1. Introduction
Statins (
Figure 1) are the inhibitors of
de novo cholesterol synthesis in the cell due to their ability to competitively inhibit the β-hydroxy-β-methylglutaryl coenzyme A (HMG-CoA) reductase. They represent the most prescribed drugs in the treatment of cardiovascular diseases [
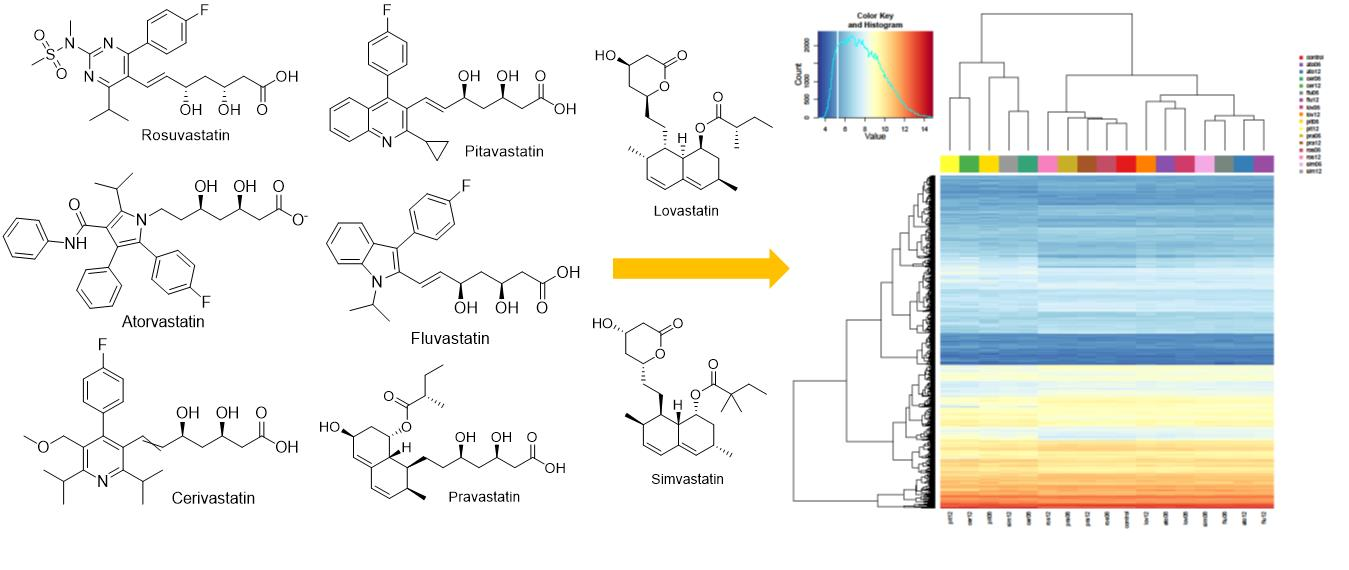
1]. Currently, there are eight existing statins, namely atorvastatin, cerivastatin, fluvastatin, lovastatin, pitavastatin, pravastatin, rosuvastatin, and simvastatin [
2]. Although all statins have the same mechanism of hypolipidemic action, they differ in their chemical structure, physico-chemical properties, pharmacokinetic effects, and effects on lipid profile [
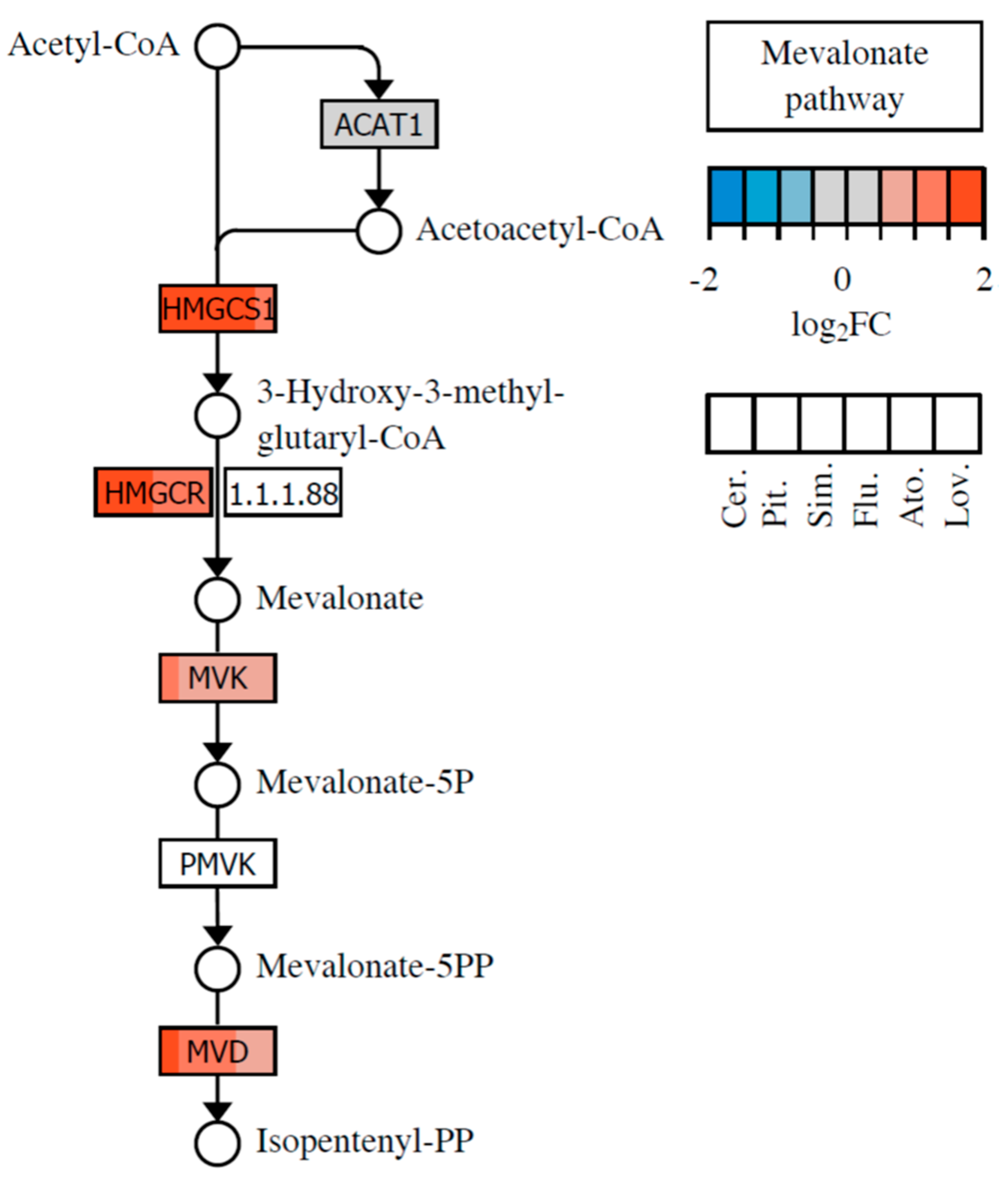
3]. The inhibition of HMG-CoA conversion to mevalonate caused by HMG-CoA reductase inhibition results in the depletion of downstream intermediates of the mevalonate pathway.
Mevalonate is the precursor of, i.e., farnesyl pyrophosphate (FPP), geranylgeranyl pyrophosphate (GGPP), isopentenyl adenine, dolichol, and the polyisoprenoid side chains of heme A and ubiquinone, which are essential molecules that play a vital role in almost any cell process [
3]. From these, FPP and GGPP play an important role in the posttranslational modification of cellular proteins involved in cell division and differentiation, gene expression, cytoskeleton formation, intracellular protein and lipid transport, and defense against pathogens [
4]. Another intermediate in the mevalonate pathway, isopentenyl adenine, is essential for proper tRNA function and protein synthesis. Dolichol acts as an important scavenger of free radicals in cell membranes, and ubiquinone is involved in mitochondrial respiration and inhibition of lipid peroxidation. A decrease in the intracellular level of ubiquinone leads to mitochondrial damage, oxidative stress, and cell damage. The involvement of the aforementioned mevalonate pathway intermediates in various cell processes explains the pleiotropic activity exhibited by statins, in addition to their hypolipidemic effect.
