
Individual DNA Methylation Pattern Shifts in Nanoparticles-Exposed Workers Analyzed in Four Consecutive Years

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Basic Characteristics of the Study Subjects
2.2. Annual Particulate Matter (PM) Monitoring, Including NP Fraction in Sampling Years
2.3. DNA Methylation Profiling During Four Consecutive Years
2.3.1. General Information on the Results
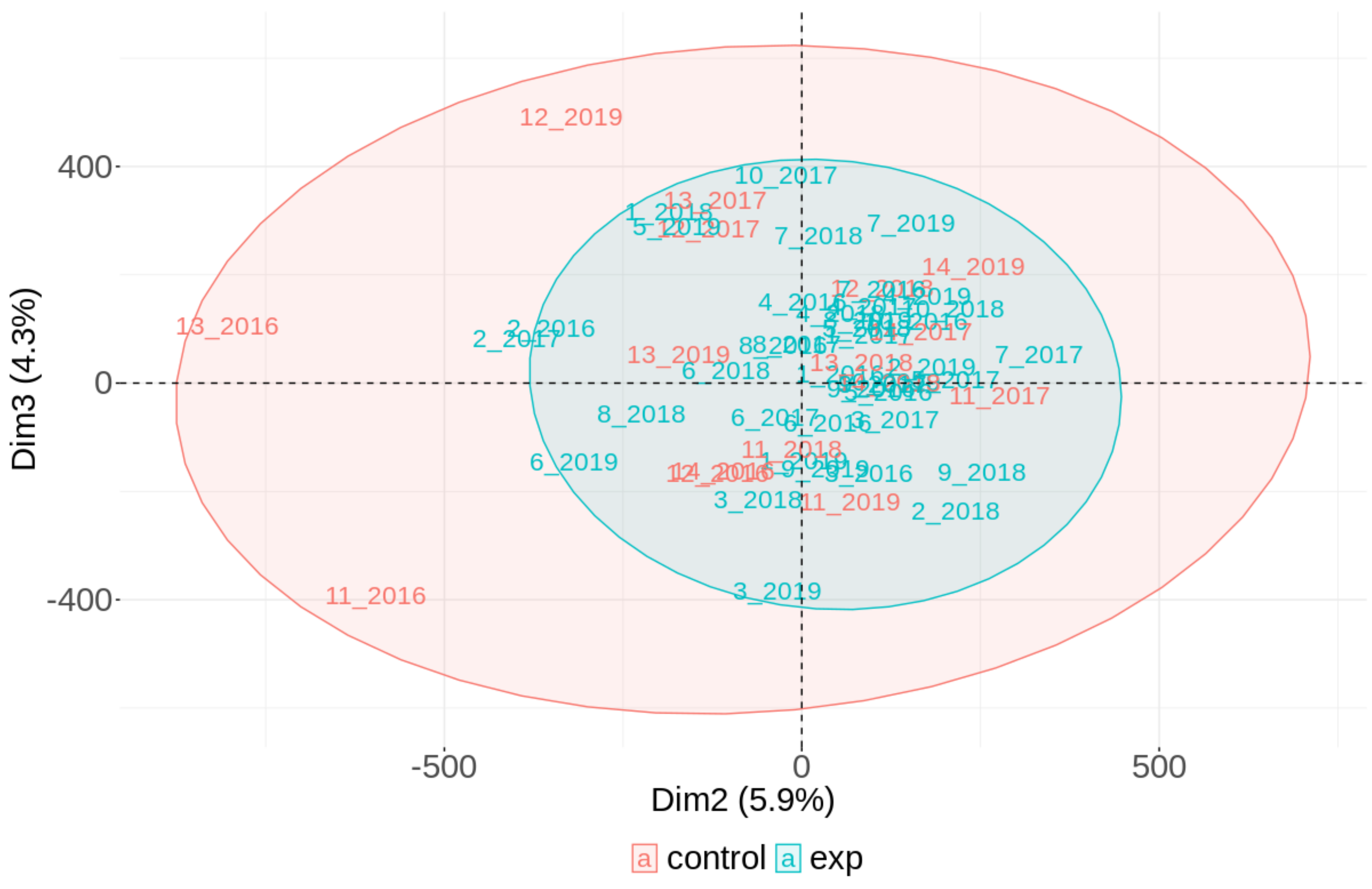
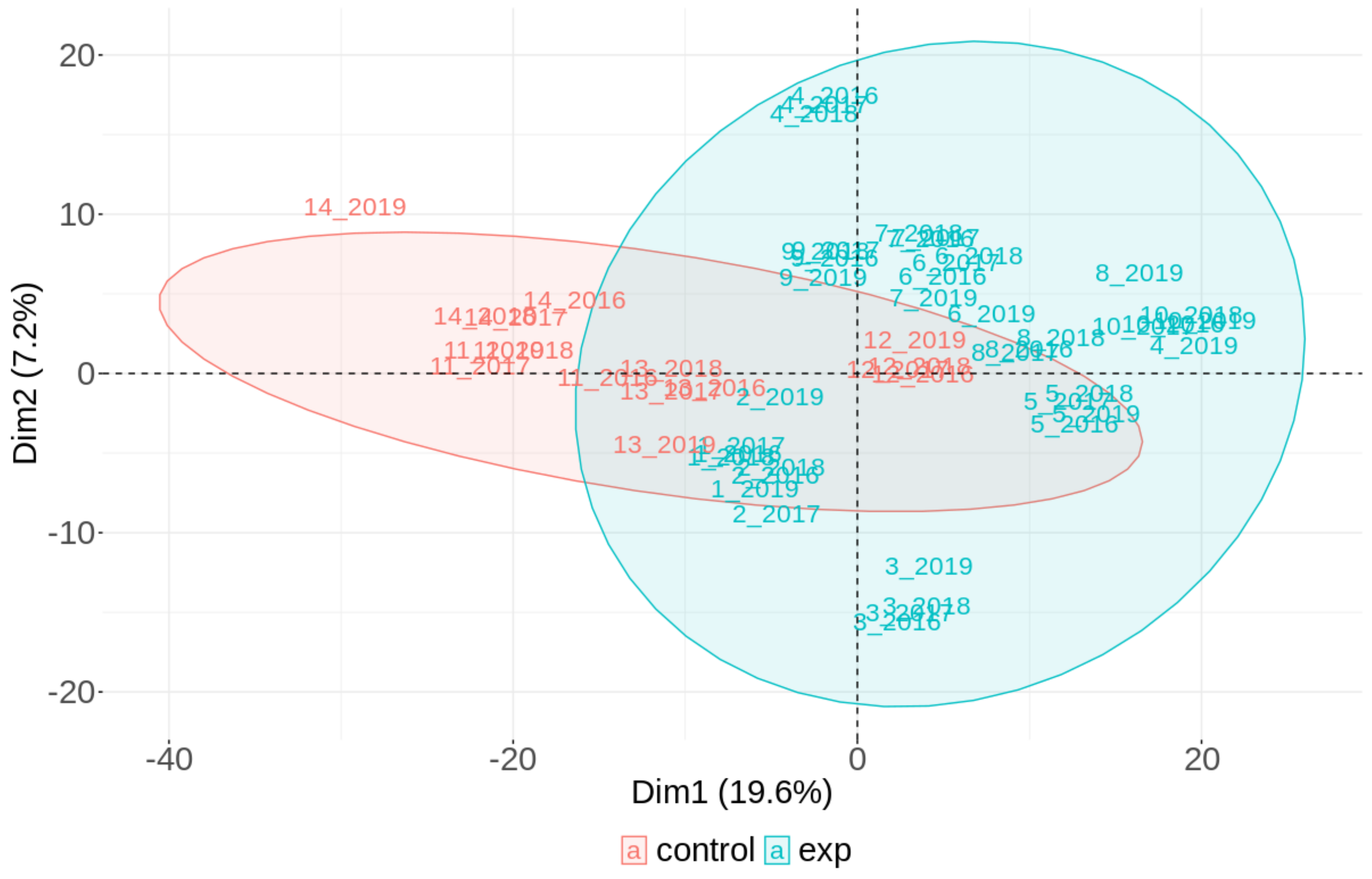
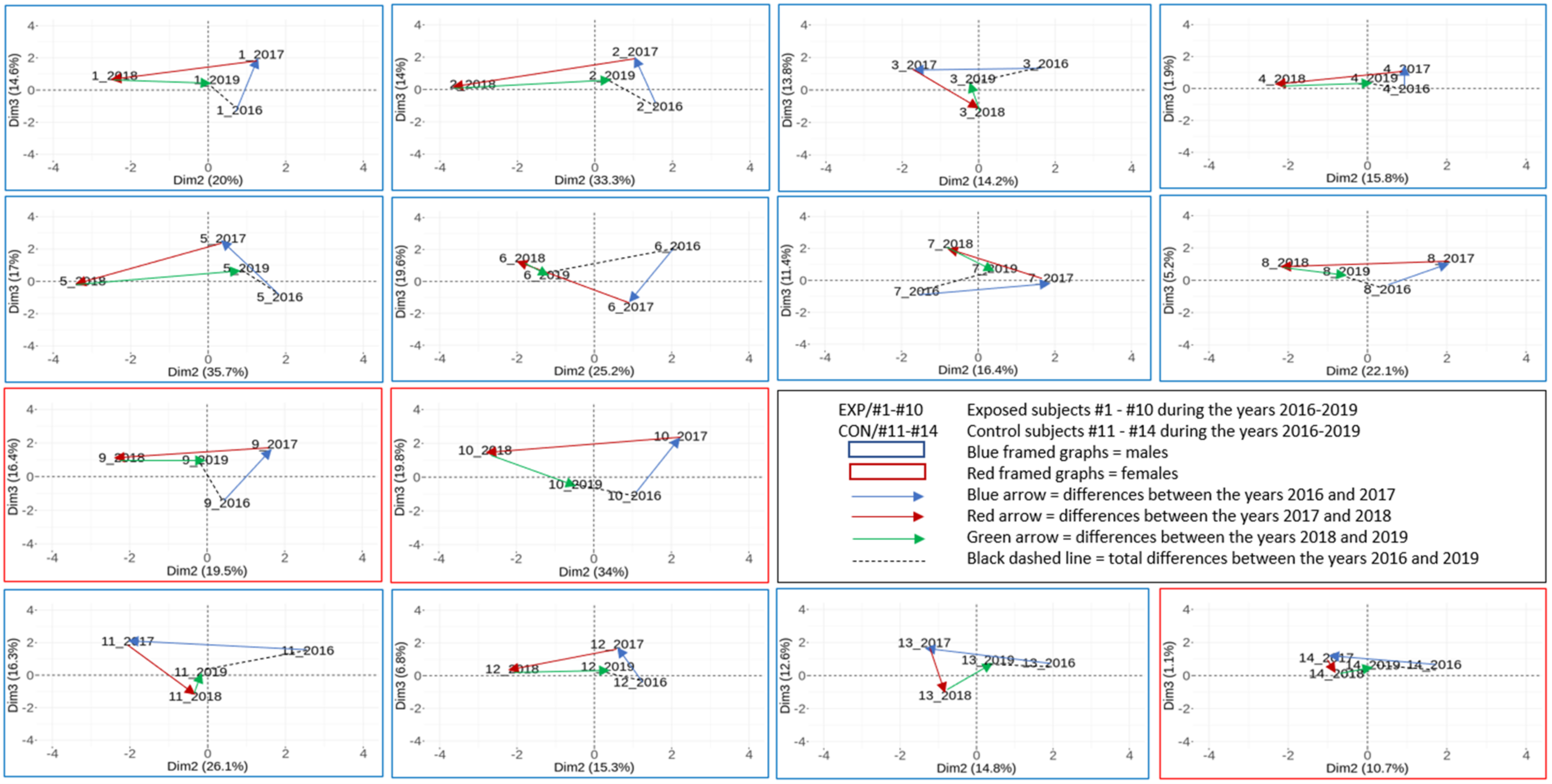
2.3.2. The Results on a Group Basis Obtained by PCA
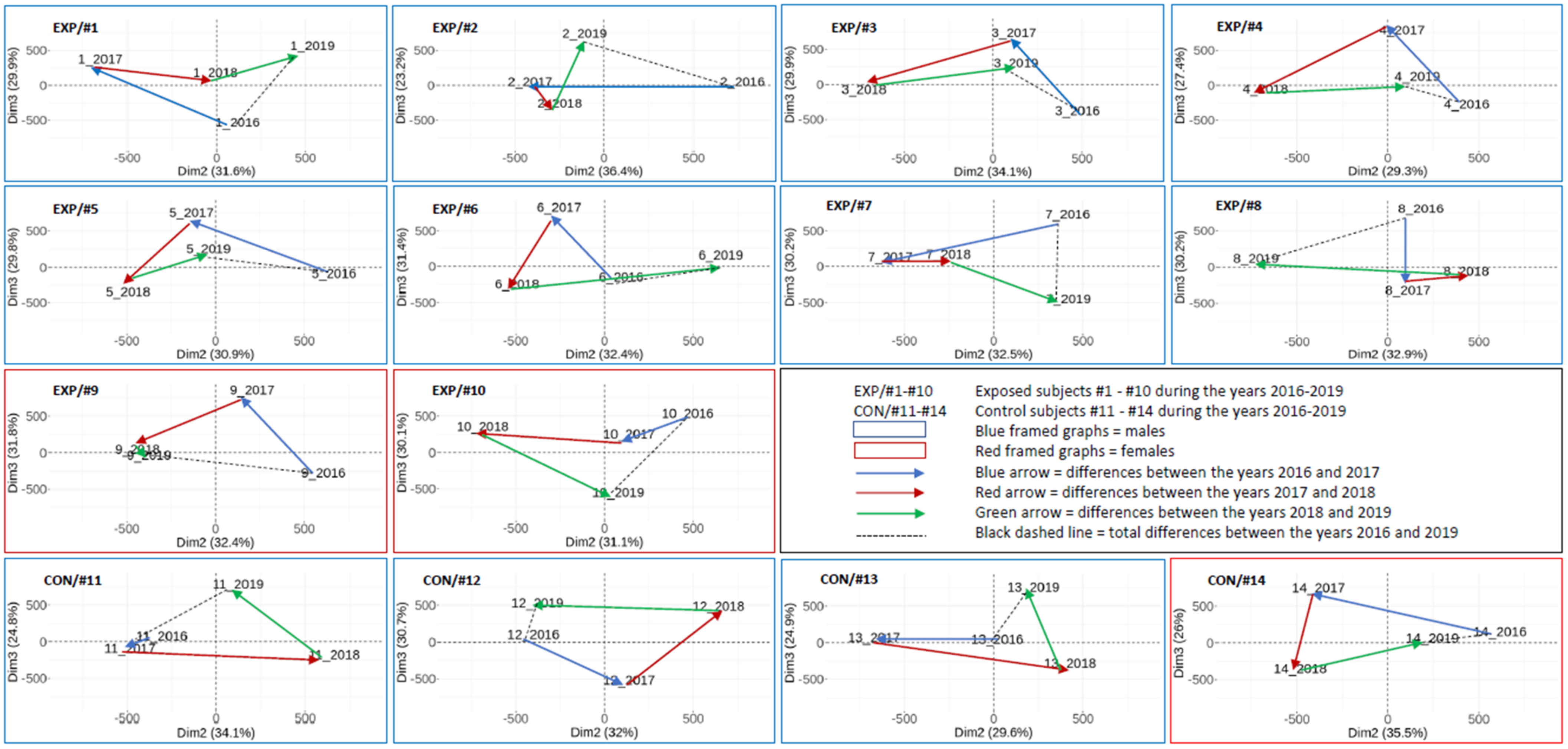
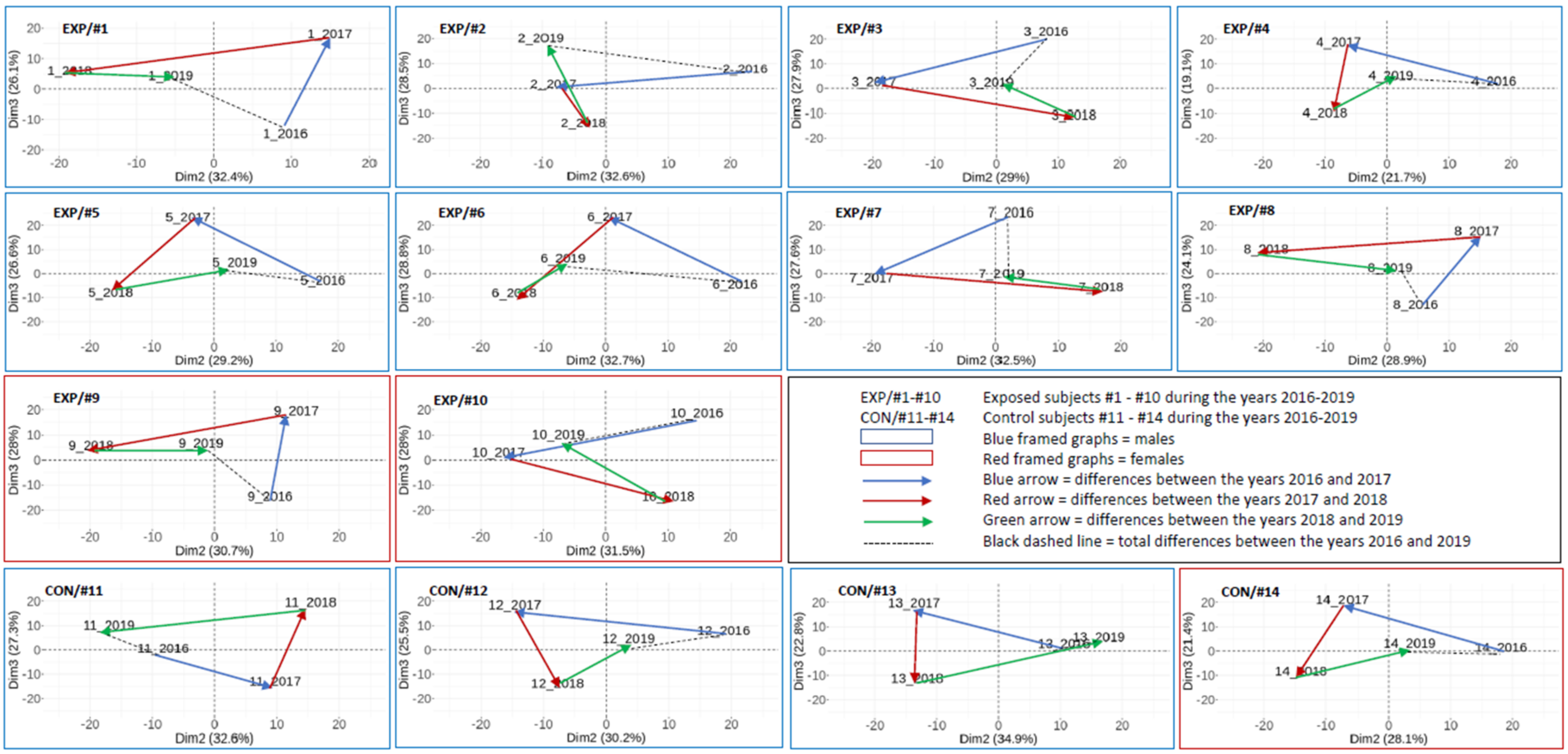
2.3.3. The Results on an Individual Basis Obtained by PCA
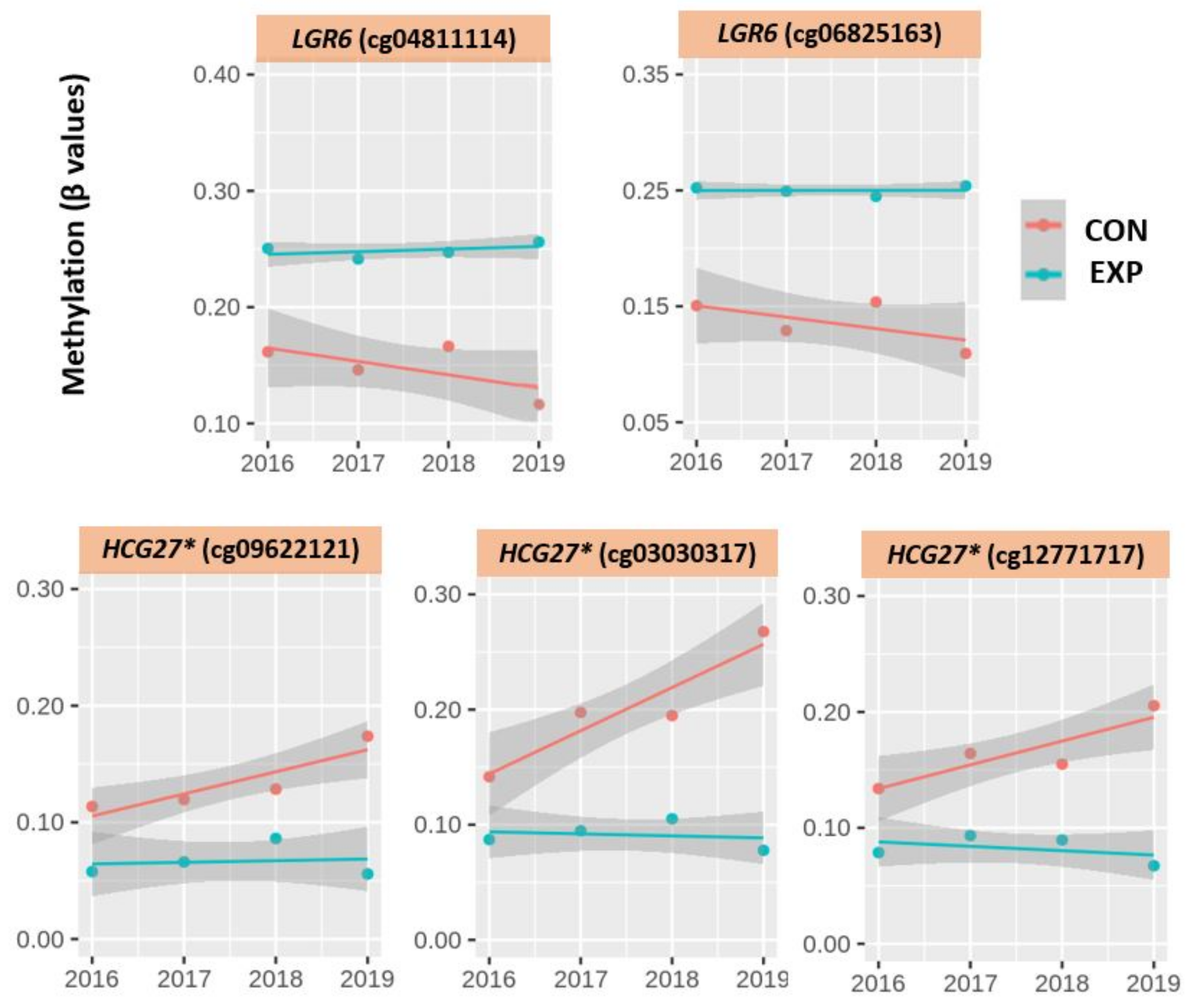
2.3.4. The Beta Values of the Most Significant CpG loci during the Four Consecutive Years
3. Discussion
3.1. Exposure Data
3.2. Overall DNA Methylation Changes in Individual Subjects
3.3. DNA Methylation Changes Related to the Most Significantly Different CpG Loci
3.4. Modification Caused by the Environment
3.5. Gaps and Challenges for Future Research
4. Materials and Methods
4.1. Study Population Selection
4.2. Annual Exposure Monitoring Related to Various Working Activities
4.3. DNA Methylation Microarray Analysis
4.4. Statistical Approaches
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
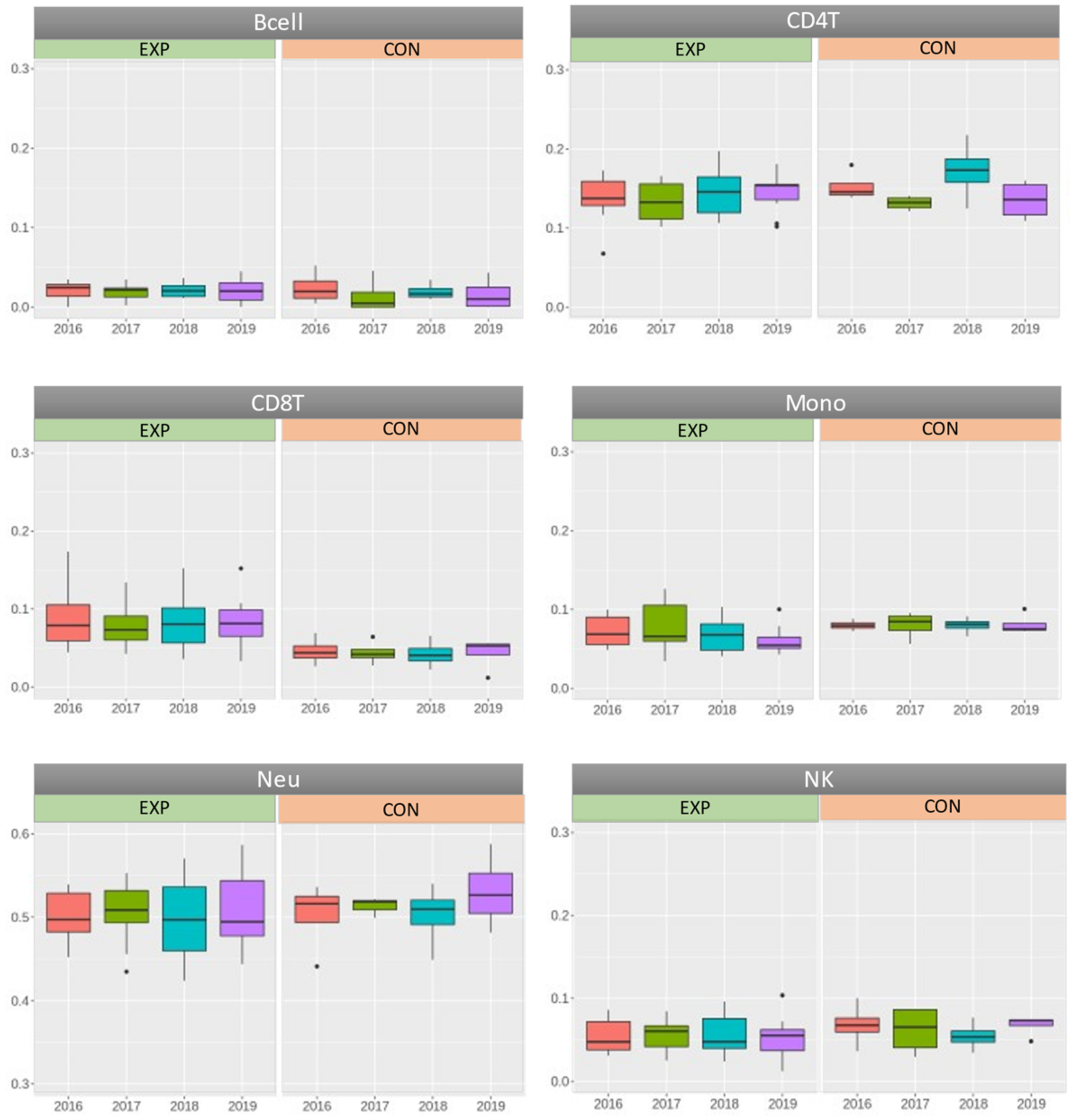
| Comparison between the Exposed and Control Subjects (p-Values Overview) | ||||||
|---|---|---|---|---|---|---|
| Years | Bcell | CD4T | CD8T | Mono | Neu | NK |
| 2016 | 0.735 | 0.356 | 0.073 | 0.483 | 0.924 | 0.352 |
| 2017 | 0.475 | 0.885 | 0.051 | 0.861 | 0.177 | 0.673 |
| 2018 | 0.743 | 0.178 | 0.053 | 0.350 | 0.833 | 0.851 |
| 2019 | 0.589 | 0.454 | 0.054 | 0.057 | 0.404 | 0.306 |
References
- Waddington, C.H. The epigenotype. Endeavour 1942, 1, 18–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waddington, C.H. Towards a Theoretical Biology. In The Basic Ideas of Biology; Edinburgh University Press: Edinburgh, UK, 1968; pp. 1–32. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [Green Version]
- Lövkvist, C.; Dodd, I.B.; Sneppen, K.; Haerter, J.O. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016, 44, 5123–5132. [Google Scholar] [CrossRef] [Green Version]
