
Reactivity of the Ethenium Cation (C2H5+) with Ethyne (C2H2): A Combined Experimental and Theoretical Study

 , , , , , and
, , , , , and
Abstract
:
1. Introduction
2. Experimental Results
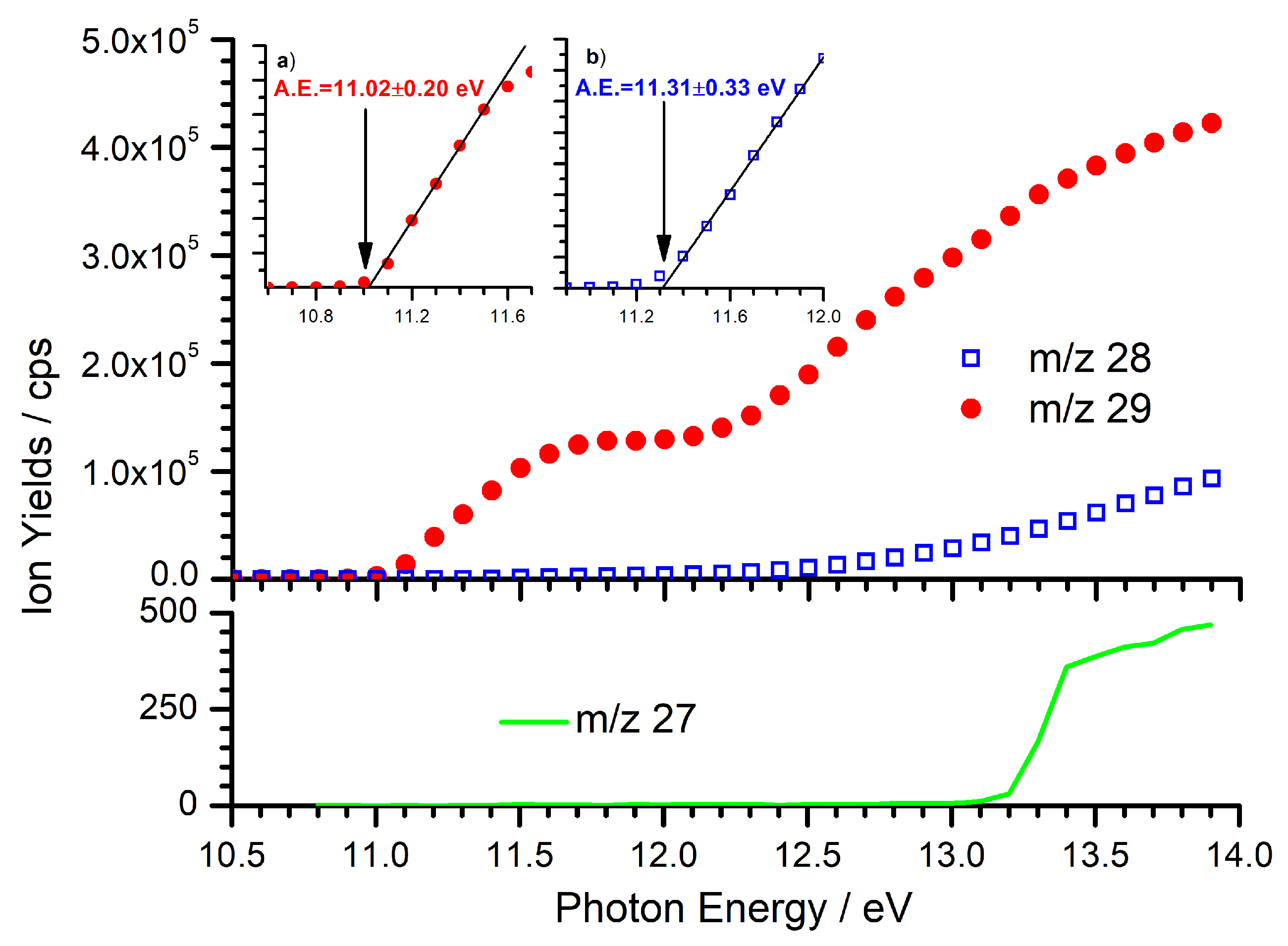
2.1. Generation of C2H5+
2.2. Reaction with C2H2

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Products | Equation | , lit. Values 1 | B97X-D/cc-pVTZ 2 | G4 3 |
|---|---|---|---|---|
| c−C3H3+ + CH4 | (4) | −1.36 0.10 4 | −1.57 | −1.36 |
| H2CCCH+ + CH4 | (5) | −0.16 0.10 4 | −0.25 | −0.19 |
| c−C3H2(CH3)+ + H2 | (6) | −1.68 0.10 5 | −1.77 | −1.57 |
| CH2CHCCH2+ + H2 | (7) | −1.01 0.10 5 | - | −0.92 |
| C2H3+ + C2H4 | (8) | +0.39 0.08 6 | +0.37 | +0.36 |
| C2H3+ + H2 + C2H2 | (9) | +2.21 0.08 6 | +2.29 | +2.09 |
| Ionic Reaction Products | Product Mass (m/z) | Equations | BR, This Work | Literature BR 1 |
|---|---|---|---|---|
| C2H3+ | 27 | (8)/(9) | 0.02 0.02 | 0.00 |
| C3H3+ | 39 | (4)/(5) | 0.76 0.05 | 0.36 |
| C4H5+ | 53 | (6)/(7) | 0.22 0.02 | 0.64 |
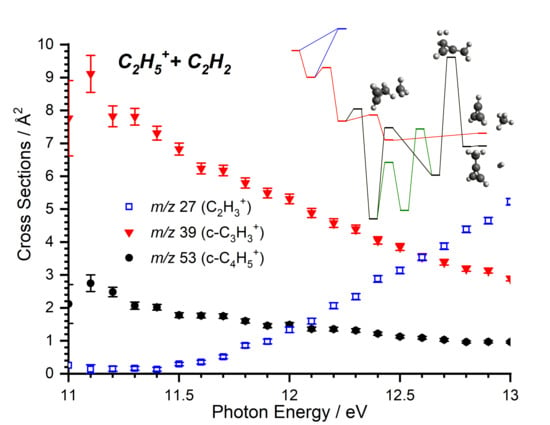
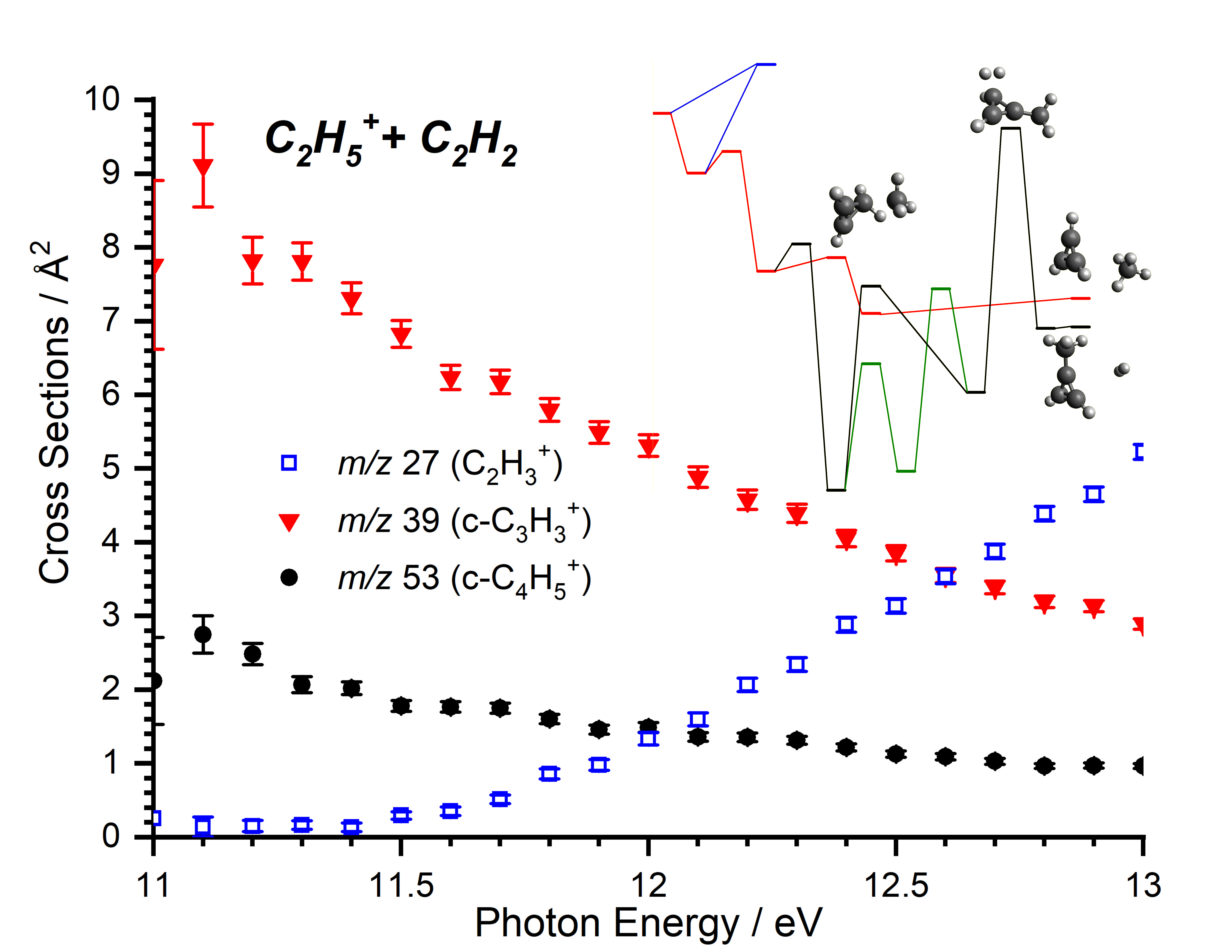
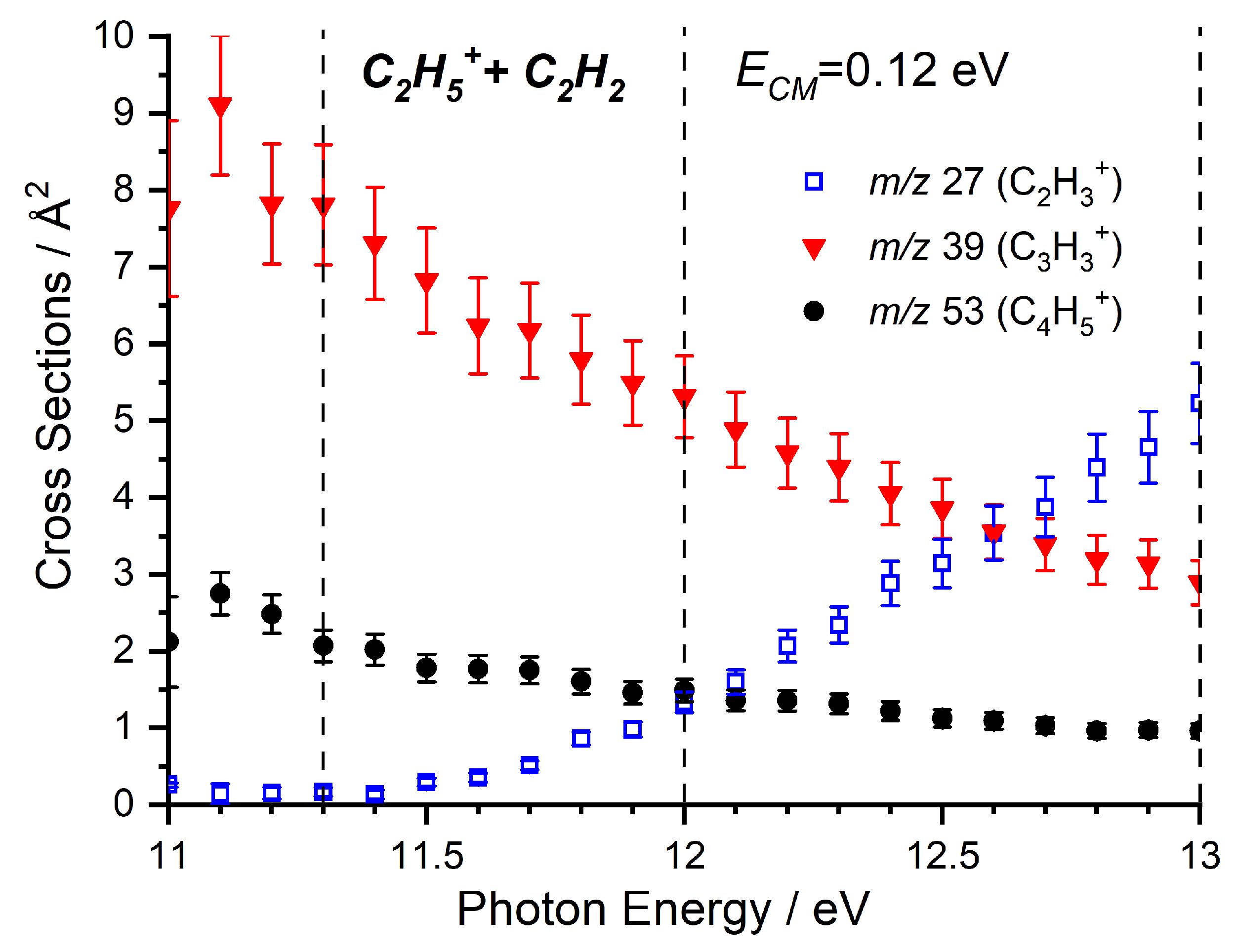
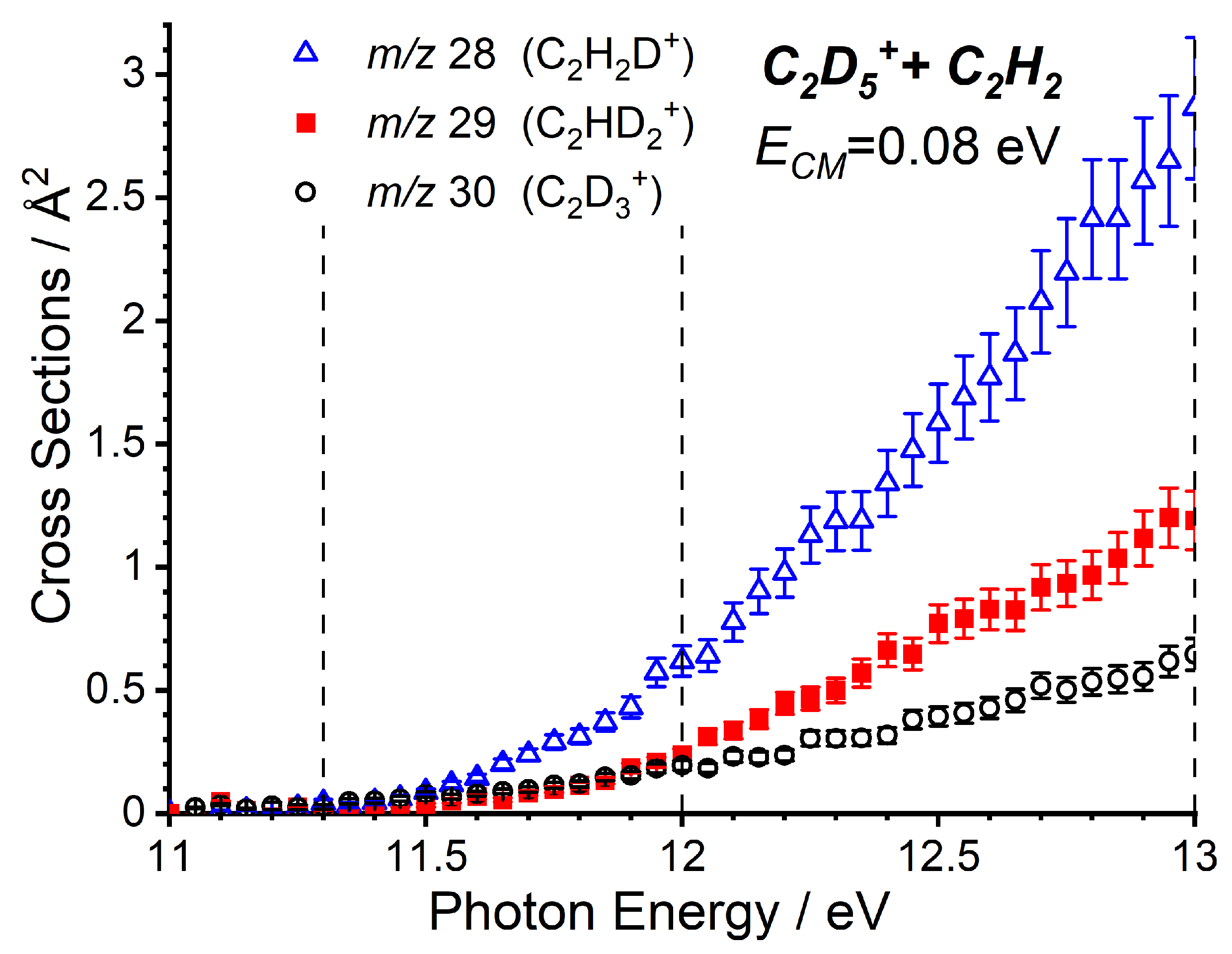
2.2.1. Cross Sections as a Function of the Photon Energy
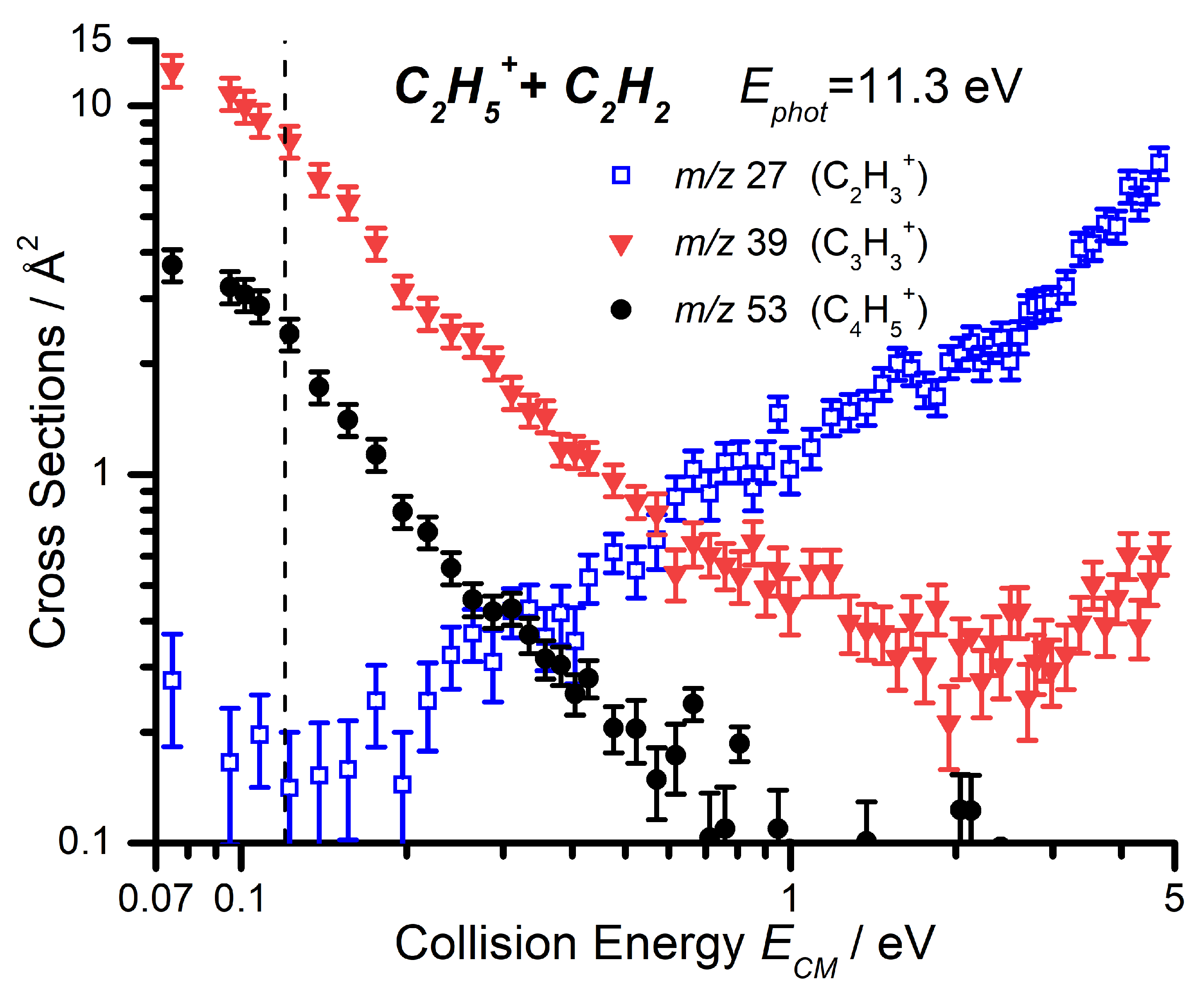
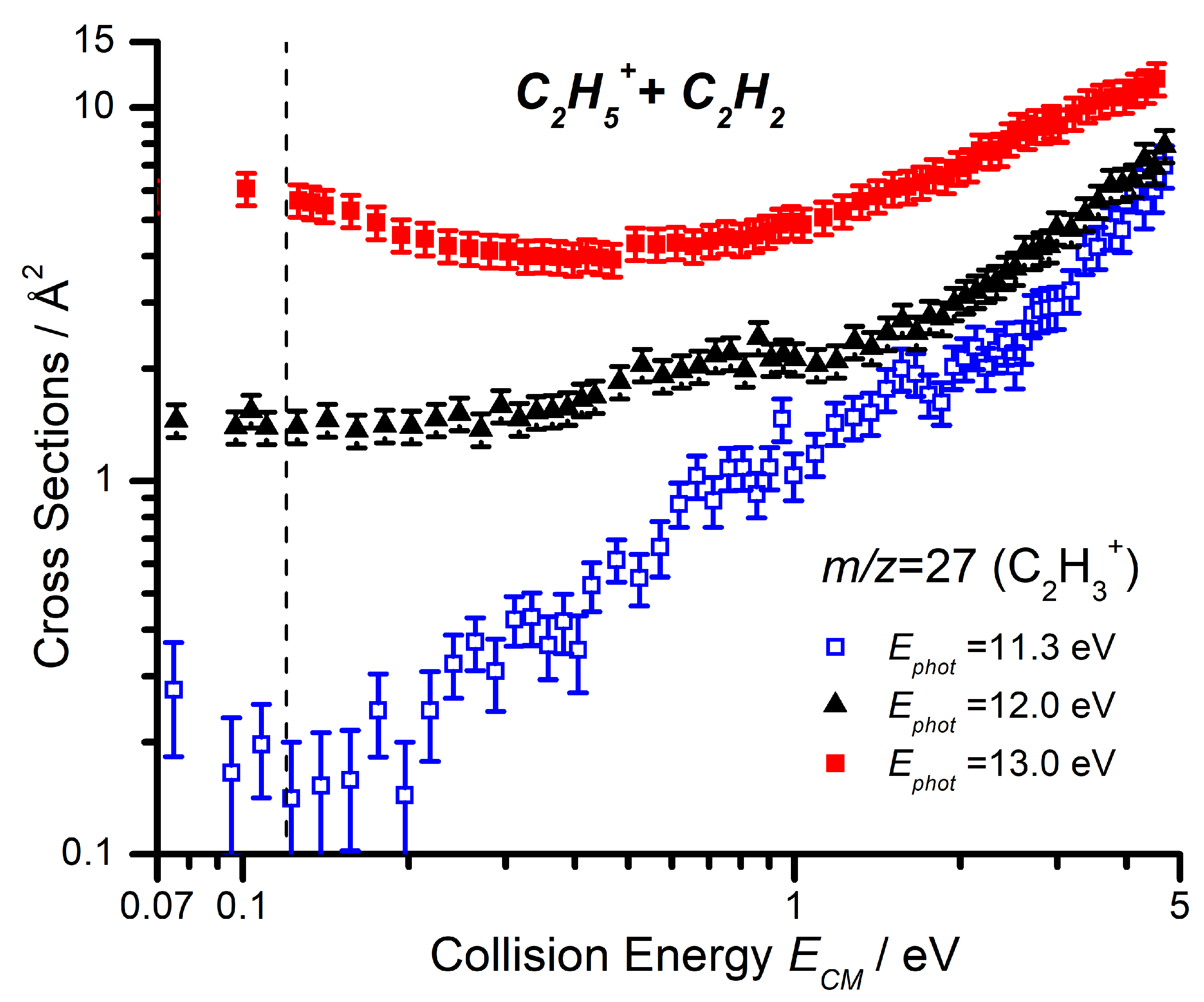
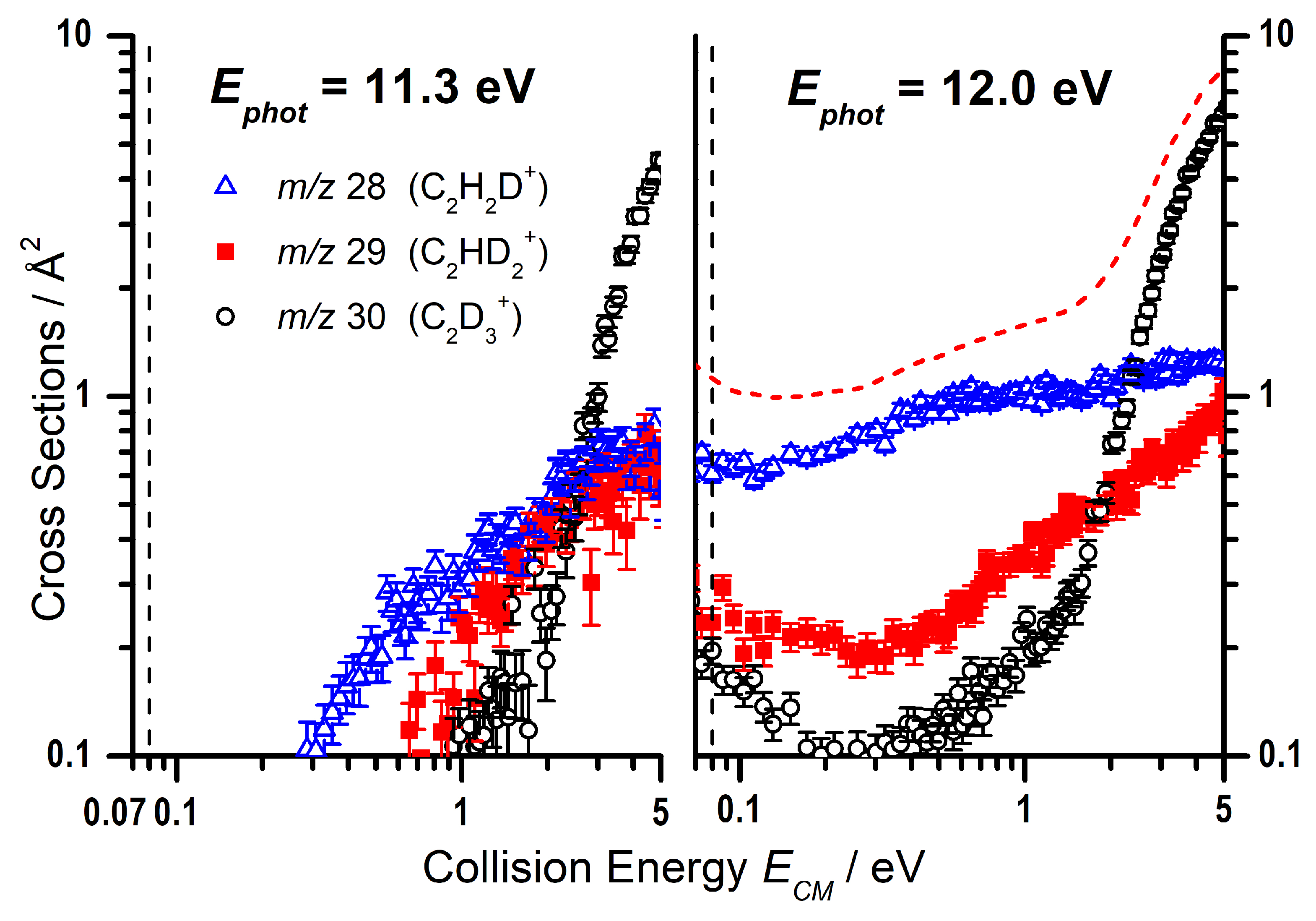
2.2.2. Cross Sections as a Function of the Collision Energy
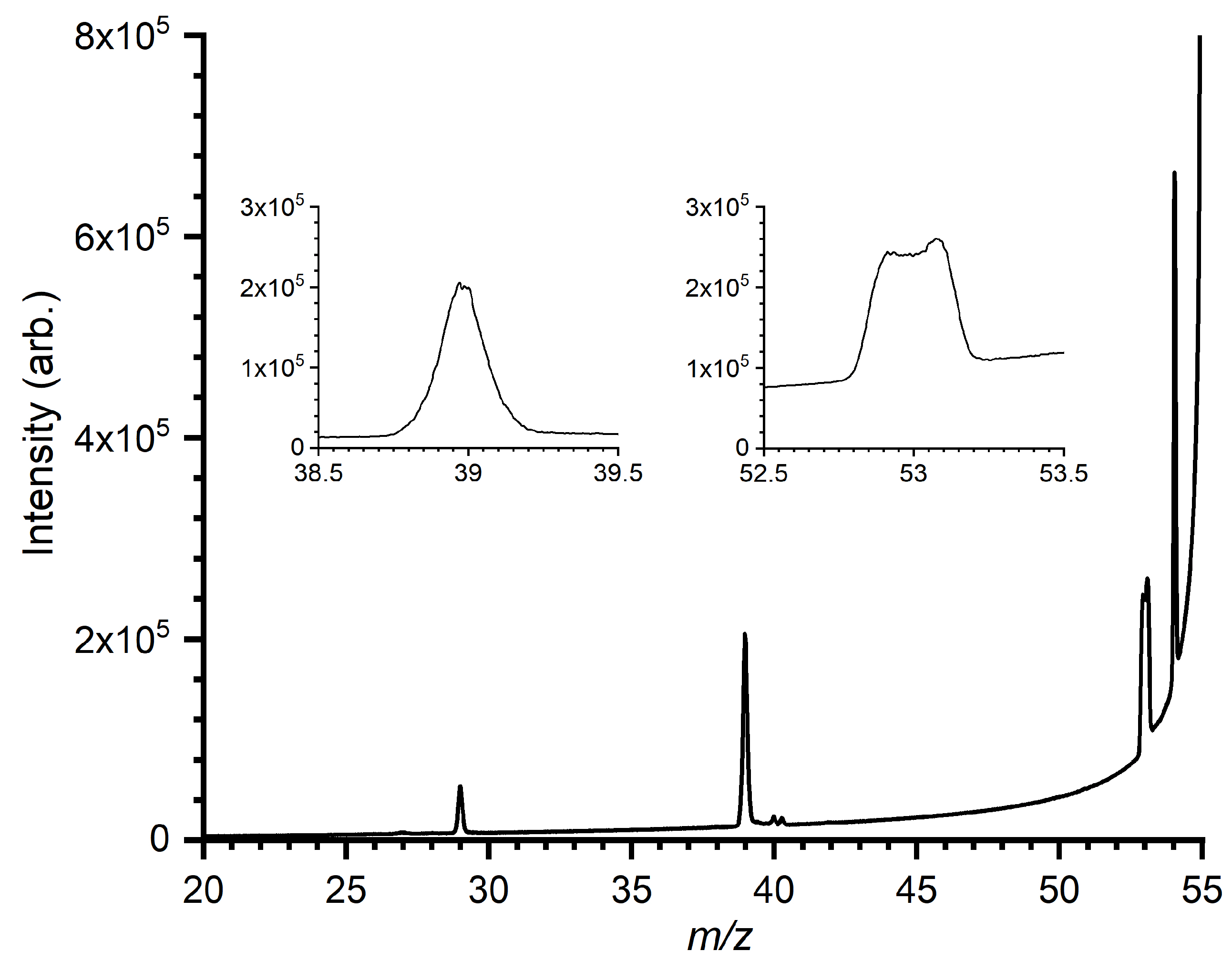
2.3. MIKE Spectra
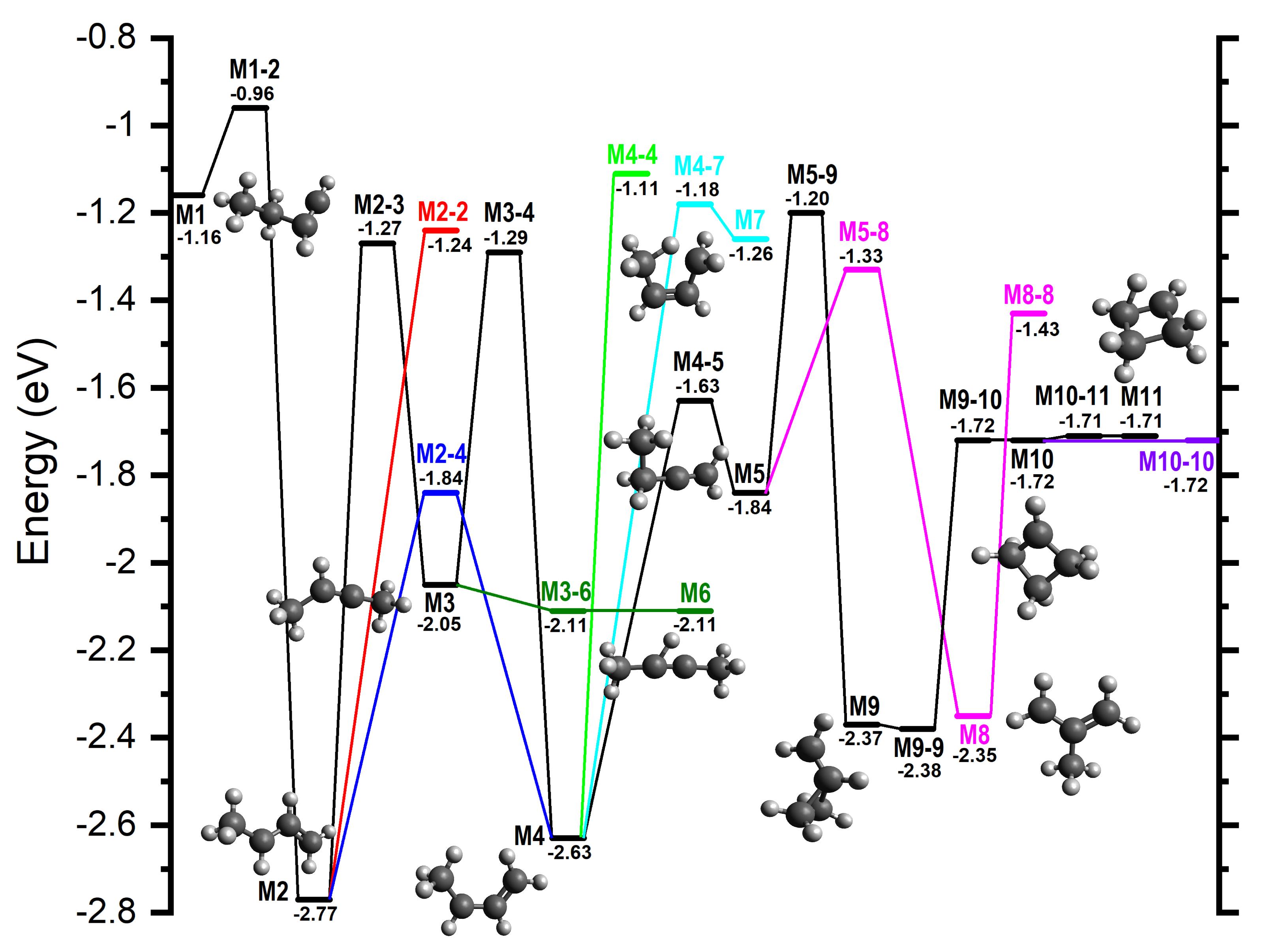
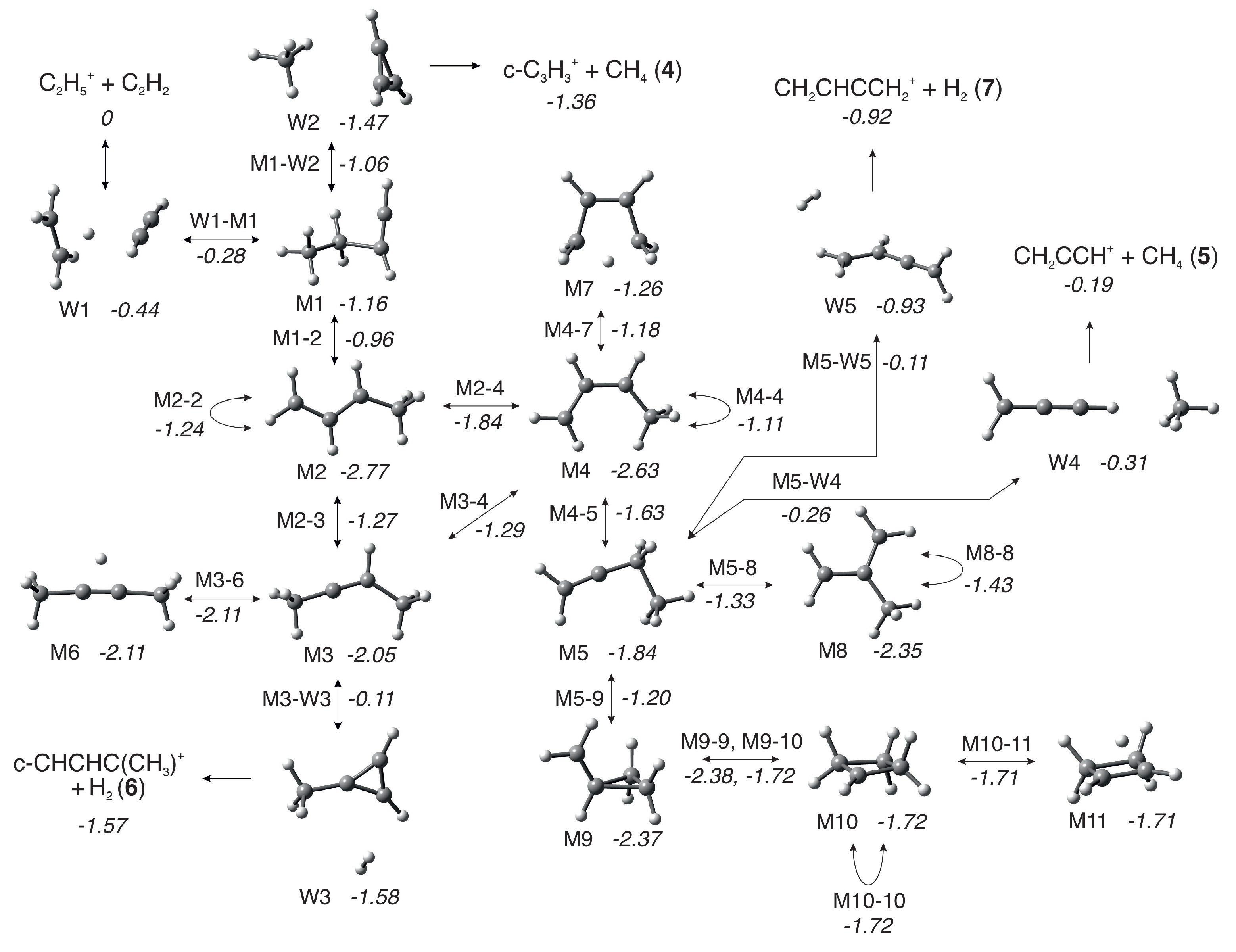
3. Computational Results
4. Discussion
4.1. Product at m/z 27: C2H3+
4.2. Products at m/z 39 ([C3H3]+) and m/z 53 ([C4H5]+)
4.2.1. Rationalization of m/z 39 and 53 Branching Ratios
4.2.2. Comparison of Computed Energy Pathways with MIKE Spectra
4.3. Comparison with Previous Results
5. Materials and Methods
5.1. Experimental Set-Up
5.2. Computational Methodology
6. Conclusions
