Identifying the Translatome of Mouse NEBD-Stage Oocytes via SSP-Profiling; A Novel Polysome Fractionation Method

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
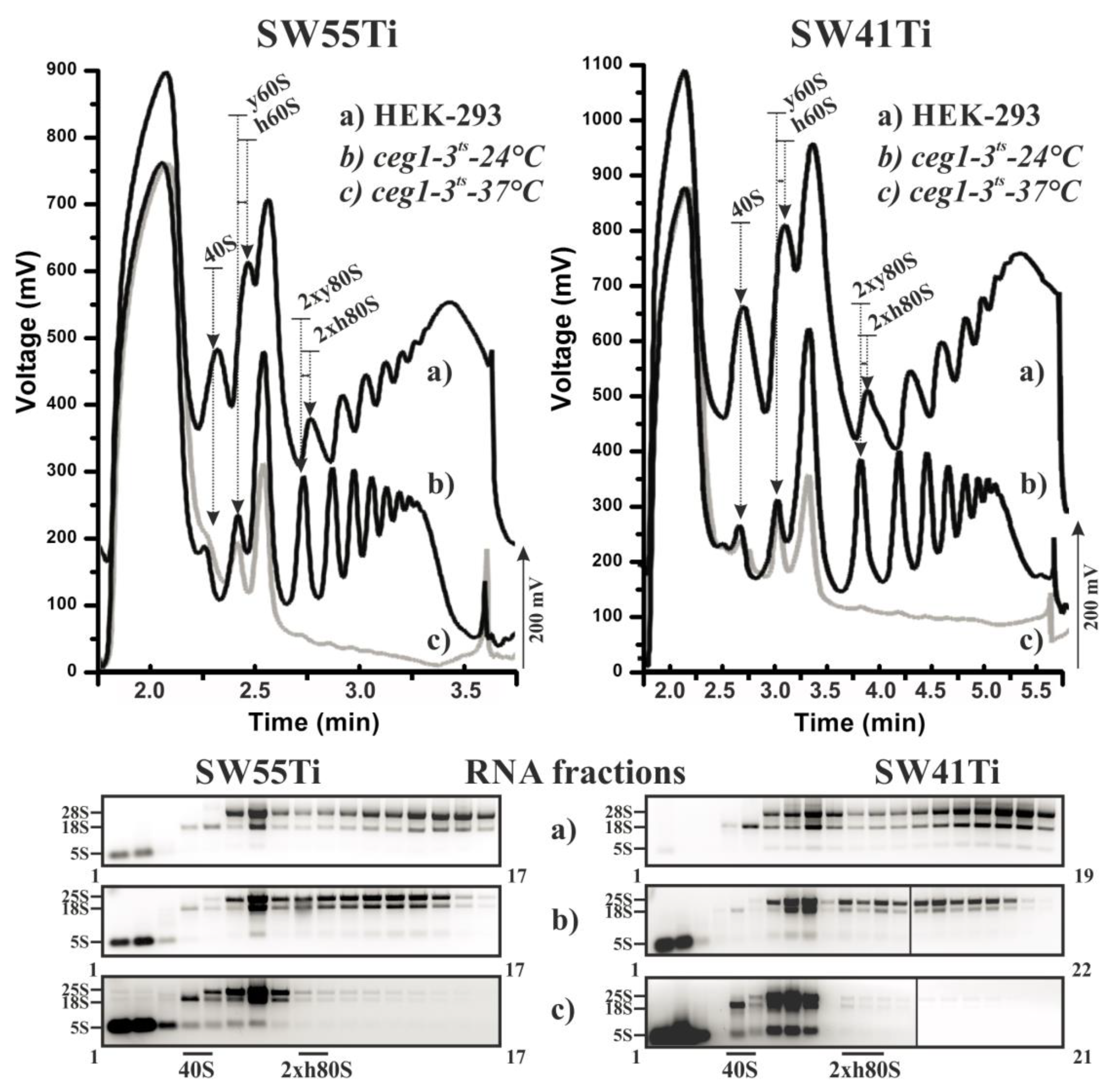
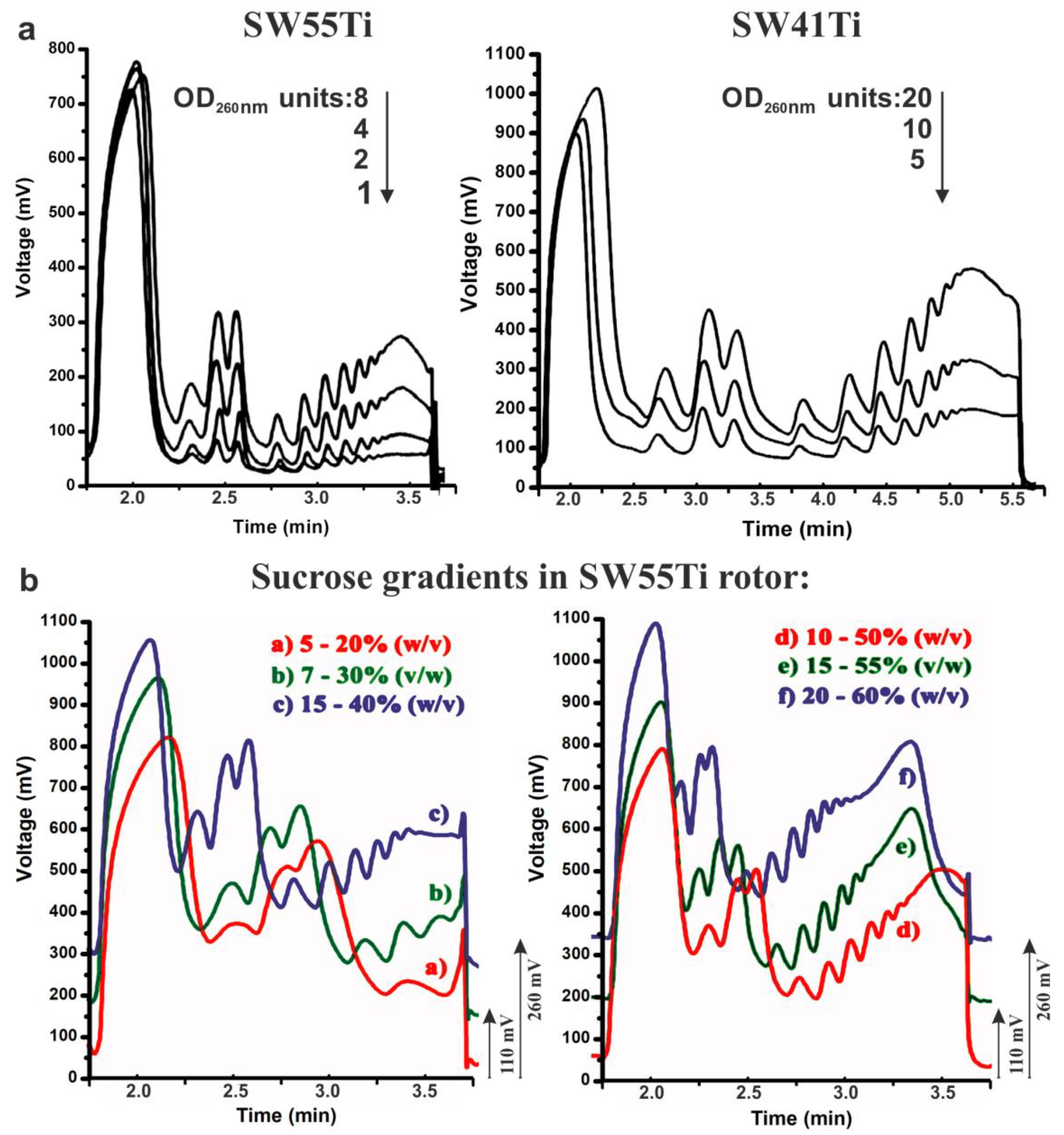
2.1. Optimisation of Polysome Profiling in SW55Ti Tubes
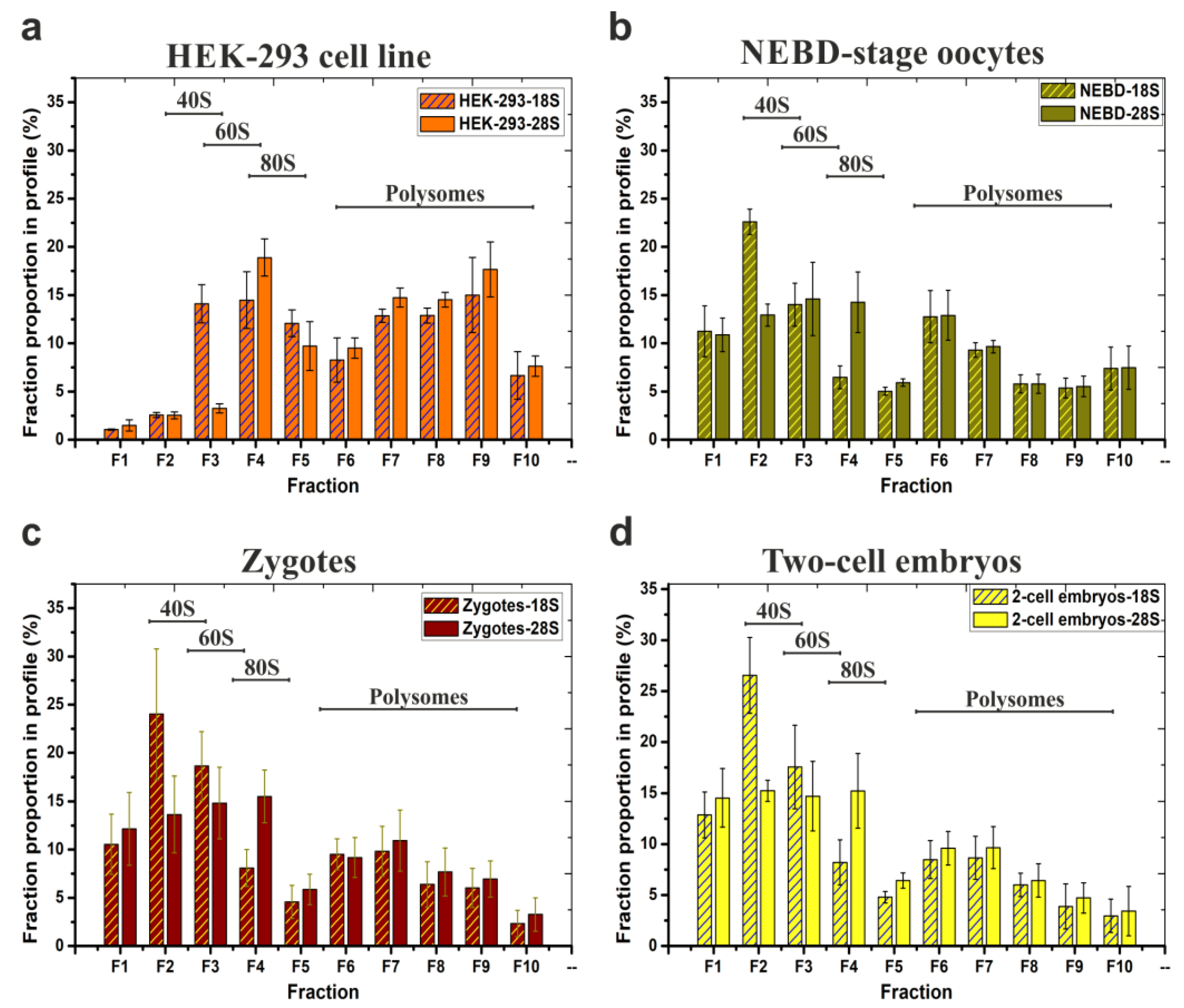
2.2. Scarce Sample Polysome Profiling (SSP-Profiling) in SW55Ti-Based Format
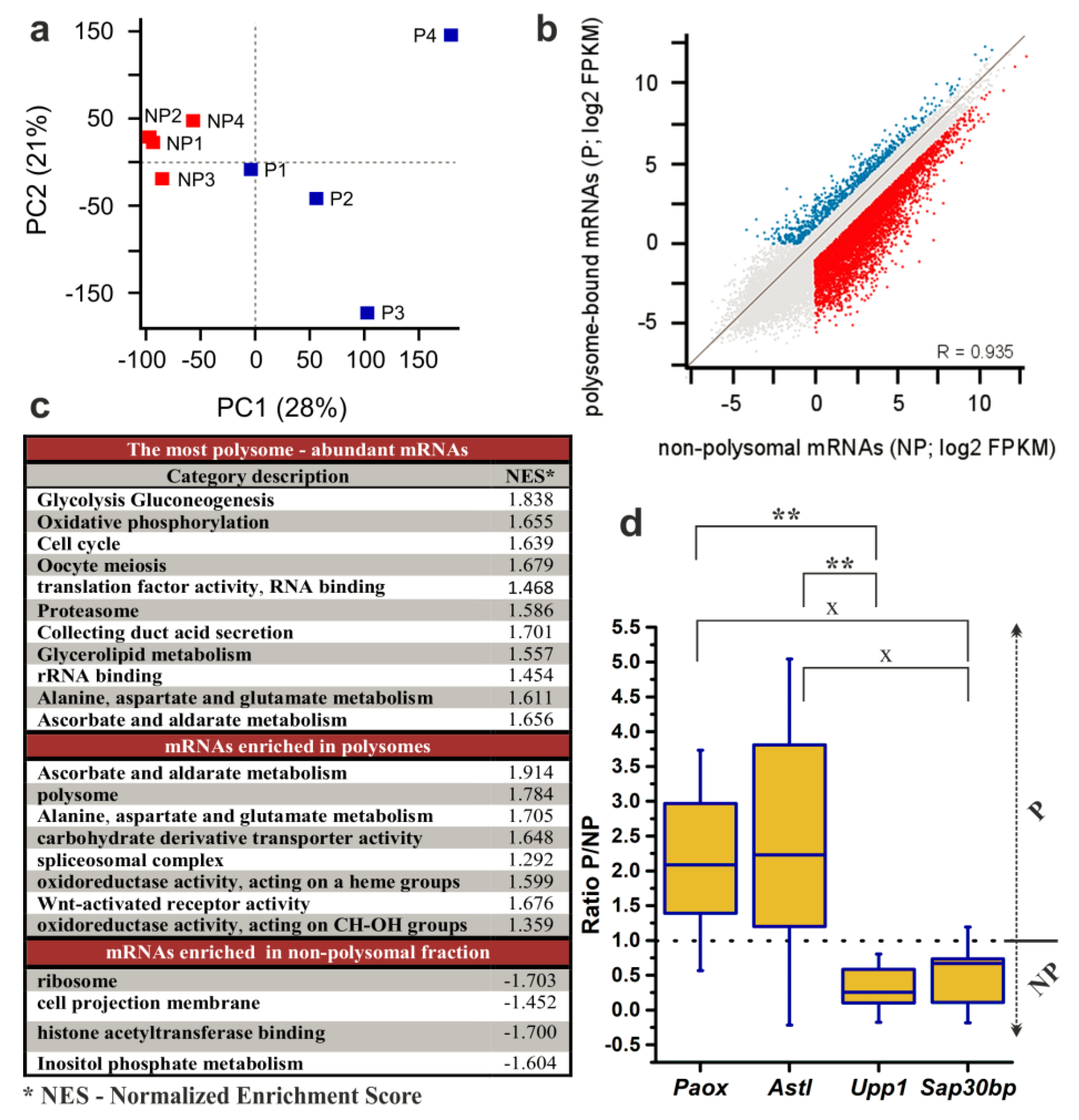
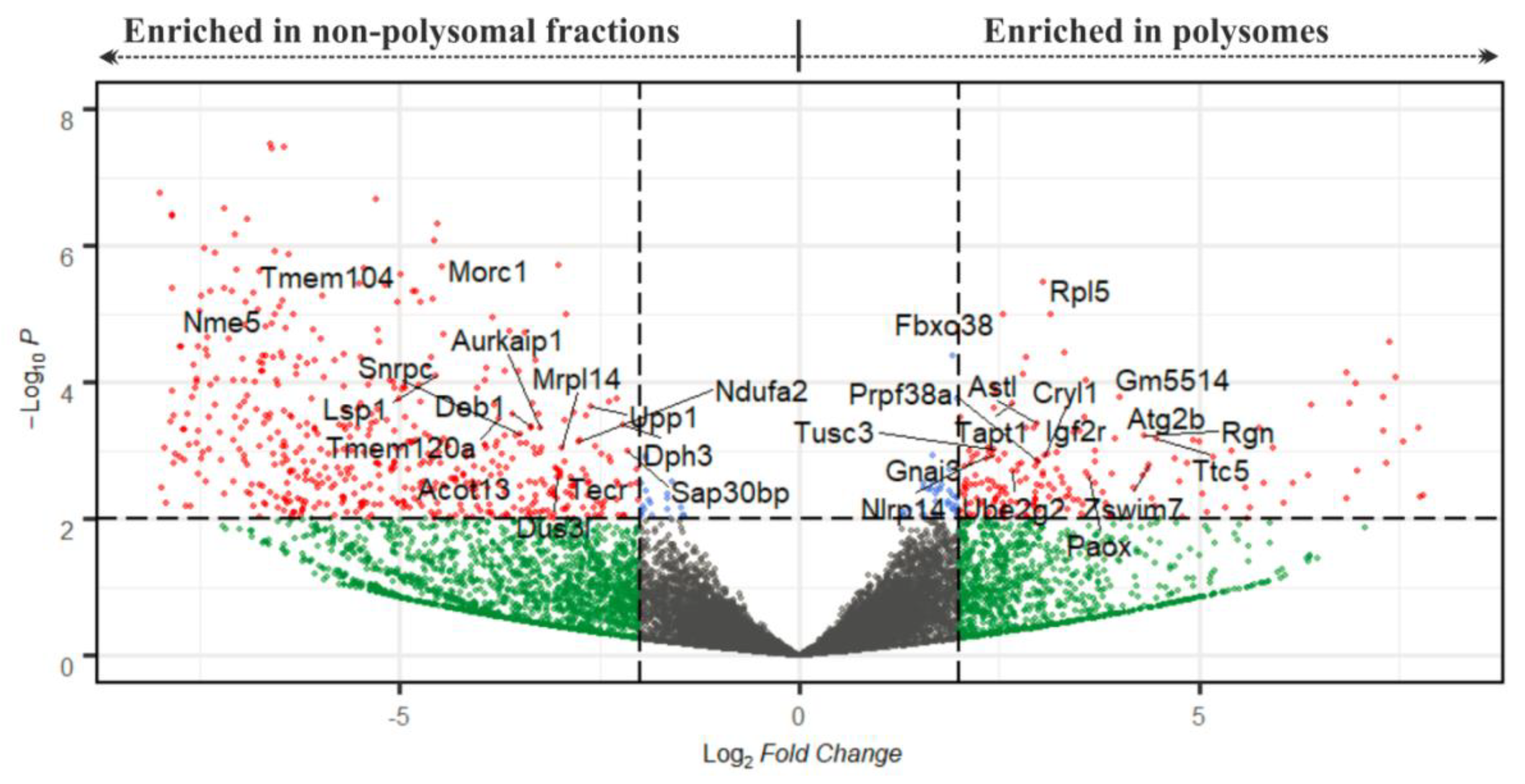
2.3. RNA-Seq Analysis to Reveal the Translatome of NEBD-Stage Oocytes
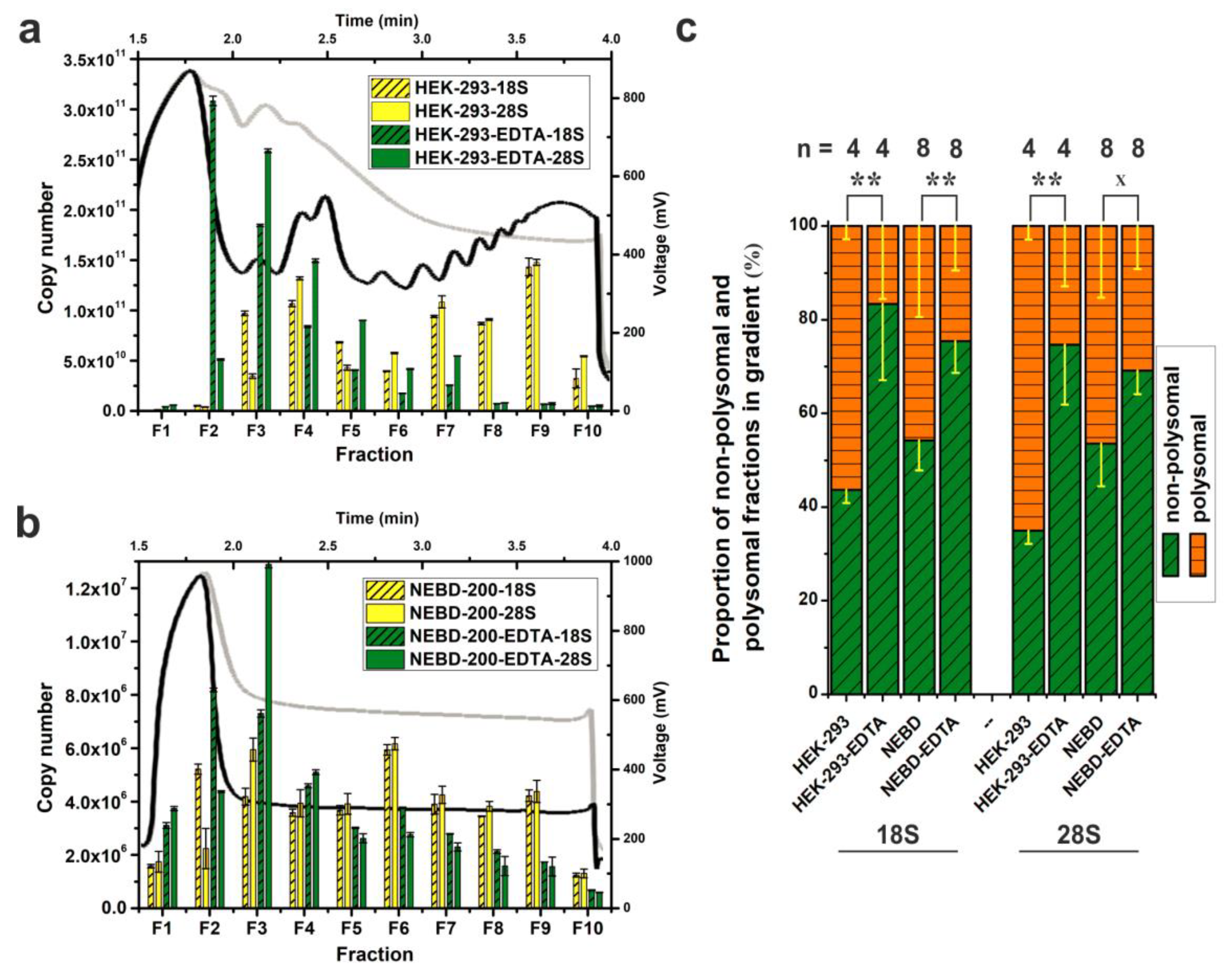
2.4. Sensitivity and Versatility of SPP-Profiling for Conventional Sample Sizes and Compatibility with Downstream Methods
3. Discussion
4. Materials and Methods
4.1. Human Cell Line, Yeast Strains and Mouse Oocyte, Zygotes and 2C-Embryos Isolation and Culture
4.2. Preparation of Lysates and Polysomal Profiling
4.3. RNA Isolation, RNA Electrophoresis and qRT-PCR
4.4. RNA-Seq Library Preparation
4.5. Bioinformatical and Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mathews, M.B.; Sonenberg, N.; Hershey, J.W. Origins and Principles of Translational control. In Translational Control in Biology and Medicine; Mathews, M.B., Sonenberg, N., Hershey, J.W., Eds.; Cold Spring Harb Lab. Press: New York, NY, USA, 2007; pp. 1–40. [Google Scholar]
- Holcik, M.; Sonenberg, N. Translational control in stress and apoptosis. Nat. Rev. Mol. Cell Biol. 2005, 6, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: The Achilles heel of pancreatic beta cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Susor, A.; Jansova, D.; Anger, M.; Kubelka, M. Translation in the mammalian oocyte in space and time. Cell Tissue Res. 2016, 363, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Warner, J.R.; Knopf, P.M.; Rich, A. A multiple ribosomal structure in protein synthesis. Proc. Natl. Acad. Sci. USA 1963, 49, 122–129. [Google Scholar] [CrossRef] [Green Version]
- Gamm, M.; Peviani, A.; Honsel, A.; Snel, B.; Smeekens, S.; Hanson, J. Increased sucrose levels mediate selective mRNA translation in Arabidopsis. BMC Plant Biol. 2014, 14, 306. [Google Scholar] [CrossRef] [Green Version]
- Ashe, M.P.; De Long, S.K.; Sachs, A.B. Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 2000, 11, 833–848. [Google Scholar] [CrossRef] [Green Version]
- Shalgi, R.; Hurt, J.A.; Krykbaeva, I.; Taipale, M.; Lindquist, S.; Burge, C.B. Widespread regulation of translation by elongation pausing in heat shock. Mol. Cell 2013, 49, 439–452. [Google Scholar] [CrossRef] [Green Version]