- Kanherkar, R.R.; Bhatia-Dey, N.; Csoka, A.B. Epigenetics across the human lifespan. Front. Cell Dev. Biol. 2014, 2, 49. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, L.; Carugno, M.; Bollati, V. Particulate matter exposure shapes DNA methylation through the lifespan. Clin. Epigenetics 2019, 11, 129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Field, A.E.; Robertson, N.A.; Wang, T.; Havas, A.; Ideker, T.; Adams, P.D. DNA Methylation Clocks in Aging: Categories, Causes, and Consequences. Mol. Cell 2018, 71, 882–895. [Google Scholar] [CrossRef] [Green Version]
- Xiao, F.-H.; Wang, H.-T.; Kong, Q.-P. Dynamic DNA Methylation during Aging: A “Prophet” of Age-Related Outcomes. Front. Genet. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Jung, S.-E.; Shin, K.-J.; Lee, H.Y. DNA methylation/based age prediction from various tissues and body fluids. BMB Rep. 2017, 50, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanz, L.A.; Kota, S.A.; Feil, R. Genome-wide DNA demethylation in mammals. Genome Biol. 2010, 11, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Seisenberger, S.J.; Peat, R.; Hore, T.A.; Santos, F.; Dean, W.; Reik, W. Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philos. Trans. R. Soc. B 2013, 368, 20110330. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Hajkova, P.; Ecker, J.R. Dynamic DNA methylation: In the right place at the right time. Science 2018, 361, 1336–1340. [Google Scholar] [CrossRef] [Green Version]
- Szyf, M. The Implication of DNA Methylation for Toxicology: Toward Toxicomethylomics, the Toxicology of DNA Methylation. Toxicol. Sci. 2011, 120, 235–255. [Google Scholar] [CrossRef] [Green Version]
- Tobi, E.W.; Slieker, R.C.; Stein, A.D.; Suchiman, H.E.; Slagboom, P.E.; van Zwet, E.W.; Heijmans, B.T.; Lumey, L.H. Early gestation as the critical time-window for changes in the prenatal environment to affect the adult human blood methylome. Int. J. Epidemiol. 2015, 44, 1211–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bateson, P.; Gluckman, P.; Hanson, M. The biology of developmental plasticity and the predictive adaptive response hypothesis. J. Physiol. 2014, 592, 2357–2368. [Google Scholar] [CrossRef] [PubMed]
- Tsang, S.-Y.; Ahmad, T.; Mat, F.W.K.; Zhao, C.; Xiao, S.; Xia, K.; Xue, H. Variation of global DNA methylation levels with age and in autistic children. Human Genom. 2016, 10, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, T.M.; Myers, J.P. Environmental Exposures and Gene Regulation in Disease Etiology. Environ. Health Perspect. 2007, 115, 1264–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidman, J.R.; Dolinoy, D.C.; Murphy, S.K.; Jirtle, R.L. Cancer Susceptibility: Epigenetic Manifestation of Environmental Exposures. Cancer J. 2007, 13, 9–16. [Google Scholar] [CrossRef]
