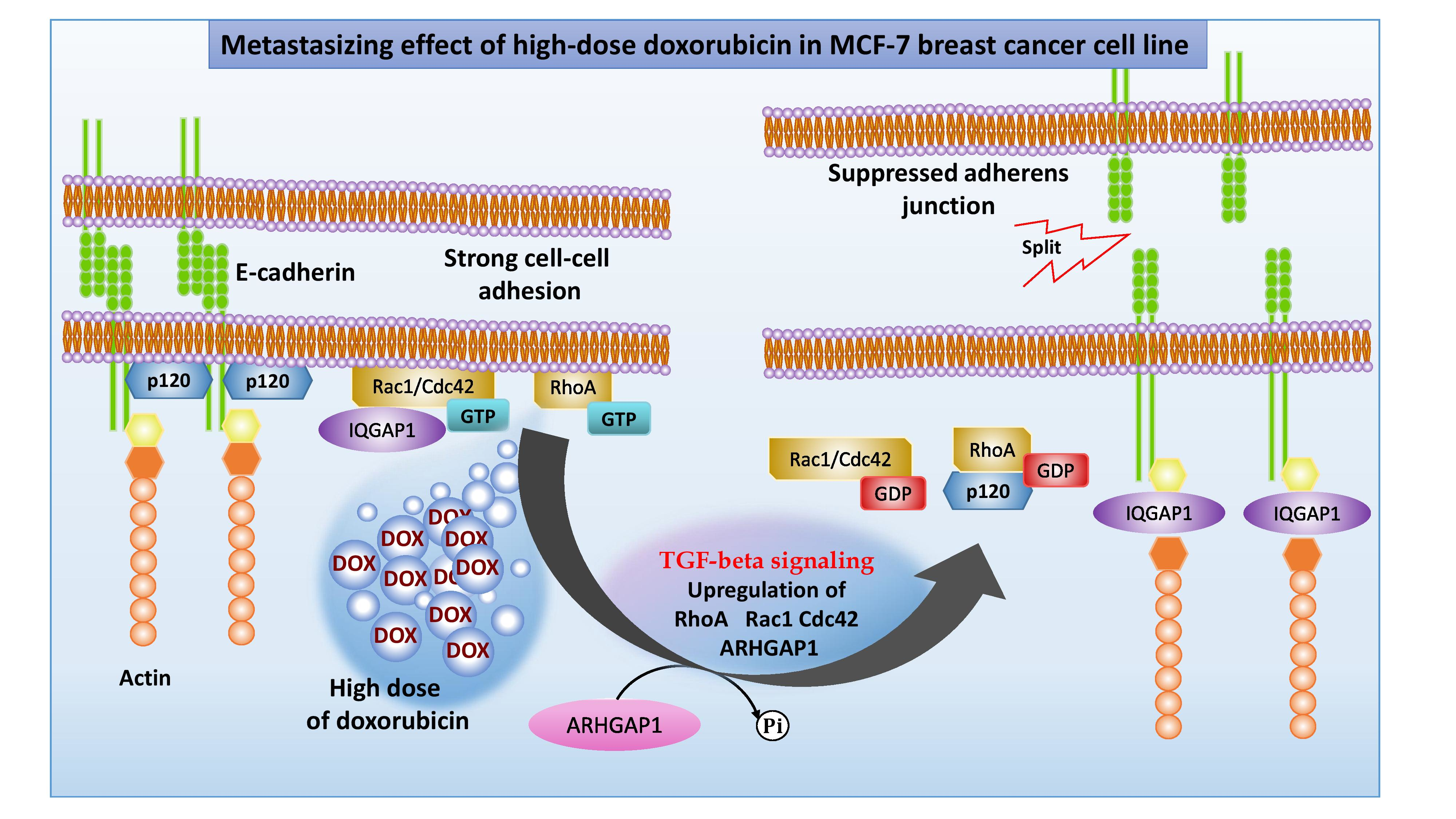
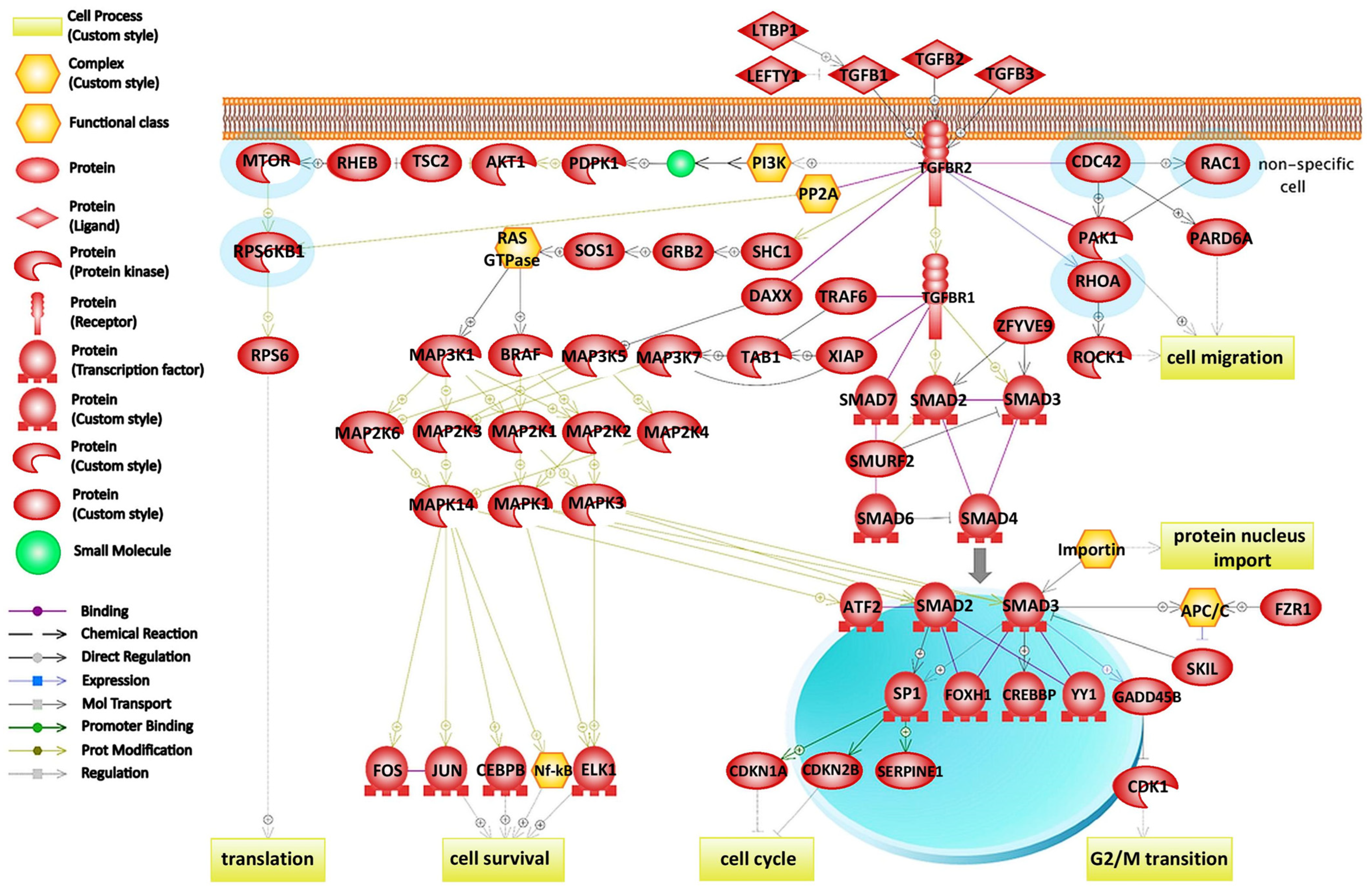
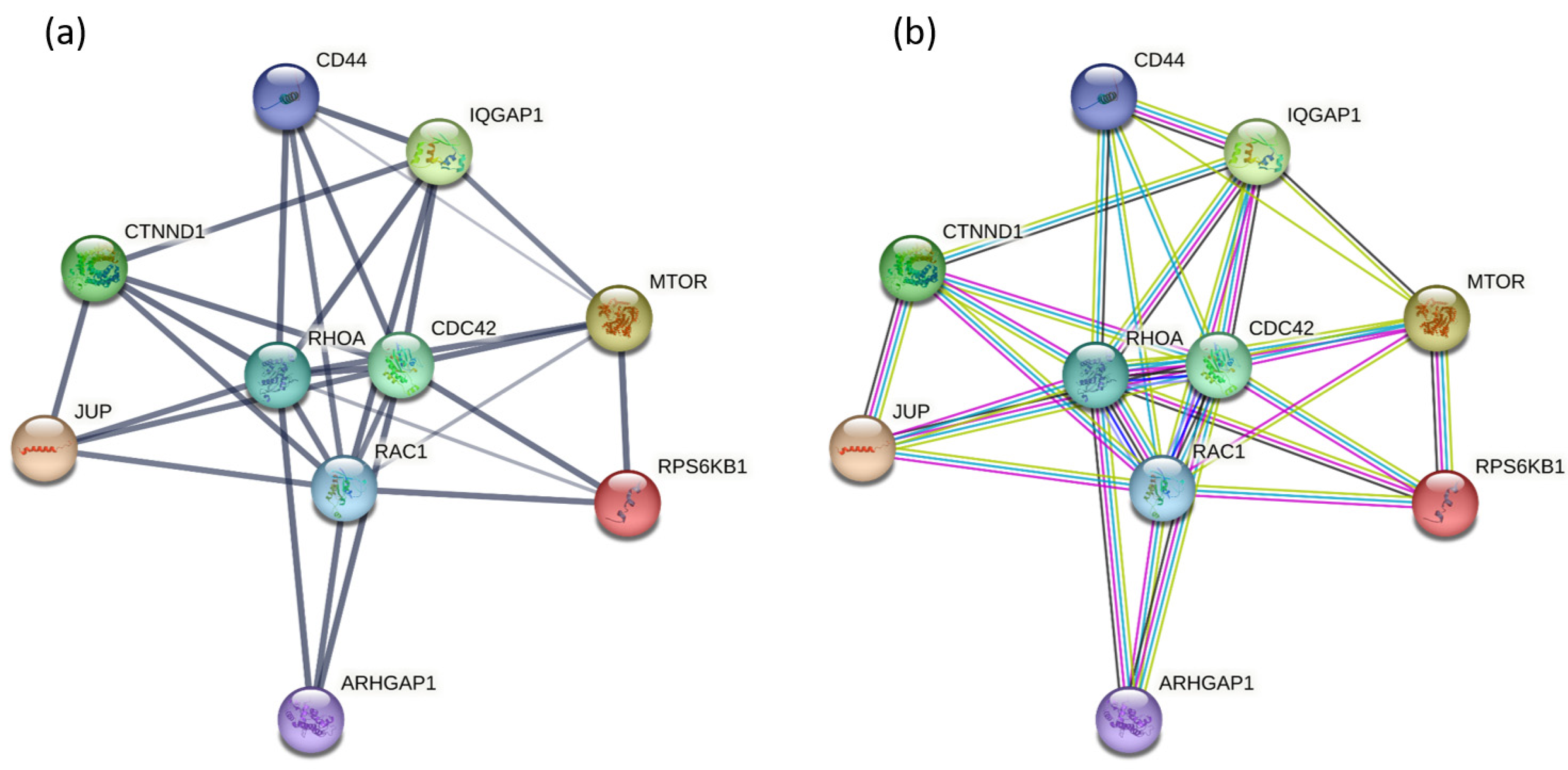
2.3. Protein–Protein Interaction Network Construction
3. Discussion
3.1. ARHGAP1 and Other Proteins Involved in EMT
3.2. Mechanism of Rho GTPase Inactivation via ARHGAP1
3.3. Role of Inactivated Rho GTPases in Adherens Junction Pathway during Weak Adhesion
3.4. Role of Rho GTPases in TGF-Beta Signaling Pathway during EMT
4. Materials and Methods
4.1. MCF-7/WT and MCF-7/DOX-1 Cultivation
4.2. Cell Lysis and Extraction of Proteins
4.3. Determination of the Protein Concentration and In-Solution Digestion
4.4. Off-Gel Fractionation
4.5. Analysis of Proteins via Nano-HPLC–ESI-MS Assembly
4.6. Protein Database Search
4.7. Protein Identification, Quantification, and Pathway Analysis
4.8. Immunohistochemistry Staining
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Buyuk, B.; Jin, S.; Ye, K. Epithelial-to-Mesenchymal Transition Signalling Pathways Responsible for Breast Cancer Metastasis. Cel. Mol. Bioeng. 2022, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhang, Z.; Zhou, C.h.; Liu, L.; Huang, C. Epithelial–mesenchymal transition: The history, regulatory mechanism, and cancer therapeutic opportunities. MedComm 2022, 3, e144. [Google Scholar] [CrossRef]