- The density of [C3H3]+ ions is increased by ∼ 30% when our new BRs are used, despite the fact that the main formation reaction for [C3H3]+ in the model is C2H4+ + C2H2→ [C3H3]+ + CH3 and not the title reaction. This is a relevant difference that should be considered in light of the fact that a change in the abundance of [C3H3]+ can induce changes in other species, such as the recently detected c−C3H2 [43].
- The density of the [C4H5]+ ion decreases by about a factor of 2 when our new BRs are used. The repercussion on the production of [C6H7]+ is lower (with a maximum change of ∼ 20%), as the main formation reaction for this ion is [C3H5]+ + CH3CCH→ [C6H7]+ + H2 rather than [C4H5]+ plus C2H4. However, it still marks a significant change to the predicted density of this key astrochemical species in Titan’s environment.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Larsson, M.; Geppert, W.D.; Nyman, G. Ion chemistry in space. Rep. Prog. Phys. 2012, 75, 066901. [Google Scholar] [CrossRef]
- Agúndez, M.; Wakelam, V. Chemistry of Dark Clouds: Databases, Networks, and Models. Chem. Rev. 2013, 113, 8710–8737. [Google Scholar] [CrossRef]
- Vigren, E.; Semaniak, J.; Hamberg, M.; Zhaunerchyk, V.; Kaminska, M.; Thomas, R.D.; af Ugglas, M.; Larsson, M.; Geppert, W.D. Dissociative recomination of nitrile ions with implications for Titan’s upper atmosphere. Planet. Space Sci. 2012, 60, 102–106. [Google Scholar] [CrossRef]
- Yuen, C.H.; Ayouz, M.A.; Balucani, N.; Ceccarelli, C.; Schneider, I.F.; Kokoouline, V. Dissociative recombination of CH2NH2+: A crucial link with interstellar methanimine and Titan ammonia. Mon. Notices Royal Astron. Soc. 2019, 484, 659–664. [Google Scholar] [CrossRef]
- Hörst, S.M. Titan’s atmosphere and climate. J. Geophys. Res. Planets 2017, 122, 432–482. [Google Scholar] [CrossRef]
- Vuitton, V.; Yelle, R.; Klippenstein, S.; Hörst, S.; Lavvas, P. Simulating the density of organic species in the atmosphere of Titan with a coupled ion-entrual photochemical model. ICARUS 2019, 324, 120–197. [Google Scholar] [CrossRef]
