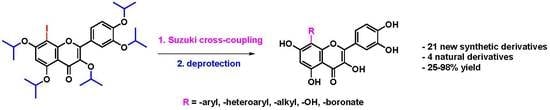
3.2.1. General Procedure A for Suzuki Cross-Coupling Reaction
8-Iodo flavonoid (1 eq) and the corresponding boronic acid (2 eq) were dissolved in a DMF/H2O mixture (9:1) in a microwave vial. The reaction mixture was degassed with nitrogen for 15 min. Pd(PPh3)4 (3 mol%) and NaOH (4 eq) were added. The reaction mixture was irradiated in a microwave reactor at 120 °C for 2 h. After the reaction mixture cooled to ambient temperature, it was filtered through a microfilter (PTFE, 0.45 µm). The reaction mixture was poured into water and extracted with dichloromethane (3 × 10 mL). The combined organic layers were washed with water (3 × 10 mL), brine (3 × 10 mL), dried over Na2SO4, and evaporated to dryness in vacuo. The residue was dissolved in dry dichloromethane (5 mL) at −10 °C. A solution of boron trichloride (10 eq, 1 M solution in dichloromethane) was added to the reaction mixture, which was then stirred at −10 °C for 30 min. The reaction mixture was then heated to 40 °C and stirred for 2 h, then cooled to 0 °C, and an excess of methanol was added. The reaction mixture was evaporated in vacuo, and the residue was purified by preparative HPLC chromatography (ASAHIPAK, 5 mL/min, MeOH isocratic) yielding the corresponding product.
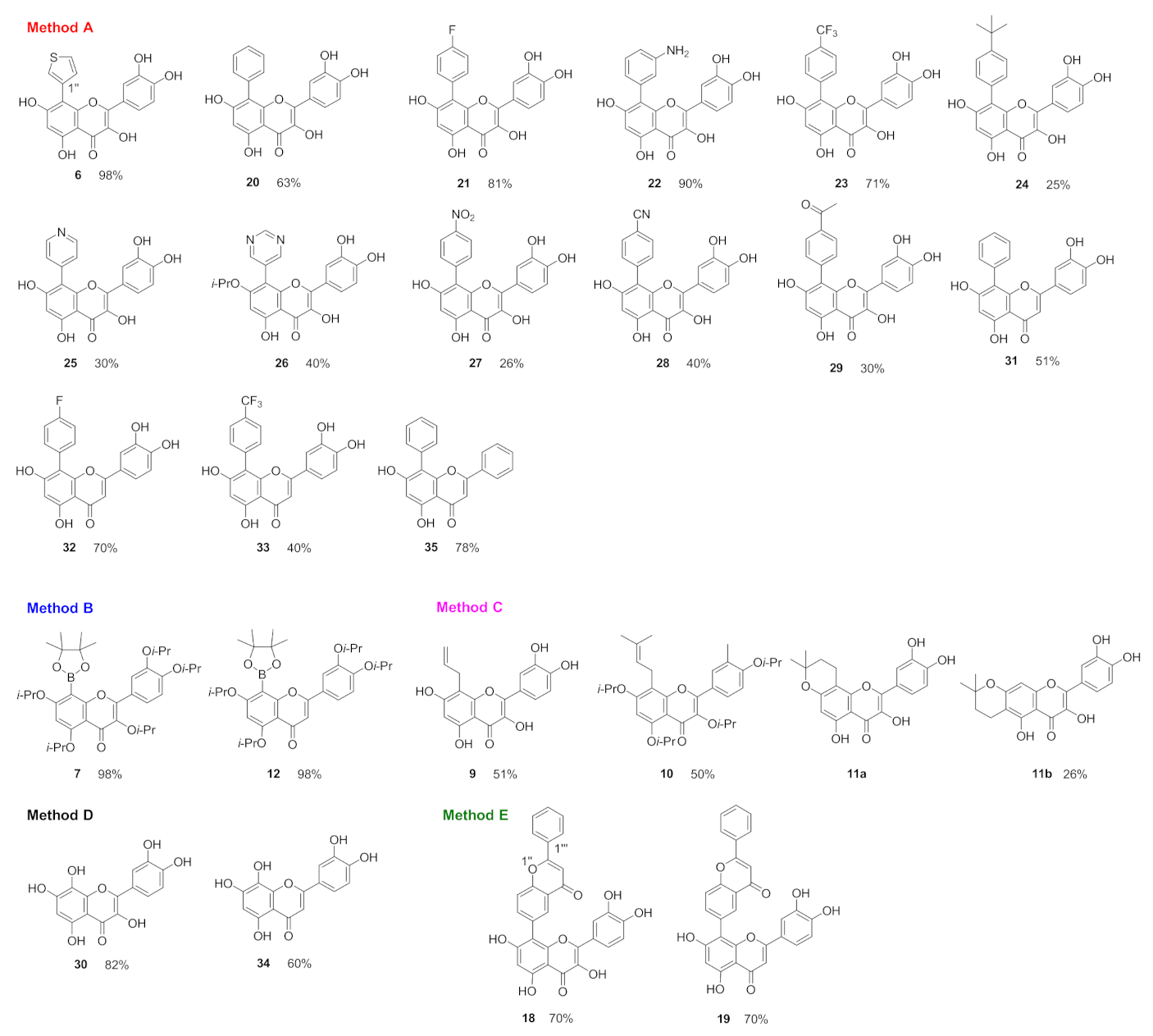
8-Thienyl quercetin (6): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5), and 3-thienylboronic acid, yielding 6 as a green solid (119 mg, 98%). HRMS (ESI, m/z): calcd for C19H11O7S [M-H]−: 383.02310, found: 383.02236. 1H NMR (500 MHz, DMSO-d6) δ 12.71 (s, 1H, HO-5), 10.84 (s, 1H, HO-7), 9.63 (s, 1H, HO-4′), 9.41 (s, 1H, HO-3), 9.15 (s, 1H, HO-3′), 7.64 (dd, J = 2.9, 1.2 Hz, 1H, H-2″), 7.61 (m, 2H, H-2′, 4″), 7.31 (dd, J = 4.9, 1.3 Hz, 1H, H-5″), 7.26 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.76 (d, J = 8.5 Hz, 1H, H-5′), 6.39 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 161.1 (C-7), 159.3 (C-5), 153.1 (C-9), 147.7 (C-4′), 147.2 (C-2), 144.9 (C-3′), 135.6 (C-3), 130.9 (C-1″), 130.2 (C-5″), 125.2 (C-4″), 124.3 (C-2″), 122.1 (C-1′), 119.7 (C-6′), 115.7 (C-2′), 115.3 (C-5′), 103.2 (C-10), 102.9 (C-8), 98.0 (C-6) ppm. IR (MeOH film): 1648 (s), 1602 (s), 1559 (s), 1507 (s), 1363 (m), 1203 (s), 1170 (s), 1109 (m), 1009 (m) cm−1. 