Based on this, it is clear that statins significantly affect basic physiological processes of cells and organs that also are connected with oncogenesis [
5]. The results of lipid-lowering therapy in animals initially indicated an increased risk of carcinogenesis [
6]. However, the dose of statins administered in that study was very high and not applicable for humans [
7]. The results of studies on tissue-specific cancer development in individuals on statins therapy (as hypolipidemic drugs) have been controversial. An increased incidence of breast cancer after statin administration was observed in one study [
8], but another study did not confirm these findings [
9]. Contrary to that, a reduced incidence of melanomas has been reported after statin administration [
9]. A similar beneficial effect of statins was observed in connection with reduced incidence of colon carcinomas [
10]. However, most of these studies were not originally designed to investigate the relationship between statin intake and cancer development, but rather between statins and cardiovascular diseases. Indeed, some reports have shown a statistically significantly lower incidence of cancer in patients receiving statin therapy, despite relatively short follow-up times and inappropriate patient selection [
11,
12,
13,
14,
15,
16]. Moreover, extensive studies conducted on 500,000 U.S. war veterans have shown that statin use is associated with a two- to fivefold lower incidence of lung [
17], breast [
18], and prostate cancers [
19]. Based on other meta-analyses, statins appear to be particularly effective in the chemoprevention of colorectal cancer [
10,
20]. While there have been several
in vitro and
in vivo studies directly targeting and confirming the marked effect of statins on the growth of a wide variety of tumor types, such as hepatocellular [
21], lung [
22], and colorectal carcinoma [
23], clinical trials targeting statin efficacy in cancer treatment have been very rare, so far. One of them showed that patients suffering from hepatocellular carcinoma exhibited statistically significantly longer survival rates for pravastatin therapy [
24] than untreated. On the other hand, beneficial effects of lovastatin therapy on the survival rate of patients suffering from glioblastoma multiforme or advanced gastric adenocarcinoma have not been observed [
25,
26]. The findings of the meta-analyses suggested an association between pravastatin treatment and cancer in elderly patients [
27] but did not support the potential role of statins in the prevention of hematological malignancies [
28] or the hypothesis that statins reduce the risk of pancreatic cancer development [
29].
The setting of the experimental conditions of the present study was based on the results of our previous published studies [
30,
31], in which we reported on the differences in the individual anticancer potential of individual statins in cells derived from pancreatic carcinoma. Moreover, we were inspired by the aforementioned findings, as well as recent reviews shedding light on statins as cancer inhibitors with several possible mechanisms of action [
32,
33,
34,
35]. Therefore, using DNA microarray analysis, we sought after statin-induced changes in the expression of genes, the products of which are connected with tumorigenesis, inhibition of tumor growth, or metastasis. We focused on the pancreatic cancer cells because, in the very recent meta-analysis of 26 studies containing more than 3 million participants and 170,000 pancreatic cancer patients, statin therapy was shown to significantly reduce the risk of pancreatic cancer [
36].
4. Discussion and Conclusions
Statins have been intensively studied drugs based on their deep impact on the human organism caused, in particular by their cholesterol-lowering activity. However, their effects also include remarkable and potentially clinically relevant antitumor effects.
Although lipids are not genetically encoded, the genome changes can reflect cholesterol homeostasis indirectly. The changes in gene expression following statin treatment studied by microarray technology have been reported since the year 2000. The interpretation of our microarray analysis indicates a significant correlation between our and previously reported results. On the other hand, many differences are resulting from distinct experimental conditions due to the type of statin used, its concentration and duration of its activity, the experimental model (the type of a cell line or organism), etc.