- Imanaka, H.; Smith, M.A. Role of photoionization in the formation of complex organic molecules in Titan’s upper atmosphere. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Coates, A.J.; Wellbrock, A.; Waite, J.H.; Jones, G.H. A new upper limit to the field-aligned potential near Titan. Geophys. Res. Lett. 2015, 42, 4676–4684. [Google Scholar] [CrossRef] [PubMed]
- Westlake, J.; Waiter, J.; Carrasco, N.; Richard, M.; Cravens, T. The role of ion-molecule reactions in the growth of heavy ions in Titan’s ionosphere. J. Geophys. Res. Space Phys. 2014, 119, 5951–5963. [Google Scholar] [CrossRef]
- Vinatier, S.; Bézard, B.; Fouchet, T.; Teanby, N.A.; de Kok, R.; Irwin, P.G.; Conrath, B.J.; Nixon, C.A.; Romani, P.N.; Flasar, F.M.; et al. Vertical abundance profiles of hydrocarbons in Titan’s atmosphere at 15S and 80N retrieved from Cassini/CIRS spectra. ICARUS 2007, 188, 120–138. [Google Scholar] [CrossRef]
- Nixon, C.; Lorenz, R.; Achterberg, R.; Buch, A.; Coll, P.; Clark, R.; Courtin, R.; Hayes, A.; Iess, L.; Johnson, R.; et al. Titan’s cold case files-Oustanding questions after Cassini-Huygens. Planet. Space Sci. 2018, 155, 50–72. [Google Scholar] [CrossRef]
- Sittler, E.C., Jr.; Cooper, J.F.; Sturner, S.J.; Ali, A. Titan’s ionospheric chemistry, fullerenes, oxygen, galactic cosmic rays and the formation of exobiological molecules on and within its surfaces and lakes. ICARUS 2020, 344, 113246. [Google Scholar] [CrossRef]
- Waite, J.; Niemann, H.; Yelle, R.; Kasprzak, W.; Cravens, T.; Luhmann, J.; McNutt, R.; Ip, W.; Gell, D.; De La Haye, V.; et al. Ion Neutral Mass Spectrometer results from the first flyby of Titan. Science 2005, 308, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Vuitton, V.; Yelle, R.; McEwan, M. Ion chemistry and N-containing molecules in Titan’s upper atmosphere. ICARUS 2007, 191, 722–742. [Google Scholar] [CrossRef]
- Vuitton, V.; Yelle, R.V.; Lavvas, P. Composition and chemistry of Titan’s thermosphere and ionosphere. Philos. Trans. Royal Soc. A-Math. Phys. Engin. Sci. 2009, 367, 729–741. [Google Scholar] [CrossRef]
- Cui, J.; Yelle, R.; Vuitton, V.; Waite, J.H., Jr.; Kasprzak, W.T.; Gell, D.; Niemann, H.; Müller-Wodarg, I.; Borgeeren, N.; Fletcher, G.; et al. Analysis of Titan’s neutral upper atmosphere from Cassini Ion Neutral Mass Spectrometer measurements. ICARUS 2009, 200, 581–615. [Google Scholar] [CrossRef]