- Swaminathan, S.; Masek, T.; Molin, C.; Pospisek, M.; Sunnerhagen, P. Rck2 is required for reprogramming of ribosomes during oxidative stress. Mol. Biol. Cell 2006, 17, 1472–1482. [Google Scholar] [CrossRef] [Green Version]
- Sydorskyy, Y.; Dilworth, D.J.; Halloran, B.; Yi, E.C.; Makhnevych, T.; Wozniak, R.W.; Aitchison, J.D. Nop53p is a novel nucleolar 60S ribosomal subunit biogenesis protein. Biochem. J. 2005, 388, 819–826. [Google Scholar] [CrossRef] [Green Version]
- Choudhuri, A.; Maitra, U.; Evans, T. Translation initiation factor eIF3h targets specific transcripts to polysomes during embryogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 9818–9823. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Nunez, R.T.; Wallace, A.; Coyne, D.; Jansson, L.; Rush, M.; Ennajdaoui, H.; Katzman, S.; Bailey, J.; Deinhardt, K.; Sanchez-Elsner, T.; et al. Modulation of nonsense mediated decay by rapamycin. Nucleic Acids Res. 2017, 45, 3448–3459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Androsavich, J.R.; Sobczynski, D.J.; Liu, X.; Pandya, S.; Kaimal, V.; Owen, T.; Liu, K.; MacKenna, D.A.; Chau, B.N. Polysome shift assay for direct measurement of miRNA inhibition by anti-miRNA drugs. Nucleic Acids Res. 2016, 44, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, D.; Fredrick, K. Analysis of polysomes from bacteria. Methods Enzymol. 2013, 530, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Seimetz, J.; Arif, W.; Bangru, S.; Hernaez, M.; Kalsotra, A. Cell-type specific polysome profiling from mammalian tissues. Methods 2018, 155, 131–139. [Google Scholar] [CrossRef]
- Kawaguchi, R.; Bailey-Serres, J. mRNA sequence features that contribute to translational regulation in Arabidopsis. Nucleic Acids Res. 2005, 33, 955–965. [Google Scholar] [CrossRef] [Green Version]
- Kopeina, G.S.; Afonina, Z.A.; Gromova, K.V.; Shirokov, V.A.; Vasiliev, V.D.; Spirin, A.S. Step-wise formation of eukaryotic double-row polyribosomes and circular translation of polysomal mRNA. Nucleic Acids Res. 2008, 36, 2476–2488. [Google Scholar] [CrossRef] [Green Version]
- McGlincy, N.J.; Ingolia, N.T. Transcriptome-wide measurement of translation by ribosome profiling. Methods 2017, 126, 112–129. [Google Scholar] [CrossRef]
- Fu, Y.; Chen, L.; Chen, C.; Ge, Y.; Kang, M.; Song, Z.; Li, J.; Feng, Y.; Huo, Z.; He, G.; et al. Crosstalk between alternative polyadenylation and miRNAs in the regulation of protein translational efficiency. Genome Res. 2018, 28, 1656–1663. [Google Scholar] [CrossRef]