8-Phenyl quercetin (20): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and phenylboronic acid, affording 20 as a yellow solid (70 mg, 63%). HRMS (ESI, m/z): calcd for C21H13O7 [M-H]−: 377.06668, found: 377.06616. 1H NMR (400 MHz, DMSO-d6) δ 12.68 (s, 1H, HO-5), 10.72 (s, 1H, HO-7), 9.60 (s, 1H, HO-4′), 9.40 (s, 1H, HO-3), 9.08 (s, 1H, HO-3′), 7.55 (d, J = 2.2 Hz, 1H, H-2′), 7.50–7.35 (m, 5H, H-2″, 3″,4″, 5″, 6″), 7.09 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.69 (d, J = 8.5 Hz, 1H, H-5′), 6.41 (s, 1H, H-6) ppm. 13C NMR (100 MHz, DMSO-d6) δ 176.1 (C-4), 160.8 (C-7), 159.4 (C-5), 153.0 (C-9), 147.6 (C-4′), 147.0 (C-2), 144.9 (C-3′), 135.6 (C-3), 132.0 (C-1″), 131.1 (C-3″, 5″), 127.8 (C-2″, 6″), 127.1 (C-4″), 122.1 (C-1′), 119.5 (C-6′), 115.7 (C-2′), 115.2 (C-5′), 107.6 (C-8), 103.1 (C-10), 98.0 (C-6) ppm. IR (MeOH film): 1650 (m), 1624 (sh), 1598 (m), 1560 (m), 1541 (m), 1490 (m), 1193 (s), 1022 (m), 999 (s), 701 (m), 686 (m) cm−1.
8-(4-Fluorophenyl) quercetin (21): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-fluorophenylboronic acid, affording 21 as a green solid (99.5 mg, 81%). HRMS (ESI, m/z): calcd for C21H12O7F [M-H]−: 395.05725, found: 395.05661. 1H NMR (400 MHz, DMSO-d6) δ 12.69 (s, 1H, HO-5), 10.77 (s, 1H, HO-7), 9.60 (s, 1H, HO-4′), 9.40 (s, 1H, HO-3), 9.11 (s, 1H, HO-3′), 7.49 (m, 3H, H-2″, 6″, 2′), 7.30 (m, 2H, H-3″, 5″), 7.13 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.73 (d, J = 8.5 Hz, 1H, H-5′), 6.40 (s, 1H, H-6) ppm. 13C NMR (100 MHz, DMSO-d6) δ 176.1 (C-4), 161.28 (d, J = 243.5 Hz, C-4″), 160.8 (C-7), 159.5 (C-5), 153.0 (C-9), 147.6 (C-4′), 147.1 (C-2), 144.9 (C-3′), 135.6 (C-3), 133.1 (d, J = 8.1 Hz, C-2″, 6″), 128.2 (d, J = 3.0 Hz, C-1″), 122.1 (C-1′), 119.6 (C-6′), 115.6 (C-2′), 115.3 (C-5′), 114.9 (d, J = 21.2 Hz, C-3″, 5″), 106.5 (C-8), 103.1 (C-10), 98.0 (C-6) ppm. 19F NMR (376 MHz, DMSO-d6) δ-115.15 ppm. IR (MeOH film): 1652 (m), 1620 (sh), 1598 (m), 1551 (m), 1506 (s), 1192 (s), 1160 (m), 1022 (m), 1000 (s), 763 (m) cm−1.