- Ali, A.; Sittler, E.C., Jr.; Chornay, D.; Rowe, B.R.; Puzzarini, C. Organic chemistry in Titan’s upper atmosphere and its astrobiological consequences: I. Views towards Cassini plasma spectrometer (CAPS) and ion neutral mass spectrometer (INMS) experiments in space. Planet. Space Sci. 2015, 109, 46–63. [Google Scholar] [CrossRef]
- Spilker, L. Cassini-Huygens’ exploration of the Saturn system: 13 years of discovery. Science 2019, 364, 1046–1051. [Google Scholar] [CrossRef] [PubMed]
- Haythornthwaite, R.P.; Coates, A.J.; Jones, G.H.; Wellbrock, A.; Waite, J.H.; Vuitton, V.; Lavvas, P. Heavy Positive Ion Groups in Titan’s Ionosphere from Cassini Plasma Spectrometer IBS Observations. Planet. Sci. J. 2021, 2, 26. [Google Scholar] [CrossRef]
- Cravens, T.E.; Robertson, I.P.; Waite, J.H., Jr.; Yelle, R.V.; Kasprzak, W.T.; Keller, C.N.; Ledvina, S.A.; Niemann, H.B.; Luhmann, J.G.; McNutt, R.L.; et al. Composition of Titan’s ionosphere. Geophys. Res. Lett. 2006, 33, L07105. [Google Scholar] [CrossRef]
- Waite, J.H., Jr.; Perryman, R.S.; Perry, M.E.; Miller, K.E.; Bell, J.; Cravens, T.E.; Glein, C.R.; Grimes, J.; Hedman, M.; Cuzzi, J.; et al. Chemical interactions between Saturn’s atmosphere and its rings. Science 2018, 362, eaat2382. [Google Scholar] [CrossRef]
- Moore, L.; Cravens, T.E.; Mueller-Wodarg, I.; Perry, M.E.; Waite, J.H., Jr.; Perryman, R.; Nagy, A.; Mitchell, D.; Persoon, A.; Wahlund, J.E.; et al. Models of Saturn’s Equatorial Ionosphere Based on In Situ Data From Cassini’s Grand Finale. Geophys. Res. Lett. 2018, 45, 9398–9407. [Google Scholar] [CrossRef]
- Cravens, T.E.; Moore, L.; Waite, J.H., Jr.; Perryman, R.; Perry, M.; Wahlund, J.E.; Persoon, A.; Kurth, W.S. The Ion Composition of Saturn’s Equatorial Ionosphere as Observed by Cassini. Geophys. Res. Lett. 2019, 46, 6315–6321. [Google Scholar] [CrossRef]