- Floor, S.N.; Doudna, J.A. Tunable protein synthesis by transcript isoforms in human cells. eLife 2016, 5. [Google Scholar] [CrossRef]
- Sterne-Weiler, T.; Martinez-Nunez, R.T.; Howard, J.M.; Cvitovik, I.; Katzman, S.; Tariq, M.A.; Pourmand, N.; Sanford, J.R. Frac-seq reveals isoform-specific recruitment to polyribosomes. Genome Res. 2013, 23, 1615–1623. [Google Scholar] [CrossRef] [Green Version]
- Peng, X.; Emiliani, F.; Smallwood, P.M.; Rattner, A.; Lei, H.; Sabbagh, M.F.; Nathans, J. Affinity capture of polyribosomes followed by RNAseq (ACAPseq), a discovery platform for protein-protein interactions. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Aviner, R.; Hofmann, S.; Elman, T.; Shenoy, A.; Geiger, T.; Elkon, R.; Ehrlich, M.; Elroy-Stein, O. Proteomic analysis of polyribosomes identifies splicing factors as potential regulators of translation during mitosis. Nucleic Acids Res. 2017, 45, 5945–5957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castelli, L.M.; Talavera, D.; Kershaw, C.J.; Mohammad-Qureshi, S.S.; Costello, J.L.; Rowe, W.; Sims, P.F.; Grant, C.M.; Hubbard, S.J.; Ashe, M.P.; et al. The 4E-BP Caf20p Mediates Both eIF4E-Dependent and Independent Repression of Translation. PLoS Genet. 2015, 11, e1005233. [Google Scholar] [CrossRef] [PubMed]
- de Sousa Abreu, R.; Penalva, L.O.; Marcotte, E.M.; Vogel, C. Global signatures of protein and mRNA expression levels. Mol. BioSyst. 2009, 5, 1512–1526. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Cui, Y.; Jin, J.; Guo, J.; Wang, G.; Yin, X.; He, Q.Y.; Zhang, G. Translating mRNAs strongly correlate to proteins in a multivariate manner and their translation ratios are phenotype specific. Nucleic Acids Res. 2013, 41, 4743–4754. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, B.; Nikolay, R.; Spahn, C.M.T.; Zhang, G. Translatomics: The Global View of Translation. Int. J. Mol. Sci. 2019, 20, 212. [Google Scholar] [CrossRef] [Green Version]
- Chasse, H.; Boulben, S.; Costache, V.; Cormier, P.; Morales, J. Analysis of translation using polysome profiling. Nucleic Acids Res. 2017, 45, e15. [Google Scholar] [CrossRef] [Green Version]
- Masek, T.; Valasek, L.; Pospisek, M. Polysome analysis and RNA purification from sucrose gradients. Methods Mol. Biol. 2011, 703, 293–309. [Google Scholar] [CrossRef]