- Rossnerova, A.; Honkova, K.; Pelclova, D.; Zdimal, V.; Hubacek, J.A.; Chvojkova, I.; Vrbova, K.; Rossner, P., Jr.; Topinka, J.; Vlckova, S.; et al. DNA methylation profiles in a group of workers occupationally exposed to nanoparticles. Int. J. Mol. Sci. 2020, 21, 2420. [Google Scholar] [CrossRef] [Green Version]
- Markunas, C.A.; Xu, Z.; Harlid, S.; Wade, P.A.; Lie, R.T.; Taylor, J.A.; Wilcox, A.J. Identification of DNA Methylation Changes in Newborns Related to Maternal Smoking during Pregnancy. Environ. Health Perspect. 2014, 122, 1147–1153. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, E.P.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [Green Version]
- Van Roekel, E.H.; Dugué, P.-A.; Jung, C.-H.; Joo, J.-E.; Makalic, E.; Wong, E.M.; English, D.R.; Southey, M.C.; Giles, G.G.; Lynch, B.M.; et al. Physical Activity, Television Viewing Time and DNA Methylation in Peripheral Blood. Med. Sci. Sports Exerc. 2019, 51, 490–498. [Google Scholar] [CrossRef] [PubMed]
- Rossnerova, A.; Tulupova, E.; Tabashidze, N.; Schmuczerova, J.; Dostal, M.; Rossner, P., Jr.; Gmuender, H.; Sram, R.J. Factor affecting the 27K DNA methylation pattern in asthmatic and healthy children from locations with various environments. Mutat. Res. 2013, 741-742, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C.H.; Feinberg, J.I.; Brown, S.C.; Lurmann, F.W.; Croen, L.A.; Hertz-Picciotto, I.; Newschaffer, C.J.; Freinberg, A.P.; Fallin, M.D.; Volk, H.E. Epigenetic marks of prenatal air pollution exposure found in multiple tissues relevant for child health. Environ. Int. 2019, 126, 363–376. [Google Scholar] [CrossRef]
- Rider, C.F.; Carlsten, C. Air pollution and DNA methylation: Effects of exposure in humans. Clin. Epigenet. 2019, 11, 131. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Zhao, B.; Su, Y.; Zhang, Y.; Chen, J.; Wu, W.; Cheng, Q.; Guo, X.; Zhao, Z.; Ke, X.; et al. Association of prenatal organochlorine pesticide-dichlorodiphenyltrichloroethane exposure with fetal genome-wide DNA methylation. Life Sci. 2018, 200, 81–86. [Google Scholar] [CrossRef]
- Van Der Plaat, D.A.; De Jong, K.; De Vries, M.; Van Diemen, C.C.; Nedeljkovic, I.; Amin, N.; Kromhout, H.; Vermeulen, R.C.H.; Postma, D.S.; Van Duijn, C.M.; et al. Occupational exposure to pesticides is associated with differential DNA methylation. Occup. Environ. Med. 2018, 75, 427–435. [Google Scholar] [CrossRef]