Previously, we have demonstrated substantial differences in cancer cell antiproliferative effects of all commercially available statins in an experimental model of human pancreatic cancer [
31]. Various statins exhibited significantly different inhibitory efficacy, and we have also observed notable differences in statin sensitivity between pancreatic cancer cell lines, concerning the level of differentiation and harboring of G12C-activating mutation in the
KRAS gene. Cerivastatin, pitavastatin, and simvastatin were the most effective, whereas rosuvastatin and pravastatin were the least effective, and their effectiveness correlated with the properties of the cell lines tested (the level of differentiation, and presence of G12C
KRAS mutation) [
31]. In the present study, we observed that depletion of farnesylated K-Ras protein caused by the three most effective statins led to significant upregulation of gene-encoding K-Ras.
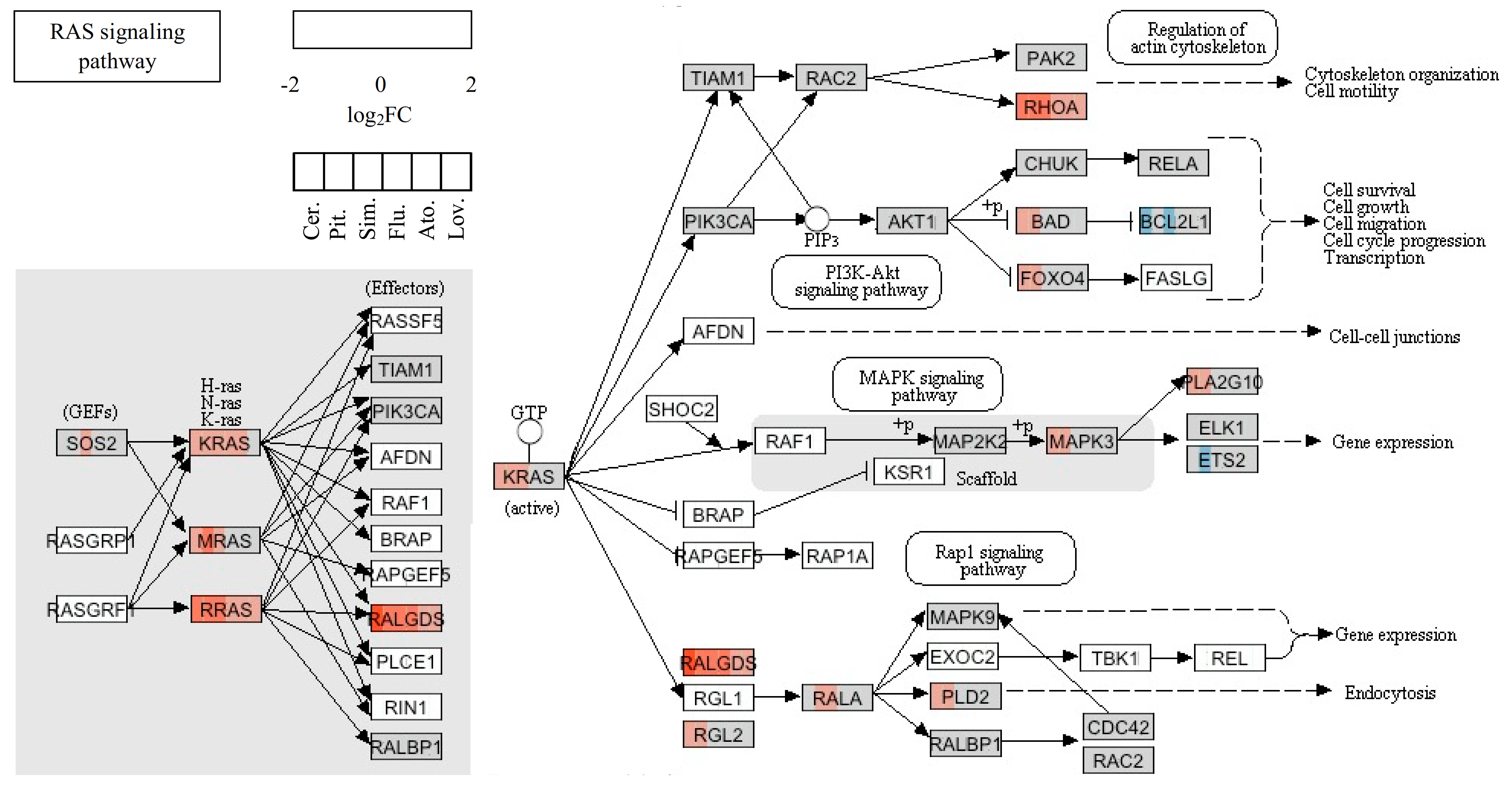
The pleiotropic effect of statins is comprehensible, since K-Ras is related to many functional pathways, such as the MAPK signaling pathway, the ErbB signaling pathway, dorso-ventral axis formation, axon guidance, the VEGF signaling pathway, tight junction, gap junction, natural killer cell-mediated cytotoxicity, the T-cell receptor signaling pathway, the B-cell receptor signaling pathway, the Fc epsilon RI signaling pathway, long-term potentiation, long-term depression, regulation of actin cytoskeleton, the insulin signaling pathway, and the GnRH signaling pathway. Moreover, the upregulation of KRAS after the statin treatment seemed to be a result of the unavailability of farnesylated K-Ras protein for cell signaling due to the mevalonate pathway inhibition. Similarly, this mechanism could also explain the upregulation of other Ras and Ras-related proteins induced by statin treatment (
Table 3). However, the products of the mevalonate pathway are required not only for post-translational modifications of many proteins, but also for regulation of the transcription of many proteins, including proteins of the cytoskeleton. For example, expression of keratin 13 is known to be regulated by nuclear receptor ligands such as retinoids, 1α, 25-dihydroxy vitamin D3, or estrogen [
65]. Except for the metabolism of lipids (biosynthesis of steroids and sphingolipid metabolism), the inhibition of the mevalonate pathway by statins directly affected the energy metabolism due to ubiquinone depletion, as ubiquinone is involved in mitochondrial respiration [
80].