8-(3-Aminophenyl) quercetin (22): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 3-aminophenylboronic acid, yielding 22 as a green solid (111 mg, 90%). HRMS (ESI, m/z): calcd for C21H14O7N [M-H]−: 392.07758, found: 392.07751. 1H NMR (500 MHz, DMSO-d6) δ 12.71 (bs, 1H, HO-5), 11.01 (s, 1H, HO-7), 9.95 (bs, 2H, NH2), 9.66 (bs, 1H, HO-4′), 9.44 (bs, 1H, HO-3), 9.15 (bs, 1H, HO-3′), 7.61–7.53 (m, 2H, H-5″, 2′), 7.44 (d, J = 7.6 Hz, 1H, H-6″), 7.37 (s, 1H, H-2″), 7.32 (d, J = 7.9 Hz, 1H, H-4″), 7.07 (dd, J = 8.4, 2.2 Hz, 1H, H-6′), 6.75 (d, J = 8.5 Hz, 1H, H-5′), 6.47 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 160.8 (C-7), 159.8 (C-5), 152.9 (C-9), 147.7 (C-4′), 147.1 (C-2), 144.9 (C-3′), 135.7 (C-3), 133.6 (C-1″), 129.2 (C-5″, 6″), 124.7 (C-2″), 121.1 (C-4″), 122.0 (C-1′), 119.5 (C-6′), 115.7 (C-2′), 115.5 (C-5′), 106.3 (C-8), 103.1 (C-10), 98.0 (C-6) ppm. IR (KBr pellet): 1653 (s), 1624 (s), 1601 (s), 1559 (m), 1515 (s), 1491 (m), 1203 (s), 1003 (m), 999 (s), 759 (m), 698 (m) cm−1.
8-(4-Trifluoromethylphenyl) quercetin (23): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-(trifluoromethyl)phenylboronic acid, affording 23 as a yellow solid (144.3 mg, 71%). HRMS (ESI, m/z): calcd for C22H14O7F3 [M + H]+: 447.06861, found: 447.06877. 1H NMR (500 MHz, DMSO-d6) δ 12.74 (s, 1H, HO-5), 10.95 (s, 1H, HO-7), 9.59 (s, 1H, HO-4′), 9.45 (s, 1H, HO-3), 9.10 (s, 1H, HO-3′), 7.86–7.79 (m, 2H, H-3″, 5″), 7.74–7.69 (m, 2H, H-2″, 6″), 7.49 (d, J = 2.2 Hz, 1H, H-2′), 7.07 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.70 (d, J = 8.5 Hz, 1H, H-5′), 6.42 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 160.8 (C-7), 160.1 (C-5), 153.0 (C-9), 147.7 (C-4′), 147.2 (C-2), 144.9 (C-3′), 136.6 (C-1″), 135.7 (C-3), 132.0 (C-2″, 6″), 127.5 (q, J = 31.6 Hz, C-4″), 124.7 (q, J = 3.8 Hz, C-3″, 5″), 124.5 (q, J = 272.5 Hz, CF3), 122.0 (C-1′), 119.5 (C-6′), 115.6 (C-2′), 115.2 (C-5′), 106.1 (C-8), 103.1 (C-10), 98.0 (C-6) ppm. 19F NMR (470 MHz, DMSO-d6) δ-60.84 ppm. IR (MeOH film): 1651 (m), 1624 (sh), 1605 (m), 1563 (m), 1511 (m), 1326 (s), 1189 (m), 1009 (m), 1002 (m) cm−1.
8-(4-tert-Butylphenyl) quercetin (24): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-(tert-butylphenyl)phenylboronic acid, affording 24 as a yellow solid (33.7 mg, 25%). HRMS (ESI, m/z): calcd for C25H21O7 [M-H]−: 433.12928, found: 433.12852. 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H, HO-5), 10.69 (s, 1H, HO-7), 9.56 (s, 1H, HO-4′), 9.40 (s, 1H, HO-3), 9.05 (s, 1H, HO-3′), 7.59 (d, J = 2.2 Hz, 1H, H-2′), 7.50 (d, J = 8.4 Hz, 1H, H-3″, 5″), 7.37 (d, J = 8.3 Hz, 1H, H-2″, 6″), 7.05 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.66 (d, J = 8.5 Hz, 1H, H-5′), 6.40 (s, 1H, H-6), 1.36 (s, 9H, CH3 -tBu) ppm. 13C NMR (100 MHz, DMSO-d6) δ 176.1 (C-4), 160.8 (C-7), 159.3 (C-5), 153.1 (C-9), 149.4 (C-1″), 147.6 (C-4′), 146.9 (C-2), 144.9 (C-3′), 135.6 (C-3), 130.8 (C-2″, 6″), 129.0 (C-4″), 124.6 (C-3″, 5″), 122.1 (C-1′), 119.5 (C-6′), 115.9 (C-2′), 115.1 (C-5′), 107.5 (C-8), 103.1 (C-10), 98.0 (C-6), 34.4 (Cq-tBu), 31.2 (CH3 -tBu) ppm. IR (MeOH film): 1650 (m), 1624 (sh), 1602 (m), 1555 (m), 1512 (s), 1391 (m),1367 (m), 1191 (s), 1022 (m), 999 (s) cm−1.