- Gooding, A.; Schiemann, W.P. Epithelial–Mesenchymal Transition Programs and Cancer Stem Cell Phenotypes: Mediators of Breast Cancer Therapy Resistance. Mol. Cancer Res. 2020, 18, 1257–1270. [Google Scholar] [CrossRef]
- Crosas-Molist, E.; Samain, R.; Kohlhammer, L.; Orgaz, J.L.; George, S.L.; Maiques, O.; Barcelo, J.; Sanz-Moreno, V. Rho GTPase Signaling in Cancer Preogression and Dissemination. Physiol. Rev. 2022, 102, 455–510. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Cong, W.; Zhou, S.; Shi, Y.; Dai, W.; Zhang, H.; Wang, X.; He, B.; Zhang, Q. Small GTPases: Structure, biological function and its interaction with nanoparticles. Asian J. Pharm. Sci. 2019, 14, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P. Activated Rho GTPases in Cancer—The Beginning of a New Paradigm. Int. J. Mol. Sci. 2018, 19, 3949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondal, S.; Hsiao, K.; Goueli, S.A. A Homogenous Bioluminescent System for Measuring GTPase, GTPase Activating Protein, and Guanine Nucleotide Exchange Factor Activities. Drug Develop. Technolog. 2015, 13, 444–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nature 2002, 420, 629–635. [Google Scholar] [CrossRef]