- Serigano, J.; Hörst, S.M.; He, C.; Gautier, T.; Yelle, R.V.; Koskinen, T.T.; Koskinen, M.G.; Trainer, M.J.R. Compositional Measurements of Saturn’s Upper Atmosphere and Rings from Cassini INMS: An Extended Analysis of Measurements from Cassini’s Grand Finale Orbits. J. Geophys. Res. Planets 2022, 127, e2022JE007238. [Google Scholar] [CrossRef]
- Bagenal, F.; Horanyi, M.; McComas, D.J.; McNutt, R.L., Jr.; Elliott, H.A.; Hill, M.E.; Brown, L.E.; Delamere, P.A.; Kollmann, P.; Krimigis, S.M.; et al. Pluto’s interaction with its space environment: Solar wind, energetic particles, and dust. Science 2016, 351, aad9045. [Google Scholar] [CrossRef]
- Gladstone, G.R.; Stern, S.A.; Ennico, K.; Olkin, C.B.; Weaver, H.A.; Young, L.A.; Summers, M.E.; Strobel, D.F.; Hinson, D.P.; Kammer, J.A.; et al. The atmosphere of Pluto as observed by New Horizons. Science 2016, 351, aad8866. [Google Scholar] [CrossRef]
- Lellouch, E.; Butler, B.; Moreno, R.; Gurwell, M.; Lavvas, P.; Bertrand, T.; Fouchet, T.; Strobel, D.F.; Moullet, A. Pluto’s atmosphere observations with ALMA: Spatially-resolved maps of CO and HCN emission and first detection of HNC. ICARUS 2022, 372, 114722. [Google Scholar] [CrossRef]
- Beth, A.; Altwegg, K.; Balsiger, H.; Berthelier, J.J.; Combi, M.R.; De Keyser, J.; Fiethe, B.; Fuselier, S.A.; Galand, M.; Gombosi, T.I.; et al. ROSINA ion zoo at Comet 67P. Astron. Astroph. 2020, 642. [Google Scholar] [CrossRef]
- Betz, A.L. Ethylene in IRC+10216. Astrophys. J. 1981, 244, L103–L105. [Google Scholar] [CrossRef]
- Hinkle, K.H.; Wallance, L.; Richter, M.J.; Cernicharo, J. Ethylene in the circumstellar envelope of IRC+10216. Proc. Int. Astron. Union 2008, 4, 161–162. [Google Scholar] [CrossRef]
- Steffl, A.J.; Young, L.A.; Strobel, D.F.; Kammer, J.A.; Evans, J.S.; Stevens, M.H.; Schindhelm, R.N.; Parker, J.W.; Stern, S.A.; Weaver, H.A.; et al. Pluto’s Ultraviolet Spectrum, Surface Reflectance, and Airglow Emissions. Astron. J. 2020, 159, 274. [Google Scholar] [CrossRef]
- Kammer, J.A.; Gladstone, G.R.; Young, L.A.; Steffl, A.J.; Parker, J.W.; Greathouse, T.K.; Retherford, K.D.; Versteeg, M.H.; Strobel, D.F.; Summers, M.E.; et al. New Horizons Observations of an Ultraviolet Stellar Occultation and Appulse by Pluto’s Atmosphere. Astron. J. 2020, 159, 26. [Google Scholar] [CrossRef]