8-(4-Pyridinyl) quercetin (25): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-pyridinylboronic acid, affording 25 as a yellow solid (35 mg, 30%). HRMS (ESI, m/z): calcd for C20H14O7N [M + H]+: 380.07648, found: 380.07682. 1H NMR (500 MHz, DMSO-d6) δ 12.74 (s, 1H, HO-5), 11.43 (s, 1H, HO-7), 9.83 (s, 1H, HO-4′), 9.43 (s, 1H, HO-3), 9.18 (s, 1H, HO-3′), 8.69–8.63 (m, 2H, H-3″, 5″), 7.54–7.46 (m, 3H, H-2″, 6″, 2′), 7.15 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.79 (d, J = 8.5 Hz, 1H, H-5′), 6.63 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.0 (C-4), 161.1 (C-7), 160.3 (C-5), 152.8 (C-9), 149.2 (C-3″, 5″), 147.8 (C-4′), 147.2 (C-2), 144.9 (C-3′), 140.4 (C-1″), 135.7 (C-3), 126.2 (C-2″, 6″), 121.9 (C-1′), 119.6 (C-6′), 115.7 (C-2′), 115.5 (C-5′), 104.8 (C-8), 103.0 (C-10), 98.2 (C-6) ppm. IR (KBr pellet): 1635 (m), 1598 (m), 1560 (m), 1490 (m), 1200 (s),1133 (m), 1002 (m) cm−1.
8-(5-Pyrimidino)-7-O-isopropyl quercetin (26): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and pyrimidine-5-boronic acid, yielding 26 as a yellow solid (50 mg, 40%). HRMS (ESI, m/z): calcd for C22H17O7N2 [M-H]−: 421.10412, found: 421.10372. 1H NMR (500 MHz, DMSO-d6) δ 12.91 (s, 1H, HO-5), 9.67 (s, 1H, HO-4′), 9.57 (s, 1H, HO-3), 9.19 (s, 1H, HO-3′), 9.17 (s, 1H, H-4″), 8.93 (s, 2H, H-2″, 6″), 7.47 (d, J = 2.2 Hz, 1H, H-2′), 7.08 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.74 (d, J = 8.5 Hz, 1H, H-5′), 6.72 (s, 1H, H-6), 4.85 (hept, J = 6.0 Hz, 1H, -CH), 1.24 (d, J = 6.0 Hz, 6H, -CH3) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.2 (C-4), 161.5 (C-7), 160.1 (C-5), 158.4 (C-2″, 6″), 156.9 (C-2″), 152.7 (C-9), 147.8 (C-4′), 147.7 (C-2), 145.0 (C-3′), 135.9 (C-3), 126.4 (C-1″), 121.8 (C-1′), 119.5 (C-6′), 115.6 (C-2′), 115.4 (C-5′), 103.6 (C-8), 102.3 (C-10), 96.1 (C-6), 71.4 (CH), 21.6 (CH3) ppm.
8-(4-Nitrophenyl) quercetin (27): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-nitrophenylboronic acid, affording 27 as a green solid (34 mg, 26%). HRMS (ESI, m/z): calcd for C21H12O9N [M-H]−: 422.05175, found: 422.05107. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (s, 1H, HO-5), 11.09 (s, 1H, HO-7), 9.57 (s, 1H, HO-4′), 9.47 (s, 1H, HO-3), 9.15 (s, 1H, HO-4′), 8.33 (d, J = 8.8 Hz, 2H, H-3″, 5″), 7.80 (d, J = 8.8 Hz, 2H, H-2″, 6″), 7.42 (d, J = 2.2 Hz, 1H, H-2′), 7.19 (dd, J = 8.4, 2.2 Hz, 1H, H-6′), 6.75 (d, J = 8.5 Hz, 1H, H-5′), 6.43 (s, 1H, H-6) ppm. 13C NMR (100 MHz, DMSO-d6) δ 176.1 (C-4), 160.8 (C-7), 160.5 (C-5), 152.9 (C-9), 147.7 (C-4′), 147.3 (C-2), 146.3 (C-4″), 144.9 (C-3′), 139.6 (C-1″), 135.8 (C-3), 132.5 (C-2″, 6″), 123.0 (C-3″, 5″), 122.0 (C-1′), 119.9 (C-6′), 115.4 (C-2′), 115.3 (C-5′), 105.5 (C-8), 103.2 (C-10), 98.03 (C-6) ppm. IR (CHCl3 film): 1653 (m), 1595 (m), 1558 (m), 1514 (m), 1345 (s), 1196 (m), 1022 (m), 999 (s), 699 (m), 680 (m) cm−1.