- Ahn, Y.H.; Gibbons, D.L.; Chakravarti, D.; Creighton, C.J.; Rizvi, Z.H.; Adams, H.P.; Pertsemlidis, A.; Gregory, P.A.; Wright, J.A.; Goodall, G.J.; et al. ZEB1 drives prometastatic actin cytoskeletal remodeling by downregulating {miR}-34a expression. J. Clin. Investig. 2012, 122, 3170–3183. [Google Scholar] [CrossRef] [Green Version]
- Clay, M.R.; Halloran, M.C. Rho activation is apically restricted by Arhgap1 in neural crest cells and drives epithelial-to-mesenchymal transition. Development 2013, 140, 3198–3209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Huang, J.; Wu, S.; Jiang, S.; Liang, L.; Liu, Y.; Liu, W.; Xie, L.; Tao, Y.; Cong, L. The roles of GTPase-activating proteins in regulated cell death and tumor immunity. J. Hematol. Oncol. 2021, 14, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Hollier, B.G.; Evans, K.; Mani, S.A. The Epithelial-to-Mesenchymal Transition and Cancer Stem Cells: A Coalition against Cancer Therapies. J. Mammary Gland Biol. Neoplasia. 2009, 14, 29–43. [Google Scholar] [CrossRef]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta Biomembr. 2008, 1778, 660–669. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Kaibuchi, K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat. Rev. Mol. Cell Biol. 2001, 2, 887–897. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voulgari, A.; Pintzas, A. Epithelial-mesenchymal transition in cancer metastasis: Mechanisms, markers and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta Rev. Cancer. 2009, 1796, 75–90. [Google Scholar] [CrossRef] [PubMed]