Genetic heterogeneity is an important factor that affects the sensitivity of particular cancer cells to the antiproliferative/proapoptotic effects of statins. Despite the level of cancer cell differentiation, pancreatic cancer cell lines harboring activating K-Ras mutation were more sensitive to the antiproliferative effect of statins than cells harboring wild-type K-Ras [
31]. Similarly, Wong et al. demonstrated that only 8 of 17 multiple myeloma cell lines evaluated were sensitive to lovastatin-induced apoptosis, while resistant cell lines had different genetic profiles [
81].
The first effect of statins, specifically simvastatin, studied by microarray analysis was focused on actomyosin contraction, gap formation, and barrier dysfunction produced by thrombin. The experiment was performed in human pulmonary artery endothelial cells (EC) treated with 5 µM simvastatin for 24 h. Several genes related to thrombin-mediated cytoskeletal dynamics and barrier regulation, including caldesmon and the thrombin receptor PAR-1, were dramatically downregulated. In addition, ITGB4, a protein known to be involved in cell–cell adhesion, was dramatically upregulated.
RhoA and
RhoC genes were also upregulated similarly to Rac1 and specific GEFs, potential regulators of preferential Rho GTPase activity. In contrast, the RhoGDP dissociation inhibitor was downregulated, which was interpreted as a compensatory response [
82]. Consistent with these data, our results also indicated that statins directly affected the expression of specific genes related to the Rho GTPase signaling and cytoskeletal regulation. However, from all the Rho family members, only
RhoB was significantly upregulated by all effective statins (pravastatin and rosuvastatin were not effective in our microarray study), and
RhoA was significantly upregulated only by the most effective statins (cerivastatin, pitavastatin, and simvastatin). No changes were observed in the expression of genes encoding specific GEFs. Also, downregulation of caldesmon or the thrombin receptor PAR-1 was not observed in our study. This could vary in different types of cell lines evaluated.
Interestingly, a gene encoding the integrin beta 4 subunit of a receptor for the laminins; i.e.,
Itgb4, was the most significantly upregulated gene (fold change—7.57) of all the tested genes [
83]. The reason for the enhanced transcription of a gene involved in the biology of invasive carcinoma is unclear. Another notable finding is that no changes were observed in the expression of the
RhoB gene after treatment of EC with 5 µM simvastatin [
84], whereas in our study, the
RhoB gene was the more affected gene compared to the Itgb4 gene after treatment with all effective statins.