8-(4-Cyanophenyl) quercetin (28): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-cyanophenylboronic acid, affording 28 as a green solid (50 mg, 40%). HRMS (ESI, m/z): calcd for C22H12O7N [M-H]−: 402.06192, found: 402.06151. 1H NMR (500 MHz, DMSO-d6) δ 12.78 (s, 1H, HO-5), 11.01 (s, 1H, HO-7), 9.60 (s, 1H, HO-4′), 9.46 (s, 1H, HO-3), 9.15 (s, 1H, HO-3′), 7.93 (m, 2H, H-3″, 5″), 7.70 (m, 2H, H-2″, 6″), 7.45 (d, J = 2.2 Hz, 1H, H-2′), 7.14 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.75 (d, J = 8.5 Hz, 1H, H-5′), 6.41 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 160.7 (C-7), 160.3 (C-5), 152.9 (C-9), 147.7 (C-4′), 147.3 (C-2), 144.9 (C-3′), 137.5 (C-1″), 135.8 (C-3), 132.2 (C-3″, 5″), 131.7 (C-2″, 6″), 122.0 (C-1′), 119.7 (C-6′), 119.1 (CN), 115.4 (C-2′), 115.4 (C-5′), 109.6 (C-4″), 105.9 (C-8), 103.2 (C-10), 98.0 (C-6) ppm. IR (CHCl3 film): 2222 (m), 1655 (m), 1614 (sh), 1594 (s), 1545 (m), 1514 (m), 1354 (s), 1192 (s), 1002 (m) cm−1.
8-(4-Acetylphenyl) quercetin (29): General method A was followed using 8-iodo-3,3′,4′,5,7-penta-O-isopropylquercetin (5) and 4-acetylphenylboronic acid, affording 29 as a yellow solid (37 mg, 30%). HRMS (ESI, m/z): calcd for C23H15O8 [M-H]−: 419.07724, found: 419.07688. 1H NMR (500 MHz, DMSO-d6) δ 12.76 (s, 1H, HO-5), 10.94 (s, 1H, HO-7), 9.61 (s, 1H, HO-4′), 9.47 (s, 1H, HO-3), 9.14 (s, 1H, HO-3′), 8.09–8.03 (m, 2H, H-3″,5″), 7.67–7.61 (m, 2H, H-2″, 6″), 7.50 (d, J = 2.2 Hz, 1H, H-2′), 7.13 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.72 (d, J = 8.5 Hz, 1H, H-5′), 6.42 (s, 1H, H-6), 2.64 (s, 3H, -CH3) ppm. 13C NMR (126 MHz, DMSO-d6) δ 197.8 (C=O), 176.1 (C-4), 160.8 (C-7), 160.0 (C-5), 153.0 (C-9), 147.7 (C-4′), 147.3 (C-2), 145.1 (C-3′), 137.3 (C-4″), 135.8 (C-3), 135.4 (C-1″), 131.5 (C-2″,6″), 127.8 (C-3″, 5″), 122.0 (C-1′), 119.7 (C-6′), 115.7 (C-2′), 115.4 (C-5′), 106.5 (C-8), 103.2 (C-10), 98.1 (C-6), 26.9 (CH3). IR (MeOH film): 1671 (sh), 1650 (m), 1624 (sh), 1602 (s), 1560 (m), 1519 (m), 1195 (s), 1022 (m), 838 (m) cm−1.
8-Phenyl luteolin (31): General method A was followed using 8-iodo-3′,4′,5,7-tetra-O-isopropylluteolin (13) and phenylboronic acid, yielding 31 as a yellow solid (40 mg, 51%). HRMS (ESI, m/z): calcd for C21H13O6 [M-H]−: 361.07176, found: 361.07149. 1H NMR (500 MHz, DMSO-d6) δ 13.19 (s, 1H, HO-5), 10.82 (s, 1H, HO-7), 10.03 (s, 1H, HO-4′), 9.16 (s, 1H, HO-3′), 7.53–7.42 (m, 4H, H-2″, 3″, 5″, 6″), 7.40 (m, 1H, H-4″), 7.09 (d, J = 2.3 Hz, 1H, H-2′), 7.06 (dd, J = 8.3, 2.3 Hz, 1H, H-6′), 6.74 (d, J = 8.3 Hz, 1H, H-5′), 6.68 (s, 1H, H-3), 6.40 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 182.1 (C-4), 164.2 (C-2), 161.2 (C-7), 160.3 (C-5), 154.1 (C-9), 149.7 (C-4′), 145.6 (C-3′), 132.0 (C-1″), 131.1 (C-2″, 6″), 127.9 (3″, 5″), 121.7 (C-1′), 118.4 (C-6′), 115.6 (C-5′), 113.8 (C-2′), 108.0 (C-8), 103.7 (C-10), 102.6 (C-3), 98.7 (C-6) ppm. IR (MeOH film): 1651 (s), 1610 (m), 1583 (s), 1560 (m), 1520 (m), 836 (s), 701 (m), 686 (m) cm−1.