- Zöller, M. CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 2011, 11, 254–267. [Google Scholar] [CrossRef]
- Leon, G.; MacDonagh, L.; Finn, S.P.; Cuffe, S.; Barr, M.P. Cancer stem cells in drug resistant lung cancer: Targeting cell surface markers and signalling pathways. Pharmacol. Ther. 2016, 158, 71–90. [Google Scholar] [CrossRef]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-Mesenchymal Transition and Breast Cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Tsou, S.H.; Chen, T.M.; Hsiao, H.T.; Chen, Y.H. A critical dose of doxorubicin is required to alter the gene expression profiles in MCF-7 cells acquiring multidrug resistance. PLoS ONE 2015, 10, e0116747. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.K.; Cheon, S.; Kim, H.; Lee, K.-M.; Ryu, H.S.; Han, D. Discovery of Proteins Responsible for Resistance to Three Chemotherapy Drugs in Breast Cancer Cells Using Proteomics and Bioinformatics Analysis. Molecules 2022, 27, 1762. [Google Scholar] [CrossRef] [PubMed]
- Dorland, J.M. Variable Response to Chemotherapeutics by a Subpopulation of MCF—7 Breast Cancer Cells. Honor. Theses. 2016, pp. 1–39. Available online: http://hdl.handle.net/2152/37808 (accessed on 7 July 2023).