- Potireddy, S.; Vassena, R.; Patel, B.G.; Latham, K.E. Analysis of polysomal mRNA populations of mouse oocytes and zygotes: Dynamic changes in maternal mRNA utilization and function. Dev. Biol. 2006, 298, 155–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scantland, S.; Grenon, J.P.; Desrochers, M.H.; Sirard, M.A.; Khandjian, E.W.; Robert, C. Method to isolate polyribosomal mRNA from scarce samples such as mammalian oocytes and early embryos. BMC Dev. Biol. 2011, 11, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ringquist, S.; Cho, A.H.; Rondeau, G.; Welsh, J. High-throughput polyribosome fractionation. Nucleic Acids Res. 2004, 32, e79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuh, M.; Ellenberg, J. Self-organization of MTOCs replaces centrosome function during acentrosomal spindle assembly in live mouse oocytes. Cell 2007, 130, 484–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collado-Fernandez, E.; Picton, H.M.; Dumollard, R. Metabolism throughout follicle and oocyte development in mammals. Int. J. Dev. Biol. 2012, 56, 799–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gahurova, L.; Tomizawa, S.I.; Smallwood, S.A.; Stewart-Morgan, K.R.; Saadeh, H.; Kim, J.; Andrews, S.R.; Chen, T.; Kelsey, G. Transcription and chromatin determinants of de novo DNA methylation timing in oocytes. Epigenetics Chromatin 2017, 10, 25. [Google Scholar] [CrossRef] [PubMed]
- Susor, A.; Kubelka, M. Translational Regulation in the Mammalian Oocyte. Results Probl. Cell Differ. 2017, 63, 257–295. [Google Scholar] [CrossRef]
- Stebbins-Boaz, B.; Hake, L.E.; Richter, J.D. CPEB controls the cytoplasmic polyadenylation of cyclin, Cdk2 and c-mos mRNAs and is necessary for oocyte maturation in Xenopus. EMBO J. 1996, 15, 2582–2592. [Google Scholar] [CrossRef]
- Igea, A.; Mendez, R. Meiosis requires a translational positive loop where CPEB1 ensues its replacement by CPEB4. EMBO J. 2010, 29, 2182–2193. [Google Scholar] [CrossRef] [Green Version]
- Eliscovich, C.; Peset, I.; Vernos, I.; Mendez, R. Spindle-localized CPE-mediated translation controls meiotic chromosome segregation. Nat. Cell Biol. 2008, 10, 858–865. [Google Scholar] [CrossRef]
- Belloc, E.; Pique, M.; Mendez, R. Sequential waves of polyadenylation and deadenylation define a translation circuit that drives meiotic progression. Biochem. Soc. Trans. 2008, 36, 665–670. [Google Scholar] [CrossRef] [Green Version]
- Susor, A.; Jansova, D.; Cerna, R.; Danylevska, A.; Anger, M.; Toralova, T.; Malik, R.; Supolikova, J.; Cook, M.S.; Oh, J.S.; et al. Temporal and spatial regulation of translation in the mammalian oocyte via the mTOR-eIF4F pathway. Nat. Commun. 2015, 6, 6078. [Google Scholar] [CrossRef] [Green Version]