- Lacy, J.H.; Evans, N.J.; Achtermann, J.M.; Bruce, D.E.; Arens, J.F.; Carr, J. Discovery of Interstellar Acetylene. Astrophys. J. Lett. 1989, 342, L43. [Google Scholar] [CrossRef]
- Ridgway, S.T.; Hall, D.N.; Kleinmann, S.G.; Weinberger, D.A.; Wojslaw, R.S. Circumstellar acetylene in the infrared spectrum of IRC +10 216. Nature 1976, 264, 345–346. [Google Scholar] [CrossRef]
- Kostiuk, T.; Espenak, F.; Deming, D.; Mumma, M.J.; Zipoy, D.; Keady, J. Study of Velocity Structure in IRC +10216 Using Acetylene Line Profiles. Bull. Am. Astron. Soc. 1985, 17, 570. [Google Scholar]
- Keady, J.J.; Ridgway, S.T. The IRC +10216 Circumstellar Envelope. III—Infrared Molecular Line Profiles. Astrophys. J. 1993, 406, 199–214. [Google Scholar] [CrossRef]
- Evans, N.J.; Lacy, J.H.; Carr, J.S. Infrared Molecular Spectroscopy toward the Orion IRc2 and IRc7 Sources—A new Probe of Physical Conditions and Abundances in Molecular Clouds. Astrophys. J. 1989, 342, L43–L46. [Google Scholar] [CrossRef]
- Carrasco, N.; Alcaraz, C.; Dutuit, O.; Plessis, S.; Thissen, R.; Vuitton, V.; Yelle, R.; Pernot, P. Sensitivity of a Titan ionospheric model to the ion-molecule reaction parameters. Planet. Space Sci. 2008, 56, 1644–1657. [Google Scholar] [CrossRef]
- Kim, J.; Anicich, V.; Huntress, W. Product distributions and rate constants for the reactions of CH3+, CH4+, C2H2+, C2H3+, C2H4+, and C2H6+ ions with CH4, C2H2, C2H4, and C2H6. J. Phys. Chem. 1977, 81, 1798–1805. [Google Scholar] [CrossRef]
- Anicich, V. Evaluated Bimolecular Ion-Molecule Gas Phase Kinetics of Positive Ions for Use in Modelling Planetary Atmospheres Cometary Comae/Interstellar Clouds. J. Phys. Chem. Ref. Data 1993, 22, 1469–1569. [Google Scholar] [CrossRef]
- Anicich, V. An Index of the Literature for Bimolecular Gas Phase Cation-Molecule Reaction Kinetics; JPL Open Repository: Pasadena, CA, USA, 2003; Available online: https://hdl.handle.net/2014/7981 (accessed on 1 May 2023).