- Zeng, Z.; Huo, X.; Zhang, Y.; Hylkema, M.N.; Wu, Y.; Xu, X. Differential DNA methylation in newborns with maternal exposure to heavy metals from an e-waste recycling area. Environ. Res. 2019, 171, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Liou, S.H.; Wu, W.T.; Liao, H.Y.; Chen, C.Y.; Tsai, C.Y.; Jung, W.T.; Lee, H.L. Global DNA methylation and oxidative stress biomarkers in workers exposed to metal oxide nanoparticles. J. Hazard. Mater. 2017, 331, 329–335. [Google Scholar] [CrossRef]
- Martins, J.; Czamara, D.; Sauer, S.; Rex-Haffner, M.; Dittrich, K.; Dörr, P.; de Punder, K.; Overfeld, J.; Knop, A.; Dammering, F.; et al. Childhood adversity correlates with stable changes in DNA methylation trajectories in children and converges with epigenetic signatures of prenatal stress. Neurobiol. Stress 2021, 15, 1–13. [Google Scholar] [CrossRef]
- Aronica, L.; Levine, A.J.; Brennan, K.; Mi, J.; Gardner, C.; Haile, R.W.; Hitchins, M.P. A systematic review of studies of DNA methylation in the context of a weight loss intervention. Epigenomics 2017, 9, 769–787. [Google Scholar] [CrossRef]
- Öner, D.; Ghosh, M.; Bové, H.; Moisse, M.; Boeckx, B.; Duca, R.C.; Poels, K.; Luyts, K.; Putzeys, E.; Landuydt, K.V.; et al. Differences in MWCNT- and SWCNT- induced DNA methylation alterations in association with the nuclear deposition. Part. Fibre Toxicol. 2018, 15, 1–19. [Google Scholar] [CrossRef]
- Moran, S.; Arribas, C.; Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016, 8, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Belli, M.; Tabocchini, M.A. Ionizing Radiation-Induced Epigenetic Modifications and Their Relevance to Radiation Protection. Int. J. Mol. Sci. 2020, 21, e5993. [Google Scholar] [CrossRef]
- Rossnerova, A.; Pokorna, M.; Svecova, V.; Sram, R.J.; Topinka, J.; Zölzer, F.; Rossner, P., Jr. Adaptation of the human population to the environment: Current knowledge, clues from Czech cytogenetic and “omics” biomonitoring studies and possible mechanisms. Mutat. Res. 2017, 773, 188–203. [Google Scholar] [CrossRef] [PubMed]
- Rossnerova, A.; Izzotti, A.; Pulliero, A.; Bast, A.; Rattan, S.I.S.; Rossner, P., Jr. The molecular mechanisms of adaptive response related to the environmental stress. Int. J. Mol. Sci. 2020, 21, 7053. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, C.; Bacalini, M.G.; Sazzini, M.; Pirazzini, C.; Franceschi, C.; Garagnani, P.; Luiselli, D. The epigenetic side of human adaptation: Hypotheses, evidences and theories. Ann. Hum. Biol. 2015, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vineis, P.; Chatziioannou, A.; Cunliffe, V.T.; Flanagan, J.M.; Hanson, M.; Kirsch-Volders, M.; Kyrtopoulos, S. Epigenetic memory in response to environmental stressors. FASEB J. 2017, 31, 2241–2251. [Google Scholar] [CrossRef] [Green Version]