- Rivankar, S. An overview of doxorubicin formulations in cancer therapy. J. Cancer Res. Therap. 2014, 10, 853–858. [Google Scholar] [CrossRef]
- Ujah, G.A.; Nna, V.U.; Suleiman, J.B.; Eleazu, C.; Nwokocha, C.; Rebene, J.A.; Imowo, M.U.; Obi, E.O.; Amachree, C.; Udechukwu, E.C.; et al. Tert-butylhydroquinone attenuates doxorubicin-induced dysregulation of testicular cytoprotective and steroidogenic genes, and improves spermatogenesis in rats. Sci. Rep. 2021, 11, 5522. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.A.; Randolph, J.K.; Yalowich, J.C.; Ritke, M.K.; Gewirtz, D.A. Interference by doxorubicin with DNA unwinding in MCF-7 breast tumor cells. Mol. Pharmacol. 1994, 45, 649–656. [Google Scholar]
- Shin, D.; Park, J.; Han, D.; Moon, J.H.; Ryu, H.S.; Kim, Y. Identification of TUBB2A by quantitative proteomic analysis as a novel biomarker for the prediction of distant metastatic breast cancer. Clin. Proteom. 2020, 17, 16. [Google Scholar] [CrossRef]
- Vasilogianni, A.M.; Al-Majdoub, Z.M.; Achour, B.; Annie Peters, S.; Barber, J.; Rostami-Hodjegan, A. Quantitative Proteomics of Hepatic Drug-Metabolizing Enzymes and Transporters in Patients with Colorectal Cancer Metastasis. Clin. Pharmacol. Ther. 2022, 112, 699–710. [Google Scholar] [CrossRef]
- Santos, A.; Colaço, A.R.; Nielsen, A.B.; Niu, L.; Strauss, M.; Geyer, P.E.; Coscia, F.; Albrechtsen, N.J.W.; Mundt, F.; Jensen, L.J.; et al. A knowledge graph to interpret clinical proteomics data. Nat. Biotechnol. 2022, 40, 692–702. [Google Scholar] [CrossRef]
- Lyons, S.M.; Alizadeh, E.; Mannheimer, J.; Schuamberg, K.; Castle, J.; Schroder, B.; Turk, P.; Thamm, D.; Prasad, A. Changes in cell shape are correlated with metastatic potential in murine and human osteosarcomas. Biol. Open. 2016, 12, 289–299. [Google Scholar] [CrossRef] [Green Version]
- Agnihotri, N.; Kumar, S.; Mehta, K. Tissue transglutaminase as a central mediator in inflammation-induced progression of breast cancer. Breast Cancer Res. 2013, 15, 202. [Google Scholar] [CrossRef]
- Gong, X.; An, Z.; Wang, Y.; Guan, L.; Fang, W.; Strömblad, S.; Jiang, Y.; Zhang, H. Kindlin-2 controls sensitivity of prostate cancer cells to cisplatin-induced cell death. Cancer Lett. 2010, 299, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Guttilla, I.K.; Phoenix, K.N.; Hong, X.; Tirnauer, J.S.; Claffey, K.P.; White, B.A. Prolonged mammosphere culture of MCF-7 cells induces an EMT and repression of the estrogen receptor by microRNAs. Breast Cancer Res. Treat. 2011, 132, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Schlienger, S.; Campbell, S.; Pasquin, S.; Gaboury, L.; Claing, A. ADP-ribosylation factor 1 expression regulates epithelial-mesenchymal transition and predicts poor clinical outcome in triple-negative breast cancer. Oncotarget 2016, 7, 15811–15827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Wu, J.; Wang, Y.; Zhao, T.; Ma, B.; Liu, Y.; Fang, W.; Zhu, W.G.; Zhang, H. Kindlin 2 forms a transcriptional complex with β-catenin and TCF4 to enhance Wnt signaling. EMBO Rep. 2012, 13, 750–758. [Google Scholar] [CrossRef] [Green Version]