- Mandt, K.E.; Gell, D.A.; Perry, M.; Waite, J.H., Jr.; Crary, F.A.; Young, D.; Magee, B.A.; Westlake, J.H.; Cravens, T.; Kasprzak, W.; et al. Ion densities and composition of Titan’s upper atmosphere derived from the Cassini Ion Neutral Mass Spectrometer: Analysis methods and comparison of measured ion densities to photochemical model simulations. J. Geophys. Res. Planets 2012, 117, E10006. [Google Scholar] [CrossRef]
- Nixon, C.A.; Thelen, A.E.; Cordiner, M.A.; Kisiel, Z.; Charnley, S.B.; Molter, E.M.; Serigano, J.; Irwin, P.G.J.; Teanby, N.A.; Kuan, Y.J. Detection of Cyclopropenylidene on Titan with ALMA. Astron. J. 2020, 160, 205. [Google Scholar] [CrossRef]
- Vuitton, V.; Yelle, R.V.; Cui, J. Formation and distribution of benzene on Titan. J. Geophys. Res. Space Phys. 2008, 113, E05007. [Google Scholar] [CrossRef]
- Misina, M.; Pokorny, P. Energy and composition of the ion flux in microwave electron cyclotron resonanceyradio frequency methane plasma. Surf. Coat. Technol. 2003, 173–174, 914–917. [Google Scholar] [CrossRef]
- Okita, A.; Suda, Y.; Ozeki, A.; Sugawara, H.; Sakai, Y.; Oda, A.; Nakamura, J. Predicting the amount of carbon in carbon nanotubes grown by CH4 rf plasmas. J. Appl. Phys. 2006, 99. [Google Scholar] [CrossRef]
- Heijkers, S.; Aghaei, M.; Bogaerts, A. Plasma-Based CH4 Conversion into Higher Hydrocarbons and H2: Modelling to Reveal the Reaction Mechanisms of Different Plasma Sources. J. Phys. Chem. C 2020, 124, 7016–7030. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Okumoto, M.; Nakayama, A.; Suzuki, E. Plasma Reforming and Coupling of Methane with Carbon Dioxide. Energy Fuels 2001, 15, 1295–1299. [Google Scholar] [CrossRef]
- Inayat, A.; Tariq, R.; Khan, Z.; Ghenai, C.; Kamil, M.; Jamil, F.; Shanableh, A. A comprehensive review on advanced thermochemical processes for bio-hydrogen production via mircowave and plasma technologies. Biomass Convers. Biorefin. 2020, 13, 8593–8602. [Google Scholar] [CrossRef]
- Khoja, A.H.; Tahir, M.; Amin, N.A.S. Recent developments in non-thermal catalytic DBD plasma reactor for dry reforming of methane. Energy Convers. Manag. 2019, 183, 529–560. [Google Scholar] [CrossRef]
- De Bie, C.; van Dijk, J.; Bogaerts, A. The Dominant Pathways for the Conversion of Methane into Oxygenates and Syngas in an Atmospheric Pressure Dielectric Barrier Discharge. J. Phys. Chem. C 2015, 119, 22331–22350. [Google Scholar] [CrossRef]
- Wang, W.; Berthelot, A.; Zhang, Q.; Bogaerts, A. Modelling of plasma-based dry reforming: How do uncertainties in the input data affect the calculation results? J. Phys. D-Appl. Phys. 2018, 51, 204003. [Google Scholar] [CrossRef]
- Chen, T.Y.; Taneja, T.S.; Rousso, A.C.; Yang, S.; Kolemen, E.; Ju, Y. Time-resolved in situ measurements and predictions of plasma-assisted methane reforming in a nanosecond-pulsed discharge. Proc. Combust. Inst. 2021, 38, 6533–6540. [Google Scholar] [CrossRef]
- Zhang, L.; Heijkers, S.; Wang, W.; Martini, L.M.; Tosi, P.; Yang, D.; Fang, Z.; Bogaerts, A. Dry reforming of methane in a nanosecond repetitively pulsed discharge: Chemical kinetics modeling. Plasma Sources Sci. Technol. 2022, 31, 055014. [Google Scholar] [CrossRef]
- Richardson, V.; Ascenzi, D.; Sundelin, D.; Alcaraz, C.; Romanzin, C.; Thissen, R.; Guillemin, J.C.; Polášek, M.; Tosi, P.; Žabka, J.; et al. Experimental and Computational Studies on the Reactivity of Methanimine Radical Cation (H2CNH+) and its Isomer Aminomethulene (HCNH2+) with C2H2. Front. Astron. Space Sci. 2021, 8, 158. [Google Scholar] [CrossRef]
- Sundelin, D.; Ascenzi, D.; Richardson, V.; Alcaraz, C.; Polášek, M.; Romanzin, C.; Thissen, R.; Tosi, P.; Žabka, J.; Geppert, W.D. The reactivity of methanimine radical cation (H2CNH+) and its isomer aminomethylene (HCNH2+) with C2H4. Chem. Phys. Lett. 2021, 777, 138677. [Google Scholar] [CrossRef]
- Maitre, P.A.; Bieniek, M.S.; Kechagiopoulos, P.N. Modelling excited species and their role on kinetic pathways in the non-oxidative coupling of methane by dielectric barrier discharge. Chem. Eng. Sci. 2021, 234, 116399. [Google Scholar] [CrossRef]
- Gardiner, S.H.; Karsili, T.N.V.; Lipciuc, M.L.; Wilman, E.; Ashfold, M.N.R.; Vallance, C. Fragmentation dynamics of the ethyl bromide and ethyl iodide cations: A velocity-map imaging study. Phys. Chem. Chem. Phys. 2014, 16, 2167–2178. [Google Scholar] [CrossRef]
- Borkar, S.; Sztáray, B. Self-Consistent Heats of Formation for the Ethyl Cation, Ethyle Bromide, and Ethyl Iodide from Threshold Photoelectron Photoion Coincidence Spectroscopy. J. Phys. Chem. A. 2010, 114, 6117–6123. [Google Scholar] [CrossRef]
- Baer, T.; Song, Y.; Liu, J.; Chen, W.; Ng, C.Y. Pulsed field ionization-photoelectron photoion coincidence spectroscopy with synchrotron radiation: The heat of formation of the C2H5+ ion. Faraday Discuss. 2000, 115, 137–145. [Google Scholar] [CrossRef]
- Miller, B.E.; Baer, T. Kinetic energy release distribution in the fragmentation of energy-selected vinyl and ethyl bromide ions. Chem. Phys. 1984, 85, 39–45. [Google Scholar] [CrossRef]
- Traeger, J.C.; McLoughlin, R.G. Absolute heats of formation for gas-phase cations. J. Am. Chem. Soc. 1981, 103, 3647–3652. [Google Scholar] [CrossRef]
- Xu, D.; Price, R.J.; Huang, J.; Jackson, W.M. Photodissociation of the Ethyl Bromide Cation at 355 nm by Means of TOF-MS and Ion Velocity Imaging Techniques. Z. FüR Phys. Chem. 2001, 215, 253–271. [Google Scholar] [CrossRef]
- Kramida, A.; Ralchenko, Y.; Reader, J.; NIST ASD Team. NIST Atomic Spectra Database (Version 5.11). 2023. Available online: https://physics.nist.gov/asd (accessed on 1 May 2023).
- Kimura, K.; Katsumatu, S.; Achiba, Y.; Yamaziki, T.; Iwata, S. Handbook of He(I) Photoelectron Spectra of Fundamental Organic Molecules; Japan Scientific Societies Press und Halstead Press: Tokyo, Japan; New York, NY, USA, 1981. [Google Scholar]
- Holmes, J.; Aubry, C.; Mayer, P. Assigning Structures to Ions in Mass Spectrometry; CRC Press, Taylor and Francis Group: Boca Raton, FL, USA, 2006. [Google Scholar] [CrossRef]
- Cunje, A.; Lien, M.H.; Hopkinson, A.C. The C4H5+ Potential Energy Surface. Structure, Relative Energies, and Enthalpies of Formation of Isomers of C4H5+. J. Org. Chem. 1996, 61, 5212–5220. [Google Scholar] [CrossRef]
- Catani, K.J.; Muller, G.; da Silva, G.; Bieske, E.J. Electronic spectrum and photodissociation chemistry of the linear methyl propargyl cation H2C4H3+. J. Chem. Phys. 2017, 146, 044307. [Google Scholar] [CrossRef]
- Ruscic, B.; Bross, D. Active Thermochemical Tables (ATcT) Values Based on ver. 1.130 of the Thermochemical Network. 2022. Available online: https://doi.org/10.17038/CSE/1997229 (accessed on 1 May 2023).
- Linstrom, P.J.; Mallard, W.G. NIST Chemistry WebBook-Standard Reference Database n. 69. Available online: https://doi.org/10.18434/T4D303 (accessed on 1 May 2023).
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01, 2016, Gaussian Inc. Wallingford CT. Available online: https://gaussian.com/ (accessed on 1 May 2023).