- Hoang, T.T.; Sikdar, S.; Xu, C.-J.; Lee, M.K.; Cardwell, J.; Forno, E.; Imboden, M.; Jeong, A.; Madore, A.-M.; Qi, C.; et al. Epigenome-wide association study of DNA methylation and adult asthma in the Agricultural Lung Health Study. Eur. Respir. J. 2020, 56, 1–187. [Google Scholar] [CrossRef]
- Cortesi, E.; Ventura, J.J. Lgr6: From Stemness to Cancer Progression. J. Lung Health Dis. 2019, 3, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Raslan, A.A.; Yoon, J.K. R-spondins: Multi-mode WNT signaling regulators in adult stem cells. Int. J. Biochem. Cell Biol. 2019, 106, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Moszcyński, P.; Rutowski, J.; Słowiński, S.; Bern, S.; Jakus-Stoga, D. Effects of occupational exposure to mercury vapors on T-cell and NK-cell populations. Arch. Med. Res. Winter 1996, 27, 503–507. [Google Scholar]
- Guo, J.U.; Su, Y.; Zhong, C.; Ming, G.; Song, H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 2011, 145, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Talbot, N.; Kubelova, L.; Makes, O.; Ondracek, J.; Cusack, M.; Schwarz, J.; Vodicka, P.; Zikova, N.; Zdimal, V. Transformations of aerosol particles from an outdoor to indoor environment. Aerosol Air Qual. Res. 2017, 17, 653–665. [Google Scholar] [CrossRef] [Green Version]
- Pelclova, D.; Zdimal, V.; Schwarz, J.; Dvorackova, S.; Komarc, M.; Ondracek, J.; Kostejn, M.; Kacer, P.; Vlckova, S.; Fenclova, Z.; et al. Markers of oxidative stress in the exhaled breath condensate of workers handling nanocomposites. Nanomaterials 2018, 8, 611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelclova, D.; Zdimal, V.; Komarc, M.; Schwarz, J.; Ondracek, J.; Ondrackova, L.; Kostejn, M.; Vlckova, S.; Fenclova, Z.; Dvorackova, S.; et al. Three-Year Study of Markers of Oxidative Stress in Exhaled Breath Condensate in Workers Producing Nanocomposites, Extended by Plasma and Urine Analysis in Last Two Years. Nanomaterials 2020, 10, 2440. [Google Scholar] [CrossRef]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [Green Version]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef] [Green Version]
- McCartney, D.L.; Walker, R.M.; Morris, S.W.; McIntosh, A.M.; Porteous, D.J.; Evans, K.L. Identification of polymorphic and off-target probe binding sites on the Illumina Infinium MethylationEPIC BeadChip. Genom. Data 2016, 9, 22–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houseman, W.P.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, N.; Mather, K.A.; Thalamuthu, A.; Wright, M.J.; Trollor, J.N.; Ames, D.; Brodaty, H.; Schofield, P.R.; Sachdev, P.S.; Kwok, J.B. Aging, exceptional longevity and comparisons of the Hannum and Horvath epigenetic clocks. Epigenomics 2017, 9, 689–700. [Google Scholar] [CrossRef]