In the other study, the DNA microarrays were used to identify gene expression patterns in the cerebral cortex of mice treated with simvastatin (50 mg/kg b.wt.) by daily oral doses for 21 days. The maximum average concentration of simvastatin was determined as 600 pmol/g in brains. This study revealed the influence of simvastatin on the expression of several genes involved in cell growth and signaling.
C-fos,
c–myc, and
Bcl-2 were of particular interest due to the linkage of simvastatin with cell growth and apoptosis. Simvastatin significantly reduced the expression of the proto-oncogene
c-fos. On the other hand, it significantly increased the expression of the oncogene
c-myc and antiapoptotic gene
Bcl-2 [
84,
85]. In contrast to these data, neither the expression of previously mentioned genes nor of other genes attributed to apoptotic cell death (e.g., genes encoding death receptors or caspases, the only exception to which was caspase 9, which was upregulated by cerivastatin) were affected by statins in our microarray analysis. However, as demonstrated by the analysis of the most affected functional pathways, statins affected some processes related to DNA repair, such as base excision repair or mismatch repair [
30]. It is known that failure of these processes could be followed by programmed necrosis [
86]. Finally, the upregulation of the
GABARAPL gene related to autophagy was observed (
Table 5). Autophagy does not always result in programmed cell death, but it represents the important mechanism for catabolic production of ATP during nutrient stress, and also plays an important role in the turnover of proteins and organelles under nutrient-replete conditions [
87]. The statin-treated cells were in nutrient stress due to inhibition of the mevalonate pathway. From many end products of this pathway, ubiquinone is required in a process of ATP formation during oxidative phosphorylation. Moreover, oxidative phosphorylation is also reported as one of the significantly affected functional pathways by statins [
30]. Interestingly, it was published that ubiquitinated hydrophobic proteins that are prone to aggregation are kept on the surface of lipid droplets and subjected to autophagy, as well as proteasomal degradation [
88].
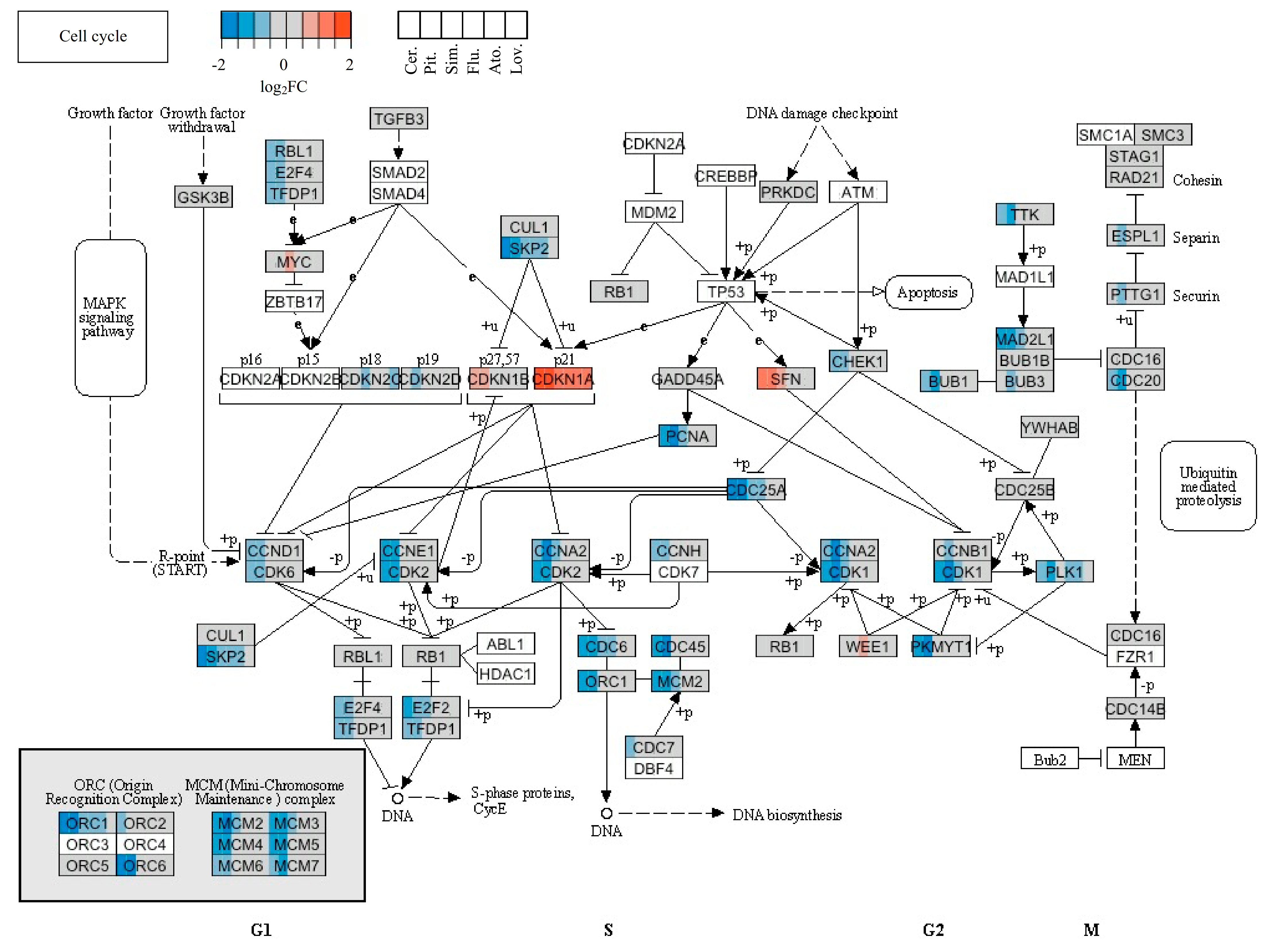
The cell-cycle arrest represents another frequently discussed event associated with statins. Many reports describe the effect of individual statins on the expression of cell-cycle-related genes. For example, changes in the expression of genes related to the cell cycle in chronic myelogenous leukemia cells K562 were described. Fifteen downregulated and 9 upregulated cell-cycle-related genes were observed in the presence of 20 µM simvastatin for 48 h. The results of flow cytometry showed that the cell cycle was arrested in the G1 phase [