- Labrecque, R.; Sirard, M.A. The study of mammalian oocyte competence by transcriptome analysis: Progress and challenges. Mol. Hum. Reprod. 2014, 20, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Li, Y.; Feng, Y.; Liu, C.; Ma, J.; Li, Y.; Xiang, H.; Ji, Y.; Cao, Y.; Tong, X.; et al. Single-cell analysis of differences in transcriptomic profiles of oocytes and cumulus cells at GV, MI, MII stages from PCOS patients. Sci. Rep. 2016, 6, 39638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suo, L.; Zhou, Y.X.; Jia, L.L.; Wu, H.B.; Zheng, J.; Lyu, Q.F.; Sun, L.H.; Sun, H.; Kuang, Y.P. Transcriptome profiling of human oocytes experiencing recurrent total fertilization failure. Sci. Rep. 2018, 8, 17890. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Bianchi, V.; Patrizio, P.; Obradors, A.; Huang, Z.; Borini, A.; Delhanty, J.D.; Wells, D. Transcriptomic profiling of human oocytes: Association of meiotic aneuploidy and altered oocyte gene expression. Mol. Hum. Reprod. 2010, 16, 570–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlic, R.; Ganesh, S.; Franke, V.; Svobodova, E.; Urbanova, J.; Suzuki, Y.; Aoki, F.; Vlahovicek, K.; Svoboda, P. Long non-coding RNA exchange during the oocyte-to-embryo transition in mice. Dna Res. Int. J. Rapid Publ. Rep. Genes Genomes 2017, 24, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Melton, C.; Suh, N.; Oh, J.S.; Horner, K.; Xie, F.; Sette, C.; Blelloch, R.; Conti, M. Genome-wide analysis of translation reveals a critical role for deleted in azoospermia-like (Dazl) at the oocyte-to-zygote transition. Genes Dev. 2011, 25, 755–766. [Google Scholar] [CrossRef] [Green Version]
- Chasse, H.; Aubert, J.; Boulben, S.; Le Corguille, G.; Corre, E.; Cormier, P.; Morales, J. Translatome analysis at the egg-to-embryo transition in sea urchin. Nucleic Acids Res. 2018, 46, 4607–4621. [Google Scholar] [CrossRef]
- Winata, C.L.; Lapinski, M.; Pryszcz, L.; Vaz, C.; Bin Ismail, M.H.; Nama, S.; Hajan, H.S.; Lee, S.G.P.; Korzh, V.; Sampath, P.; et al. Cytoplasmic polyadenylation-mediated translational control of maternal mRNAs directs maternal-to-zygotic transition. Development 2018, 145. [Google Scholar] [CrossRef] [Green Version]
- Schwer, B.; Shuman, S. Multicopy suppressors of temperature-sensitive mutations of yeast mRNA capping enzyme. Gene Expr. 1996, 5, 331–344. [Google Scholar]