- Pakala, S.B.; Singh, K.; Reddy, S.D.N.; Ohshiro, K.; Li, D.Q.; Mishra, L.; Kumar, R. TGF-β1 signaling targets metastasis-associated protein 1, a new effector in epithelial cells. Oncogene 2011, 3, 2230–2241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.; Cong, Y.; Wang, D.; Sun, Y.; Deng, L.; Liu, Y.; Martin-Trevino, R.; Shang, L.; McDermott, S.P.; Landis, M.D.; et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem. Cell Rep. 2014, 2, 78–91. [Google Scholar] [CrossRef]
- Thomson, S.; Petti, F.; Sujka-Kwok, I.; Mercado, P.; Bean, J.; Monaghan, M.; Seymour, S.L.; Argast, G.M.; Epstein, D.M. A systems view of epithelial mesenchymal-transition signaling states. Clin. Exp. Metastasis 2010, 28, 137–155. [Google Scholar] [CrossRef] [Green Version]
- Mizuuchi, Y.; Aishima, S.; Ohuchida, K.; Shindo, K.; Fujino, M.; Hattori, M.; Miyazaki, T.; Mizumoto, K.; Tanaka, M.; Oda, Y. Anterior gradient 2 downregulation in a subset of pancreatic ductal adenocarcinoma is a prognostic factor indicative of epithelial-mesenchymal transition. Lab. Investig. 2015, 95, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Ma, S.R.; Wang, W.M.; Huang, C.F.; Zhang, W.F.; Sun, Z.J. Anterior gradient protein 2 expression in high grade head and neck squamous cell carcinoma correlated with cancer stem cell and epithelial mesenchymal transition. Oncotarget 2015, 6, 8807–8821. [Google Scholar] [CrossRef] [Green Version]
- Hodge, R.G.; Ridley, A.J. Regulating Rho GTPases and their regulators. Nat. Rev. Mol. Cell Biol. 2016, 17, 496–510. [Google Scholar] [CrossRef]
- Cherfils, J.; Zeghouf, M. Regulation of Small GTPases by GEFs, GAPs, and GDIs. Physiol. Rev. 2013, 93, 269–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Z.; Zhou, D.; Yang, J.; Zhang, D. Doxorubicin promotes breast cancer cell migration and invasion via DCAF13. FEBS Open Bio. 2022, 12, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Shan, B.Q.; Wang, X.M.; Zheng, L.; Han, Y.; Gao, J.; Lv, M.-D.; Zhang, Y.; Liu, Y.-X.; Zhang, H.; Chen, H.-S.; et al. DCAF13 promotes breast cancer cell proliferation by ubiquitin inhibiting PERP expression. Cancer Sci. 2022, 113, 1587–1600. [Google Scholar] [CrossRef] [PubMed]
- Fukata, M.; Kuroda, S.; Nakagawa, M.; Kawajiri, A.; Itoh, N.; Shoji, I.; Matsuura, Y.; Yonehara, S.; Fujisawa, H.; Kikuchi, A.; et al. Cdc42 and Rac1 regulate the interaction of IQGAP1 with beta-catenin. J. Biol. Chem. 1999, 274, 26044–26050. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, S.; Fukata, M.; Nakagawa, M.; Fujii, K.; Nakamura, T.; Ookubo, T.; Izawa, I.; Nagase, T.; Nomura, N.; Tani, H.; et al. Role of IQGAP1, a target of the small GTPases Cdc42 and Rac1, in regulation of E-cadherin mediated cell-cell adhesion. Science 1998, 281, 832–835. [Google Scholar] [CrossRef]
- Kaibuchi, K.; Kuroda, S.; Amano, M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu. Rev. Biochem. 1999, 68, 459–486. [Google Scholar] [CrossRef] [Green Version]