8-(4-Fluorophenyl) luteolin (32): General method A was followed using 8-iodo-3′,4′,5,7-tetra-O-isopropylluteolin (13) and 4-fluorophenylboronic acid, yielding 32 as a green solid (70 mg, 70%). HRMS (ESI, m/z): calcd for C21H14O6F [M + H]+: 381.07689, found: 381.07652. 1H NMR (500 MHz, DMSO-d6) δ 13.19 (s, 1H, HO-5), 10.87 (s, 1H, HO-7), 10.01 (s, 1H, HO-4′), 9.22 (s, 1H, HO-3′), 7.54–7.47 (m, 2H, H-2″, 6″), 7.35–7.27 (m, 2H, H-3″, 5″), 7.09 (dd, J = 8.3, 2.3 Hz, 1H, H-6′), 7.06 (d, J = 2.3 Hz, 1H, H-2′), 6.77 (d, J = 8.2 Hz, 1H, H-5′), 6.68 (s, 1H, H-3), 6.40 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 182.1 (C-4), 164.1 (C-2), 161.35 (d, J = 243.7 Hz, C-4″), 161.2 (C-7), 160.4 (C-5), 154.1 (C-9), 149.7 (C-4′), 145.6 (C-3′), 133.1 (d, J = 8.3 Hz, C-2″, 6″), 128.2 (C-1″), 121.7 (C-1′), 118.9 (C-6′), 115.7 (C-5′), 114.9 (d, J = 21.2 Hz, C-3″, 5″), 113.7 (C-2′), 107.0 (C-8), 103.7 (C-10), 102.7 (C-3), 98.6 (C-6) ppm. IR (MeOH film): 1648 (s), 1601 (m), 1558 (m), 1511 (s), 1228 (m), 1158 (m), 1008 (m), 835 (s), 686 (m) cm−1.
8-(4-Trifluoromethylphenyl) luteolin (33): General method A was followed using 8-iodo-3′,4′,5,7-tetra-O-isopropylluteolin (13) and 4-(trifluoromethyl)phenylboronic acid, yielding 33 as a green solid (70 mg, 85%). HRMS (ESI, m/z): calcd for C22H14O6F3 [M + H]+: 431.07370, found: 431.07386. 1H NMR (500 MHz, DMSO-d6) δ 13.24 (s, 1H, HO-5), 11.02 (s, 1H, HO-7), 9.98 (s, 1H, HO-4′), 9.16 (s, 1H, HO-3′), 7.85 (d, J = 7.9 Hz, 1H, H-3″, 5″), 7.73 (d, J = 8.6 Hz, 2H, H-2″, 6″), 7.10–7.02 (m, 2H, H-2′, 6′), 6.78–6.72 (m, 1H, H-5′), 6.69 (s, 1H, H-3), 6.42 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 182.0 (C-4), 164.2 (C-2), 161.1 (C-7), 160.8 (C-5), 154.1 (C-9), 149.7 (C-4′), 145.6 (C-3′), 136.5 (C-1″), 132.0 (C-2″, 6″), 127.6 (q, J = 31.8 Hz, C-4″), 124.7 (q, J = 3.8 Hz, C-3″, 5″), 124.5 (q, J= 271.6Hz, CF3), 121.6, 118.7 (C-6′), 115.7 (C-5′), 113.8 (C-2′), 106.5 (C-8), 103.7 (C-10), 102.8 (C-3), 98.6 (C-6) ppm. 19F NMR (470 MHz, DMSO-d6) δ-60.81 ppm. IR (MeOH film): 1649 (s), 1615 (m), 1582 (m), 1556 (m), 1522 (m), 1328 (s), 1193 (s), 1278 (m), 1131 (m), 1009 (m), 835 (s) cm−1.
8-Phenyl chrysin (35): General method A was followed using 8-iodo-5,7-di-O-isopropylchrysin and phenylboronic acid, yielding 35 as a white solid (124 mg, 78%). HRMS (ESI, m/z): calcd for C21H13O4 [M-H]−: 329.08193, found: 329.08200. 1H NMR (500 MHz, DMSO-d6) δ 13.02 (s, 1H, HO-5), 10.90 (s, 1H, HO-7), 7.77–7.70 (m, 2H, H-2′, 6′), 7.54–7.38 (m, 8H, H-3′, 4′, 5′, 2″, 3″, 4″, 5″, 6″), 7.01 (s, 1H, H-3), 6.45 (s, 1H, H-6) ppm. 13C NMR (126 MHz, DMSO-d6) δ 182.4 (C-4), 163.1 (C-2), 161.5 (C-7), 160.2 (C-5), 154.1 (C-9), 132.0 (C-4′), 131.9 (C-1″), 131.1 (C-2″, 6″ or C-3″, 5″), 130.8 (C-1′), 129.0 (C-3′, 5′), 127.9 (C-3″, 5″ or C-2″, 6″), 127.2 (C-4″), 126.2 (C-2′, 6′), 108.2 (C-8), 104.8 (C-3), 104.0 (C-10), 98.8 (C-6) ppm.