- De La Fuente, R.; Viveiros, M.M.; Burns, K.H.; Adashi, E.Y.; Matzuk, M.M.; Eppig, J.J. Major chromatin remodeling in the germinal vesicle (GV) of mammalian oocytes is dispensable for global transcriptional silencing but required for centromeric heterochromatin function. Dev. Biol. 2004, 275, 447–458. [Google Scholar] [CrossRef]
- Svoboda, P.; Franke, V.; Schultz, R.M. Sculpting the Transcriptome During the Oocyte-to-Embryo Transition in Mouse. Curr. Top. Dev. Biol. 2015, 113, 305–349. [Google Scholar] [CrossRef] [PubMed]
- Rederstorff, M.; Bernhart, S.H.; Tanzer, A.; Zywicki, M.; Perfler, K.; Lukasser, M.; Hofacker, I.L.; Huttenhofer, A. RNPomics: Defining the ncRNA transcriptome by cDNA library generation from ribonucleo-protein particles. Nucleic Acids Res. 2010, 38, e113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, C.; Patil, D.P.; Kennedy, B.A.; Bakthavachalu, B.; Bundschuh, R.; Schoenberg, D.R. Identification of cytoplasmic capping targets reveals a role for cap homeostasis in translation and mRNA stability. Cell Rep. 2012, 2, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Bouckenheimer, J.; Fauque, P.; Lecellier, C.H.; Bruno, C.; Commes, T.; Lemaitre, J.M.; De Vos, J.; Assou, S. Differential long non-coding RNA expression profiles in human oocytes and cumulus cells. Sci. Rep. 2018, 8, 2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, J.M.; Silva, E.; Chitwood, J.L.; Schoolcraft, W.B.; Krisher, R.L.; Ross, P.J. Differing molecular response of young and advanced maternal age human oocytes to IVM. Hum. Reprod. 2017, 32, 2199–2208. [Google Scholar] [CrossRef]
- Capco, D.G.; Gallicano, G.I.; McGaughey, R.W.; Downing, K.H.; Larabell, C.A. Cytoskeletal sheets of mammalian eggs and embryos: A lattice-like network of intermediate filaments. Cell Motil. Cytoskelet. 1993, 24, 85–99. [Google Scholar] [CrossRef]
- Vogt, E.; Kirsch-Volders, M.; Parry, J.; Eichenlaub-Ritter, U. Spindle formation, chromosome segregation and the spindle checkpoint in mammalian oocytes and susceptibility to meiotic error. Mutat. Res. 2008, 651, 14–29. [Google Scholar] [CrossRef]
- Duncan, F.E.; Chiang, T.; Schultz, R.M.; Lampson, M.A. Evidence that a defective spindle assembly checkpoint is not the primary cause of maternal age-associated aneuploidy in mouse eggs. Biol. Reprod. 2009, 81, 768–776. [Google Scholar] [CrossRef]