- Keirsebilck, A.; Bonné, S.; Staes, K.; van Hengel, J.; Nollet, F.; Reynolds, A.; van Roy, F. Molecular Cloning of the Human p120ctnCatenin Gene (CTNND1): Expression of Multiple Alternatively Spliced Isoforms. Genomics 1998, 50, 129–146. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadis, P.Z.; Reynolds, A.B. The p120 catenin family: Complex roles in adhesion, signaling and cancer. J. Cell Sci. 2000, 113, 1319–1334. [Google Scholar] [CrossRef]
- Grosheva, I.; Shtutman, M.; Elbaum, M.; Bershadsky, D. p120 catenin affects cell motility via modulation of activity of Rho-family GTPases: A link between cell-cell contact formation and regulation of cell locomotion. J. Cell Sci. 2001, 114, 695–707. [Google Scholar] [CrossRef]
- Thoreson, M.A.; Anastasiadis, P.Z.; Daniel, J.M.; Ireton, R.C.; Wheelock, M.J.; Johnson, K.R.; Hummingbird, D.K.; Albert, B. Reynolds Selective uncoupling of p120(ctn) from E-cadherin disrupts strong adhesion. J. Cell Biol. 2000, 148, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Bellovin, D.I.; Bates, R.C.; Muzikansky, A.; Rimm, D.L.; Mercurio, A.M. Altered localization of p120 catenin during epithelial to mesenchymal transition of colon carcinoma is prognostic for aggressive disease. Cancer Res. 2005, 65, 10938–10945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagaraj, N.S.; Datta, P.K. Targetting the Transforming Growth factor-beta Signalling Pathway in Human Cancer. Expert Opin. Investig. Drugs. 2010, 5, 557–577. [Google Scholar] [CrossRef] [Green Version]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell. 2001, 12, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Edlund, S. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef] [Green Version]
- Nobes, C.D.; Hall, A.R. Rac, and Cdc42 GTPases Regulate the Assembly of Multimolecular Focal Complexes Associated with Actin Stress Fibers, Lamellipodia, and Filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ariadne Cell Process Pathway | Total Entities | Overlap | Overlapping Entities | p-Value | Jaccard Similarity |
|---|---|---|---|---|---|
| VEGF signaling | 82 | 6 | RPS6KB1, MTOR, RAC1, IQGAP1, CDC42, HSPB1 | 0.016 | 0.013 |
| p38 MAPK/MAPK14 signaling | 41 | 4 | RAC1, CDC42, HSPB1, RHOA | 0.020 | 0.010 |
| Ras–GAP regulation signaling | 47 | 4 | RAC1, CDC42, DYNC1H1, STAT1 | 0.032 | 0.010 |
| TGF-beta signaling | 75 | 5 | RPS6KB1, MTOR, RAC1, CDC42, RHOA | 0.041 | 0.011 |
| Signal Transduction Pathway | |||||
| Translation | 84 | 15 | EIF4G1, EEF1G, EEF1D, ETF1, RARS, AARS, IARS, RPS6KB1, EPRS, VARS, QARS, DARS, NARS, EIF5B, EIF4A1 | 4.28 × 10−7 | 0.034 |
| Endosomal recycling | 50 | 7 | PDCD6IP, ARF1, CDC42, RAB11A, TFRC, EHD4, DNM2 | 0.003 | 0.017 |
| Adherens junction assembly (cadherins) | 39 | 6 | CTNND1, RHOA, JUP, RAC1, CDC42, IQGAP1 | 0.004 | 0.015 |
| Golgi to endosome transport | 59 | 7 | VPS35, SNX2, STX16, NSF, NAPA, ARF1, DNM2 | 0.008 | 0.017 |
| SRCAP chromatin remodeling | 18 | 3 | RUVBL2, ACTL6A, RUVBL1 | 0.032 | 0.008 |