89]. Assmus et al. performed a microarray analysis of about 12,000 genes in endothelial progenitor cells (a primary cell line) treated with 0.1 µM atorvastatin for 10 h. The expression of cyclins and proliferating cell nuclear antigen (PCNA) was increased after atorvastatin treatment. Moreover, the expression of the cell-cycle inhibitory protein p27 was reduced [
90]. In the next study, downregulation of cyclin D1, PCNA, and c-myc, and upregulation of p21 and p19, were provided by the treatment of human breast cancer cells with cerivastatin [
91].
In our microarray analysis, the expression of genes encoding cyclin D1 was affected only by cerivastatin, cyclin A2 by pitavastatin, and cyclin E2 by both mentioned statins, similar to the
PCNA gene. Other statins did not affect the expression of genes encoding cyclins or PCNA. The expression of the
p21 gene was increased by all effective statins. Moreover, the genes associated with DNA replication, such as
ORC1L,
MCM2, or
MCM3, were downregulated by cerivastatin and pitavastatin (
Table 4). This suggests that statins blocked the progress of the cell cycle through the S phase of MiaPaCa-2 cells. The downregulation of the genes encoding histone H4 and upregulation of the gene encoding histone H2B by the three most effective statins (cerivastatin, pitavastatin, and simvastatin) also was very interesting. The effect of simvastatin on expression of the gene encoding histone was also observed in a report by Johnson-Anuna et al., in which the expression level of the gene encoding the linker histone H1.2 was increased after simvastatin therapy [
84]. Likewise, other proteins related to the S phase (SKP2, E2F2) or M phase (CDC2, CDC25B) were downregulated by cerivastatin and pitavastatin treatment of MiaPaCa-2 cells in our study, indicating that except for the G1 phase, the most effective statins blocked the cell-cycle entry into the M phase. This was not surprising, as lovastatin is used in the cell-cycle synchronization protocols [
92] and as a pharmacological tool for controlling the growth of neoplastic cells both in vitro and in vivo [
23,
93,
94]. Furthermore, lovastatin is commercially available as an inhibitor of the cell cycle in the G1 and G2/M phases (Sigma, USA). The G1 block has been attributed to the inhibition of either cytokinesis or cell spreading following cytokinesis [
95]. Several authors have also noted the retardation or arrest of the cell cycle at the G2/M transition [
93,
94,
96]. However, the mechanism of the cell-cycle arrest by statins is not exactly clear. Despite a piece of evidence that lovastatin suppresses cell proliferation through inhibition of proteasome-mediated degradation of p21 and p27 [
97], it was concluded that lovastatin neither synchronizes cells nor arrests the cells in the G1 phase of the division cycle [
98].