- Ma, J.Y.; Li, M.; Luo, Y.B.; Song, S.; Tian, D.; Yang, J.; Zhang, B.; Hou, Y.; Schatten, H.; Liu, Z.; et al. Maternal factors required for oocyte developmental competence in mice: Transcriptome analysis of non-surrounded nucleolus (NSN) and surrounded nucleolus (SN) oocytes. Cell Cycle 2013, 12, 1928–1938. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.E.; Leese, H.J.; Gosden, R.G.; Picton, H.M. Pyruvate and oxygen consumption throughout the growth and development of murine oocytes. Mol. Reprod. Dev. 2009, 76, 231–238. [Google Scholar] [CrossRef]
- Pelland, A.M.; Corbett, H.E.; Baltz, J.M. Amino Acid transport mechanisms in mouse oocytes during growth and meiotic maturation. Biol. Reprod. 2009, 81, 1041–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huo, L.J.; Fan, H.Y.; Zhong, Z.S.; Chen, D.Y.; Schatten, H.; Sun, Q.Y. Ubiquitin-proteasome pathway modulates mouse oocyte meiotic maturation and fertilization via regulation of MAPK cascade and cyclin B1 degradation. Mech. Dev. 2004, 121, 1275–1287. [Google Scholar] [CrossRef] [PubMed]
- Dun, M.D.; Smith, N.D.; Baker, M.A.; Lin, M.; Aitken, R.J.; Nixon, B. The chaperonin containing TCP1 complex (CCT/TRiC) is involved in mediating sperm-oocyte interaction. J. Biol. Chem. 2011, 286, 36875–36887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, R.P.; de Boer, K. The bottleneck: Mitochondrial imperatives in oogenesis and ovarian follicular fate. Mol. Cell. Endocrinol. 1998, 145, 81–88. [Google Scholar] [CrossRef]
- Dumollard, R.; Ward, Z.; Carroll, J.; Duchen, M.R. Regulation of redox metabolism in the mouse oocyte and embryo. Development 2007, 134, 455–465. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Peviani, A.; van der Horst, S.; Gamm, M.; Snel, B.; Bentsink, L.; Hanson, J. Extensive translational regulation during seed germination revealed by polysomal profiling. New Phytol. 2017, 214, 233–244. [Google Scholar] [CrossRef]
- Juntawong, P.; Bailey-Serres, J. Dynamic Light Regulation of Translation Status in Arabidopsis thaliana. Front. Plant Sci. 2012, 3, 66. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Morales, R.; Banday, M.; Garcia, S.; Hao, L.; Cross, G.A.; Estevez, A.M.; Bellofatto, V. The essential polysome-associated RNA-binding protein RBP42 targets mRNAs involved in Trypanosoma brucei energy metabolism. RNA 2012, 18, 1968–1983. [Google Scholar] [CrossRef] [Green Version]
- Bunnik, E.M.; Chung, D.W.; Hamilton, M.; Ponts, N.; Saraf, A.; Prudhomme, J.; Florens, L.; Le Roch, K.G. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 2013, 14, R128. [Google Scholar] [CrossRef]
- Frey, S.; Pool, M.; Seedorf, M. Scp160p, an RNA-binding, polysome-associated protein, localizes to the endoplasmic reticulum of Saccharomyces cerevisiae in a microtubule-dependent manner. J. Biol. Chem. 2001, 276, 15905–15912. [Google Scholar] [CrossRef] [Green Version]