| Group. Participant # | N | Sex a (M/F) | Age a (4-Year Range b) | BMI a (kg/m2) (4-Year Range c) | Exposure Total (4-Year Range b) | Exposure of Last Occupation (4-Year Range b) |
|---|---|---|---|---|---|---|
| Exposed (All) | 10 | 8/2 | 30–66 | 19.2–36.7 | 5–33 | 4–23 |
| #1 | M | 30–33 | 24.9–27.8 | 8–11 | 7–10 | |
| #2 | M | 31–34 | 32.9–36.2 | 6–9 | 6–9 | |
| #3 | M | 32–35 | 19.2–21.3 | 10–13 | 4–7 | |
| #4 | M | 33–36 | 25.7–28.8 | 10–13 | 7–10 | |
| #5 | M | 36–39 | 22.7–23.9 | 5–8 | 4–7 | |
| #6 | M | 49–52 | 25.2–30.3 | 30–33 | 7–10 | |
| #7 | M | 54–57 | 33.2–36.7 | 25–28 | 9–12 | |
| #8 | M | 63–66 | 24.8–26.6 | 10–13 | 8–11 | |
| #9 | F | 37–40 | 19.9–21.7 | 10–13 | 6–9 | |
| #10 | F | 50–53 | 26.2–27.9 | 25–28 | 20–23 | |
| Controls (All) | 4 | 3/1 | 35–57 | 20.1–38.9 | N/A | N/A |
| #11 | M | 39–42 | 25.6–26.7 | N/A | N/A | |
| #12 | M | 48–51 | 33.1–35.4 | N/A | N/A | |
| #13 | M | 54–57 | 36.6–38.9 | N/A | N/A | |
| #14 | F | 35–38 | 20.1–22.1 | N/A | N/A |
| Year | Processing Backgrounds | Nano PM <25–100 nm | Major PM >100–10 µm | Processed Materials |
|---|---|---|---|---|
| 2016 | Welding | 48,800 | 72,933 | Mild steel S355J2 |
| Smelting | 46,000 | 2603 | AlSi9Cu3, alloy | |
| Background in basement | 20,100 | 673 | N/A | |
| Grinding and milling | 322,000 | 204,030 | Epoxide resin with nano-SiO2 | |
| Background in ground floor | 116,000 | 168,019 | N/A | |
| 2017 | Grinding and milling | 89,900 | 4045 | Epoxide resin with nano-SiO2 and geopolymers with metakaolin, ash or basalt |
| Background in ground floor | 4680 | 4101 | N/A | |
| 2018 | Welding | 5520 | 3791 | Mild steel S355J2 |
| Background in basement | 6834 | 2370 | N/A | |
| Grinding and milling | 26,100 | 3059 | Epoxide resin with nano-SiO2 | |
| Background in ground floor | 3642 | 1680 | N/A | |
| 2019 | Welding | 3793 | 12,839 | Mild steel S355J2 |
| Background in basement | 3017 | 998 | N/A | |
| Grinding and milling | 12,830 | 1075 | Epoxide resin with nano-SiO2 | |
| Background in ground floor | 1536 | 728 | N/A |
| Beta Values in Exposed Subjects #1–#10 | Beta Values in Control Subjects #11–#14 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2016 | 2017 | 2018 | 2019 | 2016 vs. 2019 | 2016 | 2017 | 2018 | 2019 | 2016 vs. 2019 | |
| Probe Chr.: Gene | Mean ± SD Median (Range) | Mean Diff. (%) | Mean ± SD Median (Range) | Mean Diff. (%) | ||||||