| Secretory pathway: Golgi transport | 36 | 4 | RAB2A, SEC24C, SEC16A, ARF1 | 0.052 | 0.010 |
| Accession | Gene | Protein | Fold Change | t-Test (p ≤ 0.05) | MCF-7/DOX-1 Regulated |
|---|---|---|---|---|---|
| Ras-related C3 botulinum toxin substrate1 | RAC1 | RAC1_HUMAN | 13.8 | 0.003 | up |
| Cell division control protein 42 homolog | CDC42 | CDC42_HUMAN | 19.8 | 0.002 | up |
| Transforming protein RhoA | RHOA | RHOA_HUMAN | 18.9 | 0.021 | up |
| Ras GTPase-activating-like protein | IQGAP1 | IQGA1_HUMAN | 2.7 | 0.014 | up |
| Rho GTPase-activating protein 1 | ARHGAP1 | RHG01_HUMAN | 15.6 | 0.004 | up |
| Catenin gamma | JUP | PLAK_HUMAN | 2.5 | 0.023 | up |
| p120 catenin | CTNND1 | CTND1_HUMAN | 13.2 | 0.004 | up |
| Serine/threonine-pro-tein kinase mTOR | MTOR | MTOR_HUMAN | 5.1 | 0.0001 | up |
| Ribosomal protein S6 kinase beta-1 | RPS6KB1 | KS6B1_HUMAN | 3.1 | 0.040 | up |
| CD44 antigen | CD44 | CD44_HUMAN | 4.9 | 0.012 | up |
| Proteins/Chemicals Regulating Cell Processes | Total Entities | Overlap—% | MCF-7/DOX-1 | p-Value | Jaccard Similarity |
|---|---|---|---|---|---|
| Cell motility | 3208 | 70 2 | STAT1, CLIC1, FOXA1, TGM2, NAMPT, IPO7, TACSTD2, CAPN1, PKM, CDC42, AIFM1, ARL1, GNA13, HSPA2, MYO1C, APEX1, MEMO1, ATP1A1, TRIM16, DIAPH1, HNRNPK, SND1, MTA1, RUVBL1, CYFIP1, MYO1B, GNAS, G6PD, FLOT1, YWHAZ, RHOA, NSF, HUWE1, IQGAP1, MYOF, DNM2, ACLY, ATP1B1, NPM1, KDM1A, CD44, PRDX1, AGR2, FAM129B, MUC5AC, FLII, MTOR, CTNND1, PAPOLA, ANXA7, RAC1, RAB11A, DNM1L, NARS, GNB2L1, WDR1, HSPB1, RPS6KB1, S100P, TWF1, STOML2, SLC12A2, JUP, MARCKS, CUL1, KHSRP, YWHAB, GOLPH3, ARHGAP1, FERMT2 | 8.177 × 10−16 | 0.020 |
| Epithelial-to-mesenchymal transition | 2423 | 53 2 | STAT1, DNAJB6, ACTL6A, HYOU1, FOXA1, TGM2, NAMPT, TACSTD2, BLVRA, PKM, CDC42, GNA13, PRMT5, RUVBL2, MEMO1, PRDX2, TRIM16, GLRX3, MTA1, KIF5B, GNAS, ARF1, YWHAZ, SENP3, RHOA, IQGAP1, MYOF, EEF1D, ACLY, ATP1B1, DSP, CTBP2, KDM1A, CD44, PRDX1, ALDH7A1, AGR2, MTOR, CTNND1, IDH2, RAC1, DNM1L, GNB2L1, HSPB1, RPS6KB1, S100P, TWF1, CKAP4, JUP, CUL1, YWHAB, ARHGAP1, FERMT2 | 0.012 | 0.019 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Géci, I.; Bober, P.; Filová, E.; Amler, E.; Sabo, J. The Role of ARHGAP1 in Rho GTPase Inactivation during Metastasizing of Breast Cancer Cell Line MCF-7 after Treatment with Doxorubicin. Int. J. Mol. Sci. 2023, 24, 11352. https://doi.org/10.3390/ijms241411352
Géci I, Bober P, Filová E, Amler E, Sabo J. The Role of ARHGAP1 in Rho GTPase Inactivation during Metastasizing of Breast Cancer Cell Line MCF-7 after Treatment with Doxorubicin. International Journal of Molecular Sciences. 2023; 24(14):11352. https://doi.org/10.3390/ijms241411352
Chicago/Turabian StyleGéci, Imrich, Peter Bober, Eva Filová, Evžen Amler, and Ján Sabo. 2023. "The Role of ARHGAP1 in Rho GTPase Inactivation during Metastasizing of Breast Cancer Cell Line MCF-7 after Treatment with Doxorubicin" International Journal of Molecular Sciences 24, no. 14: 11352. https://doi.org/10.3390/ijms241411352