To explain the reported results, the distinct impact of statins on gene expression profiles in MiaPaCa-2 cells should be related to their inhibitory activity interfering with the mevalonate pathway. Quantum biochemistry computations indicated some variations among the attractive forces of four tested statins (atorvastatin, rosuvastatin, simvastatin, and fluvastatin) and the HMG-CoA reductase binding site. The highest binding energies was determined for atorvastatin followed by rosuvastatin, while the lowest were found for simvastatin and fluvastatin; i.e., binding energies of 320, 310, 290, and 290 kcal·mol
-1, respectively [
99]. However, in this study, cerivastatin, lovastatin, and pitavastatin were not included in the calculations. In another study, the crystal structures of the catalytic moiety of HMG-CoA reductase in a complex with six statins documented van der Waals interactions of the rigid hydrophobic moieties of the statins through a shallow nonpolar binding pocket and a part of the binding surface for CoA [
100]. These interactions prevented the binding of the substrate HMG-CoA to the active site of the enzyme. No dramatic differences were found among the numbers of binding interactions among the statins evaluated, namely: compactin, simvastatin, fluvastatin, cerivastatin, atorvastatin, and rosuvastatin. Atorvastatin, simvastatin, lovastatin, fluvastatin, and cerivastatin are relatively lipophilic and are metabolized by the cytochrome P450 system. The other lipophilic compound pitavastatin is metabolized poorly via this pathway. Very limited P450-mediated metabolization was reported also for hydrophilic pravastatin and rosuvastatin, which were only nonsignificantly metabolized. Interestingly, high systemic bioavailability was reported for both cerivastatin and pitavastatin (60% [
101] and 80% [
102], respectively), which could explain their large impact on changes in gene expression. Except for pravastatin, all the other statins were efficiently bound to plasma proteins. However, the unbound pravastatin was poorly distributed in tissues due to its high hydrophilic nature [
103].
Further, the distinct efficacy of individual statins on both antiproliferative activity and changes in gene expression could be also correlated with the statin levels inside cells. This correlation was the strongest for the least efficient statins (rosuvastatin and pravastatin); whereas for the most bioavailable statins (in particular lovastatin), this correlation was not so strong [
30]. Thus, the statin effects on whole gene expression correlated with their bioavailability, as well as the impact on cell viability, only to a limited extent. This hypothesis held for cerivastatin; however, not so for pitavastatin and lovastatin, which led us to the conclusion that other crucial factors played an important role in the differences of statin effects on pancreatic cancer cell proliferation.
In summary, differences in the efficacy of individual statins are known depending on their structure, concentration, duration of action, or microenvironment. Although tumor cells exhibit many identical properties, the effect of statins depends also on the cell type [
104]. The antitumor effect of statins is not only a function of the mechanism of their action, but also of how they are metabolized. In general, healthy cells are generally more resistant to statins than tumor cells [
105]. The use of statins in the treatment of cancer as monotherapeutics is not effective enough, but their use in combination with other therapeutic approaches would significantly increase the effectiveness of cancer treatments and patient survival [
106,
107].


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}