3.2.3. General Procedure C for Alkylation of Per-Isopropyl Derivatives
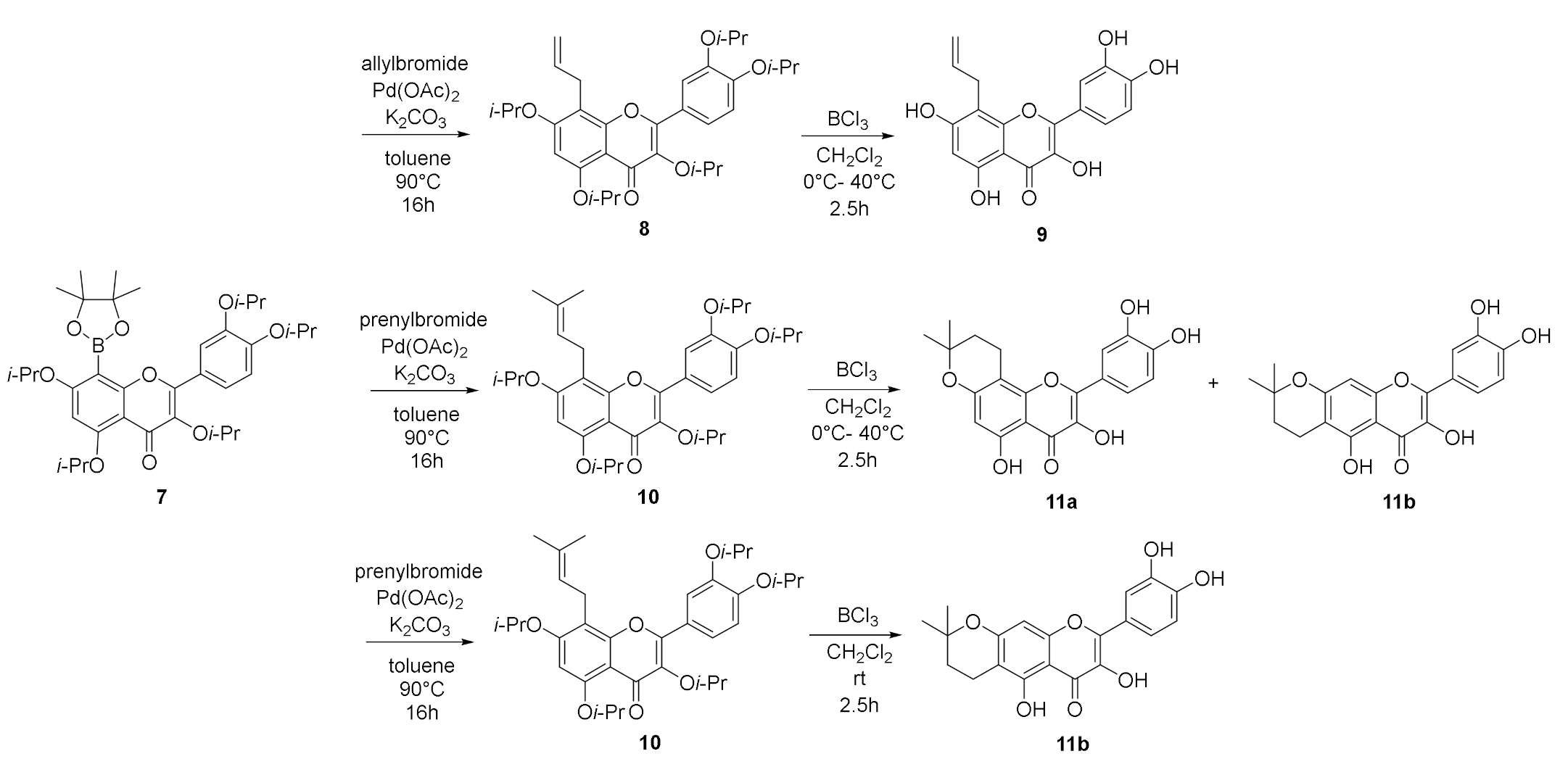
8-Boronyl flavonoid (1 eq), K2CO3 (2 eq), and Pd(OAc)2 (5 mol%) were dissolved in dry toluene. Alkyl bromide (1.5 eq) was added under an inert atmosphere. The reaction mixture was heated and stirred at 90 °C for 2 days. The reaction mixture was cooled to ambient temperature and evaporated in vacuo. The residue was dissolved in dry dichloromethane (5 mL) and cooled to −10 °C. Boron trichloride solution (10 eq, 1 M solution in dichloromethane) was added dropwise and the reaction mixture was stirred at −10 °C for 30 min. The reaction mixture was heated to 40 °C and stirred for 2 h. The reaction mixture was cooled to 0 °C and an excess of methanol was added. The reaction mixture was evaporated in vacuo, and the residue was purified by a preparative HPLC system (ASAHIPACK, 5 mL/min isocratic) to give the corresponding product.
8-Allyl quercetin (9): General method C was followed using 8-boronyl-3,3′,4′,5,7-penta-O-isopropylquercetin (7) and allyl bromide, affording 9 as a yellow solid (54 mg, 51%). HRMS (ESI, m/z): calcd for C18H13O7 [M-H]−: 341.06668, found: 341.06653. 1H NMR (500 MHz, DMSO-d6) δ 12.44 (s, 1H, HO-5), 10.74 (s, 1H, HO-7), 9.60 (s, 1H, HO-4′), 9.36 (br. s, 2H, HO-3, HO-3′), 7.71 (d, J = 2.2 Hz, 1H, H-2′), 7.56 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.89 (d, J = 8.5 Hz, 1H, H-5′), 6.30 (s, 1H, H-6), 6.00–5.89 (m, 1H, H-2″), 5.11–4.88 (m, 2H, H-3″), 3.49 (d, J = 5.9 Hz, 2H, H-1″) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 161.3 (C-7), 158.6 (C-5), 153.5 (C-9), 147.7 (C-4′), 146.7 (C-3), 145.2 (C-3′), 135.9 (C-2″), 135.6 (C-3), 122.3 (C-1′), 119.9 (C-6′), 115.7 (C-5′), 115.0 (C-2′), 114.8 (C-3″), 103.6 (C-8), 103.0 (C-10), 97.7 (C-6), 26.3 (C-1″) ppm. IR (MeOH film): 1653 (s), 1618 (m), 1555 (m), 1518 (m), 1320 (s), 1244 (s), 1159 (m), 1003 (m) cm−1.