| cg04811114 1: LGR6 | 0.25 ± 0.09 0.26 (0.13–0.37) | 0.24 ± 0.09 0.26 (0.13–0.35) | 0.24 ± 0.09 0.26 (0.13–0.34) | 0.25 ± 0.07 ** 0.27 (0.15–0.36) | 0 | 0.17 ± 0.05 0.15 (0.14–0.24) | 0.15 ± 0.01 0.15 (0.14–0.16) | 0.17 ± 0.07 0.15 (0.12–0.27) | 0.12 ± 0.02 0.13 (0.09–0.14) | −5 |
| cg06825163 1: LGR6 | 0.25 ± 0.09 * 0.28 (0.13–0.35) | 0.25 ± 0.10 * 0.27 (0.12–0.37) | 0.24 ± 0.09 0.25 (0.13–0.34) | 0.25 ± 0.07 ** 0.25 (0.15–0.36) | 0 | 0.16 ± 0.05 0.14 (0.12–0.23) | 0.13 ± 0.01 0.13 (0.12–0.15) | 0.16 ± 0.05 0.14 (0.13–0.23) | 0.11 ± 0.02 0.12 (0.08–0.13) | −4 |
| cg09622121 6: HCG27 * | 0.06 ± 0.03 0.05 (0.03–0.11) | 0.07 ± 0.04 0.05 (0.03–0.13) | 0.09 ± 0.05 0.07 (0.04–0.17) | 0.06 ± 0.02 ** 0.05 (0.03–0.09) | 0 | 0.11 ± 0.02 0.12 (0.08–0.13) | 0.11 ± 0.05 0.14 (0.04–0.14) | 0.12 ± 0.03 0.13 (0.08–0.15) | 0.16 ± 0.08 0.19 (0.05–0.23) | −5 |
| cg03030317 6: HCG27 * | 0.09 ± 0.03 0.08 (0.05–0.16) | 0.10 ± 0.04 * 0.08 (0.05–0.19) | 0.11 ± 0.05 * 0.10 (0.06–0.19) | 0.08 ± 0.02 ** 0.08 (0.06–0.13) | −1 | 0.14 ± 0.06 0.15 (0.05–0.18) | 0.18 ± 0.08 0.21 (0.06–0.25) | 0.18 ± 0.08 0.22 (0.07–0.22) | 0.25 ± 0.12 0.27 (0.08–0.36) | +11 |
| cg12771717 6: HCG27 * | 0.08 ± 0.05 0.05 (0.03–0.14) | 0.09 ± 0.05 0.09 (0.03–0.18) | 0.09 ± 0.06 0.06 (0.02–0.24) | 0.07 ± 0.03 ** 0.06 (0.04–0.13) | −1 | 0.13 ± 0.06 0.15 (0.04–0.16) | 0.15 ± 0.06 0.17 (0.06–0.21) | 0.15 ± 0.06 0.17 (0.05–0.19) | 0.19 ± 0.10 0.24 (0.04–0.25) | +6 |
| cg18467790 7: RADIL | 0.43 ± 0.14 0.42 (0.18–0.69) | 0.45 ± 0.15 0.44 (0.21–0.71) | 0.44 ± 0.15 0.41 (0.18–0.70) | 0.39 ± 0.16 0.35 (0.18–0.79) | −4 | 0.31 ± 0.08 0.28 (0.25–0.42) | 0.32 ± 0.07 0.29 (0.27–0.43) | 0.30 ± 0.07 0.28 (0.25–0.41) | 0.39 ± 0.08 0.38 (0.33–0.49) | +8 |
| cg07044115 10: FGFR2 | 0.45 ± 0.12 0.39 (0.33–0.64) | 0.47 ± 0.12 0.42 (0.32–0.65) | 0.46 ± 0.13 0.44 (0.32–0.65) | 0.44 ± 0.10 0.41 (0.33–0.65) | −1 | 0.56 ± 0.10 0.55 (0.47–0.67) | 0.54 ± 0.07 0.55 (0.46–0.61) | 0.55 ± 0.07 0.54 (0.48–0.62) | 0.55 ± 0.17 0.54 (0.40–0.73) | −1 |
| cg16653991 10: FGFR2 | 0.34 ± 0.10 0.34 (0.19–0.47) | 0.35 ± 0.09 0.36 (0.19–0.46) | 0.33 ± 0.10 0.31 (0.18–0.47) | 0.36 ± 0.09 0.39 (0.16–0.45) | +2 | 0.24 ± 0.07 0.24 (0.16–0.30) | 0.26 ± 0.08 0.26 (0.18–0.33) | 0.26 ± 0.08 0.26 (0.19–0.34) | 0.28 ± 0.16 0.28 (0.11–0.43) | +4 |
| cg10379346 10: FGFR2 | 0.41 ± 0.10 0.41 (0.29–0.58) | 0.40 ± 0.11 0.38 (0.26–0.58) | 0.41 ± 0.11 0.39 (0.29–0.57) | 0.38 ± 0.10 0.36 (0.24–0.61) | −3 | 0.47 ± 0.10 0.49 (0.35–0.55) | 0.48 ± 0.12 0.50 (0.34–0.58) | 0.46 ± 0.11 0.48 (0.33–0.56) | 0.48 ± 0.16 0.45 (0.34–0.67) | +1 |