- Minia, I.; Merce, C.; Terrao, M.; Clayton, C. Translation Regulation and RNA Granule Formation after Heat Shock of Procyclic Form Trypanosoma brucei: Many Heat-Induced mRNAs Are also Increased during Differentiation to Mammalian-Infective Forms. PLoS Negl. Trop. Dis. 2016, 10, e0004982. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tetkova, A.; Hancova, M. Mouse Oocyte Isolation, Cultivation and RNA Microinjection. Bio-protocol 2016, 6, e1729. [Google Scholar] [CrossRef] [Green Version]
- Masek, T.; Vopalensky, V.; Suchomelova, P.; Pospisek, M. Denaturing RNA electrophoresis in TAE agarose gels. Anal. Biochem. 2005, 336, 46–50. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Sivan, G.; Kedersha, N.; Elroy-Stein, O. Ribosomal slowdown mediates translational arrest during cellular division. Mol. Cell. Biol. 2007, 27, 6639–6646. [Google Scholar] [CrossRef] [Green Version]
- Frydryskova, K.; Masek, T.; Borcin, K.; Mrvova, S.; Venturi, V.; Pospisek, M. Distinct recruitment of human eIF4E isoforms to processing bodies and stress granules. BMC Mol. Biol. 2016, 17, 21. [Google Scholar] [CrossRef] [Green Version]
- Zamostna, B.; Novak, J.; Vopalensky, V.; Masek, T.; Burysek, L.; Pospisek, M. N-terminal domain of nuclear IL-1alpha shows structural similarity to the C-terminal domain of Snf1 and binds to the HAT/core module of the SAGA complex. PLoS ONE 2012, 7, e41801. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Masek, T.; del Llano, E.; Gahurova, L.; Kubelka, M.; Susor, A.; Roucova, K.; Lin, C.-J.; Bruce, A.W.; Pospisek, M. Identifying the Translatome of Mouse NEBD-Stage Oocytes via SSP-Profiling; A Novel Polysome Fractionation Method. Int. J. Mol. Sci. 2020, 21, 1254. https://doi.org/10.3390/ijms21041254
Masek T, del Llano E, Gahurova L, Kubelka M, Susor A, Roucova K, Lin C-J, Bruce AW, Pospisek M. Identifying the Translatome of Mouse NEBD-Stage Oocytes via SSP-Profiling; A Novel Polysome Fractionation Method. International Journal of Molecular Sciences. 2020; 21(4):1254. https://doi.org/10.3390/ijms21041254
Chicago/Turabian StyleMasek, Tomas, Edgar del Llano, Lenka Gahurova, Michal Kubelka, Andrej Susor, Kristina Roucova, Chih-Jen Lin, Alexander W. Bruce, and Martin Pospisek. 2020. "Identifying the Translatome of Mouse NEBD-Stage Oocytes via SSP-Profiling; A Novel Polysome Fractionation Method" International Journal of Molecular Sciences 21, no. 4: 1254. https://doi.org/10.3390/ijms21041254