- Lago, A.; Baer, T. A Photoelectron Photoion Coincidence Study of the Vinyl Bromide and Tribromoethane Ion Dissociation Dynamics: Heats of Formation of C2H3+, C2H3Br+, C2H3Br2+, and C2H3Br3. J. Phys. Chem. A. 2006, 110, 3036–3041. [Google Scholar] [CrossRef]
- Blair, A.S.; Heslin, E.J.; Harrison, A.G. Biomolecular reactions of trapped ions. IV. Reactions in gaseous ethane and mixtures with acetylene and methane-d4. J. Am. Chem. Soc. 1972, 94, 2935–2939. [Google Scholar] [CrossRef]
- Ervin, K.M.; Armentrout, P. Translational energy dependence of Ar+ + XY - ArX+ + Y (XY = H2, D2, HD) from thermal to 30 eV c.m. J. Chem. Phys. 1985, 83, 166–189. [Google Scholar] [CrossRef]
- Nicolas, C.; Alcaraz, C.; Thissen, R.; Žabka, J.; Dutuit, O. Effects of ion excitation on ion-molecule reactions of the Mars, Venus, and Earth ionospheres. Plan. Space Sci. 2002, 50, 877–887. [Google Scholar] [CrossRef]
- Richardson, V.; Valença Ferreira de Aragão, E.; He, X.; Pirani, F.; Mancini, L.; Faginas-Lago, N.; Rosi, M.; Martini, L.M.; Ascenzi, D. Fragmentation of interstellar methanol by collisions with He+: An experimental and computational study. Phys. Chem. Chem. Phys. 2022, 24, 22437–22452. [Google Scholar] [CrossRef]
- Tsikritea, A.; Diprose, J.A.; Softley, T.P.; Heazlewood, B.R. Capture theory models: An overview of their development, experimental verification, and applications to ion–molecule reactions. J. Chem. Phys. 2022, 157, 060901. [Google Scholar] [CrossRef]
- Szilágyi, Z.; Vékey, K. A simple algorithm for the calculation of kinetic energy release distributions. Eur. Mass Spectrom. 1995, 1, 507–518. [Google Scholar] [CrossRef]
- Ess, D.H.; Wheeler, S.E.; Iafe, R.G.; Xu, L.; Çelebi Ölçüm, N.; Houk, K.N. Bifurcations on Potential Energy Surfaces of Organic Reactions. Angew. Chem. Int. Ed. 2008, 47, 7592–7601. [Google Scholar] [CrossRef]
- Bruce, J.E.; Eyler, J.R. Probing Trapped Ion Energies Via Ion-Molecule Reaction Kinetics: Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1992, 3, 727–733. [Google Scholar] [CrossRef]
- Grover, R.; Decouzon, M.; Maria, P.C.; Gal, J.F. Reliability of Fourier transform-ion cyclotron resonance determinations of rate constants for ion/molecule reactions. Eur. J. Mass Spectrom. 1996, 2, 213–223. [Google Scholar] [CrossRef]
- Carpignano, R.; Operti, L.; Rabezzana, R.; Vaglio, G.A. Chemometric Approach to Evaluate the Parameters Affecting the Determination of Reaction Rate Constants by Ion Trap Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1998, 9, 938–944. [Google Scholar] [CrossRef]
- Kemper, P.R.; Bowers, M.T. An improved tandem mass spectrometer-ion-cyclotron-resonance spectrometer. Int. J. Mass Spectrom. Ion Phys. 1983, 52, 1–24. [Google Scholar] [CrossRef]
- Richardson, V.; Alcaraz, C.; Geppert, W.D.; Polášek, M.; Romanzin, C.; Sundelin, D.; Thissen, R.; Tosi, P.; Žabka, J.; Ascenzi, D. The reactivity of methanimine radical cation (H2CNH+) and its isomer aminomethylene (HCNH2+) with methane. Chem. Phys. Lett. 2021, 775, 138611. [Google Scholar] [CrossRef]
- Alcaraz, C.; Nicolas, C.; Thissen, R.; Žabka, J.; Dutuit, O. 15N+ + CD4 and O+ + 13CO2 State-Selected Ion-Molecule Reactions Relevant to the Chemistry of Planetary Ionospheres. J. Phys. Chem. A. 2004, 108, 9998–10009. [Google Scholar] [CrossRef]
- Cunha de Miranda, B.; Romanzin, C.; Chefdeville, S.; Vuitton, V.; Žabka, J.; Polášek, M.; Alcaraz, C. Reactions of State-Selected Atomic Oxygen Ions O+ (4S, 2D, 2P) with Methane. J. Phys. Chem. A. 2015, 119, 6082–6098. [Google Scholar] [CrossRef] [PubMed]
- Nahon, L.; de Oliveira, N.; Garcia, G.; Gil, J.F.; Pilette, B.; Marcouilleé, O.; Lagarde, B.; Polack, F. DESIRS: A state-of-the-art VUV beamline featuring high resolution and variable polarization for spectroscopy and dichroism at SOLEIL. J. Synchrotron Rad. 2012, 19, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Mercier, B.; Complin, M.; Prevost, C.; Bellec, G.; Thissen, R.; Dutuit, O.; Nahon, L. Experimental and theoretical study of a differentially pumped absorption gas cell used as a low energy-pass filter in the vacuum ultraviolet photon energy range. J. Vac. Sci. Technol. A 2000, 18, 2533–2541. [Google Scholar] [CrossRef]
- Minnhagen, L. Spectrum and the energy levels of neutral argon, Ar I. J. Opt. Soc. Am. 1973, 63, 1185–1198. [Google Scholar] [CrossRef]
- Teloy, E.; Gerlich, D. Integral cross sections for ion-molecule rections. I. The guided beam technique. Chem. Phys. 1974, 4, 417–427. [Google Scholar] [CrossRef]
- Mardirossian, N.; Head-Gordon, M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. [Google Scholar] [CrossRef]
- Curtiss, L.A.; Redfern, P.C.; Raghavachari, K. Gaussian-4 theory. J. Chem. Phys. 2007, 126, 084108. [Google Scholar] [CrossRef]
- Silva, W.G.D.P.; Cernicharo, J.; Schlemmer, S.; Marcelino, N.; Loison, J.C.; Agúndez, M.; Gupta, D.; Wakelam, V.; Thorwirth, S.; Cabezas, C.; et al. Discovery of H2CCCH+ in TMC-1. Astron. Astrophys. 2023, 676, L1. [Google Scholar] [CrossRef]
- Cernicharo, J.; Gottlieb, C.A.; Guelin, M.; Killian, T.C.; Paubert, G.; Thaddeus, P.; Vrtilek, J.M. Astronomical Detection of H2CCC. Astrophys. J. Lett. 1991, 368, L39. [Google Scholar] [CrossRef]
- Thaddeus, P.; Vrtilek, J.M.; Gottlieb, C.A. Laboratory and astronomical identification of cyclopropenylidene, C3H2. Astrophys. J. 1985, 299, L63–L66. [Google Scholar] [CrossRef]
- Tsai, B.P.; Werner, A.S.; Baer, T. A photoion-photoelectron coincidence (PIPECO) study of fragmentation rates and kinetic energy release in energy selected metastable ions. J. Chem. Phys. 1975, 63, 4384–4392. [Google Scholar] [CrossRef]
- Booze, J.A.; Weitzel, K.M.; Baer, T. The rates of HCl loss from energy-selected ethylchloride ions: A case of tunneling through an H-atom transfer barrier. J. Chem. Phys. 1991, 94, 3649–3656. [Google Scholar] [CrossRef]
- Kuck, D. Half a century of scrambling in organic ions: Complete, incomplete, progressive and composite atom interchange. Int. J. Mass Spectrom. 2002, 213, 101–144. [Google Scholar] [CrossRef]










| Eave, eV | ktot 1 | ktot/kL 2 |
|---|---|---|
| 0.096 | 0.14 | |
| 0.12 | 0.13 | |
| 0.52 | 0.04 | |
| 2.02 | 0.12 | |
| 4.52 | 0.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richardson, V.; Polášek, M.; Romanzin, C.; Tosi, P.; Thissen, R.; Alcaraz, C.; Žabka, J.; Ascenzi, D. Reactivity of the Ethenium Cation (C2H5+) with Ethyne (C2H2): A Combined Experimental and Theoretical Study. Molecules 2024, 29, 810. https://doi.org/10.3390/molecules29040810
Richardson V, Polášek M, Romanzin C, Tosi P, Thissen R, Alcaraz C, Žabka J, Ascenzi D. Reactivity of the Ethenium Cation (C2H5+) with Ethyne (C2H2): A Combined Experimental and Theoretical Study. Molecules. 2024; 29(4):810. https://doi.org/10.3390/molecules29040810
Chicago/Turabian StyleRichardson, Vincent, Miroslav Polášek, Claire Romanzin, Paolo Tosi, Roland Thissen, Christian Alcaraz, Ján Žabka, and Daniela Ascenzi. 2024. "Reactivity of the Ethenium Cation (C2H5+) with Ethyne (C2H2): A Combined Experimental and Theoretical Study" Molecules 29, no. 4: 810. https://doi.org/10.3390/molecules29040810