8,7-(2,2-Dimethyldihydropyrano) quercetin (sinoflavonoid NB) (11a) and 7,6-(2,2-dimethyldihydropyrano)quercetin (11b): General method C was followed using 8-boronyl-3,3′,4′,5,7-penta-O-isopropylquercetin (7) and prenyl bromide, affording a mixture of 11a and 11b as a yellow solid (22 mg, 22%). HRMS (ESI, m/z): calcd for C20H17O7 [M-H]−: 369.09798, found: 369.09768. 1H NMR (500 MHz, DMSO) δ 10.77 (s, 1H, HO-5), 7.77 (d, J = 2.2 Hz, 1H, H-2′ (11b)), 7.74 (d, J = 2.2 Hz, 1H, H-2′, (11a)), 7.61 (dd, J = 8.5, 2.2 Hz, 1H, H-6′, (11a)), 7.56 (dd, J = 8.5, 2.2 Hz, 1H, H-6′, (11b)), 6.90 (d, J = 8.5 Hz, 1H, H-5′, (11a)), 6.85 (d, J = 8.4 Hz, 1H, H-5′, (11b)), 6.31 (s, 1H, H-8, (11b)), 6.13 (s, 1H, H-6, (11a)), 2.96–2.89 (m, 2H, H-1″, (11b)), 2.85 (t, J = 6.7 Hz, 2H, H-1″, (11a)), 1.95–1.90 (m, 1H, H-2″, (11b)), 1.87 (t, J = 6.7 Hz, 2H, H-2″, (11a)), 1.65 (s, 6H, CH3, (11b)), 1.33 (s, 6H, CH3, (11a)) ppm. 13C NMR (126 MHz, DMSO) δ 176.1 (C-4, (11a)), 176.0 (C-4, (11b)), 161.4 (C-7, (11b)), 159.2 (C-5, (11a)), 158.4 (C-9, (11b)), 158.0 (C-9, (11a)), 153.5 (C-5, (11b)), 153.0 (C-5, (11a)), 147.8 (C-4′, (11a)), 147.7 (C-4′, (11b)), 146.7 (C-2, (11b)), 145.2 (C-3′, (11a)), 145.1 (C-3′, (11b)), 136.1 (C-3, (11a)), 135.6 (C-3, (11b)), 122.3 (C-1′, (11b)), 122.2 (C-1′, (11a)), 120.0 (C-6′, (11a)), 119.6 (C-6′, (11b)), 115.7 (C-2′, (11a)), 115.5 (C-2′, (11b)), 115.4 (C-5′, (11b)), 114.9 (C-5′, (11a)), 105.2 (C-6, (11b)), 103.6 (C-10, (11a)), 103.0 (C-10, (11b)), 99.7 (C-8, (11a)), 98.6 (C-6, (11a)), 97.7 (C-8, (11b)), 76.2 (C-3″′, (11a)), 72.2 (C-3″′, (11b)), 44.4 (C-2″′, (11b)), 32.1 (CH3, (11b)), 31.0 (C-2″′, (11a)), 26.3 (CH3, (11a)), 18.2 (C-1″′, (11b)), 15.7 (C-1″′, (11a)) ppm.
6,7-(2,2-Dimethyldihydropyrano) quercetin (11b): General method C was followed using 8-boronyl-3,3′,4′,5,7-penta-O-isopropylquercetin (7) and prenyl bromide, affording 8-prenyl-3,3′,4′,5,7-penta-O-isopropylquercetin quercetin (10), which was deprotected using boron trichloride at room temperature yielding 11b as a yellow solid (42 mg, 26%). HRMS (ESI, m/z): calcd for C20H17O7 [M-H]−: 369.09798, found: 369.09768. 1H NMR (500 MHz, DMSO-d6) δ 7.77 (d, J = 2.2 Hz, 1H, H-2′), 7.56 (dd, J = 8.5, 2.2 Hz, 1H, H-6′), 6.85 (d, J = 8.5 Hz, 1H, H-5′), 6.30 (s, 1H, H-8), 2.96–2.90 (m, 2H, H-1″), 1.95–1.90 (m, 2H, H-2″), 1.66 (s, 6H, CH3) ppm. 13C NMR (126 MHz, DMSO-d6) δ 176.1 (C-4), 161.4 (C-7), 158.4 (C-9), 153.5 (C-5), 147.7 (C-4′), 146.7 (C-2), 145.1 (C-3′), 135.5 (C-3), 122.3 (C-1′), 119.6 (C-6′), 115.5 (C-2′), 115.4 (C-5′), 105.2 (C-6), 103.0 (C-10), 97.7 (C-8), 72.2 (C-3″), 44.4 (C-2″), 32.1 (CH3), 18.2 (C-1″) ppm.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