| cg06791446 10: FGFR2 | 0.43 ± 0.11 0.46 (0.29–0.60) | 0.42 ± 0.10 0.41 (0.30–0.60) | 0.45 ± 0.11 0.42 (0.31–0.61) | 0.40 ± 0.10 0.37 (0.29–0.63) | −3 | 0.53 ± 0.09 0.55 (0.40–0.61) | 0.53 ± 0.10 0.54 (0.39–0.62) | 0.49 ± 0.10 0.51 (0.36 -0.59) | 0.52 ± 0.17 0.50 (0.37–0.70) | −1 |
| cg25052156 10: FGFR2 | 0.57 ± 0.10 0.54 (0.43–0.74) | 0.58 ± 0.10 0.55 (0.43–0.74) | 0.56 ± 0.10 0.52 (0.44–0.72) | 0.54 ± 0.09 0.53 (0.43–0.74) | −3 | 0.65 ± 0.12 0.67 (0.50–0.75) | 0.64 ± 0.08 0.67 (0.54–0.71) | 0.64 ± 0.08 0.64 (0.56–0.71) | 0.62 ± 0.18 0.62 (0.45–0.81) | −3 |
| cg15570860 11: TMEM9B | 0.77 ± 0.17 0.85 (0.57–0.96) | 0.78 ± 0.17 0.86 (0.56–0.97) | 0.79 ± 0.18 0.88 (0.57–0.96) | 0.79 ± 0.17 0.87 (0.58–0.97) | −2 | 0.93 ± 0.02 0.92 (0.91–0.95) | 0.91 ± 0.05 0.92 (0.84–0.96) | 0.91 ± 0.04 0.92 (0.86–0.95) | 0.95 ± 0.01 0.95 (0.94–0.97) | +2 |
| cg14764203 19: FCGBP | 0.52 ± 0.13 0.54 (0.33–0.67) | 0.51 ± 0.13 0.54 (0.33–0.70) | 0.52 ± 0.13 0.53 (0.33–0.69) | 0.53 ± 0.14 0.59 (0.32–0.68) | +1 | 0.42 ± 0.12 0.42 (0.29–0.55) | 0.41 ± 0.10 0.42 (0.30–0.52) | 0.40 ± 0.11 0.41 (0.29–0.51) | 0.44 ± 0.24 0.45 (0.16–0.68) | +2 |
| cg03635532 19: FCGBP | 0.63 ± 0.15 0.63 (0.37–0.79) | 0.61 ± 0.16 0.56 (0.42–0.79) | 0.61 ± 0.15 0.63 (0.39–0.77) | 0.65 ± 0.15 0.71 (0.41–0.80) | +2 | 0.47 ± 0.15 0.46 (0.33–0.63) | 0.53 ± 0.14 0.52 (0.41–0.68) | 0.49 ± 0.12 0.49 (0.37–0.62) | 0.51 ± 0.27 0.55 (0.18–0.77) | +4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossnerova, A.; Honkova, K.; Chvojkova, I.; Pelclova, D.; Zdimal, V.; Hubacek, J.A.; Lischkova, L.; Vlckova, S.; Ondracek, J.; Dvorackova, S.; et al. Individual DNA Methylation Pattern Shifts in Nanoparticles-Exposed Workers Analyzed in Four Consecutive Years. Int. J. Mol. Sci. 2021, 22, 7834. https://doi.org/10.3390/ijms22157834
Rossnerova A, Honkova K, Chvojkova I, Pelclova D, Zdimal V, Hubacek JA, Lischkova L, Vlckova S, Ondracek J, Dvorackova S, et al. Individual DNA Methylation Pattern Shifts in Nanoparticles-Exposed Workers Analyzed in Four Consecutive Years. International Journal of Molecular Sciences. 2021; 22(15):7834. https://doi.org/10.3390/ijms22157834
Chicago/Turabian StyleRossnerova, Andrea, Katerina Honkova, Irena Chvojkova, Daniela Pelclova, Vladimir Zdimal, Jaroslav A. Hubacek, Lucie Lischkova, Stepanka Vlckova, Jakub Ondracek, Stepanka Dvorackova, and et al. 2021. "Individual DNA Methylation Pattern Shifts in Nanoparticles-Exposed Workers Analyzed in Four Consecutive Years" International Journal of Molecular Sciences 22, no. 15: 7834. https://doi.org/10.3390/ijms22157834