
Abstract
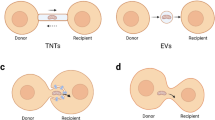
Tunnelling nanotubes (TNTs) are an emerging route of long-range intercellular communication that mediate cell-to-cell exchange of cargo and organelles and contribute to maintaining cellular homeostasis by balancing diverse cellular stresses. Besides their role in intercellular communication, TNTs are implicated in several ways in health and disease. Transfer of pathogenic molecules or structures via TNTs can promote the progression of neurodegenerative diseases, cancer malignancy, and the spread of viral infection. Additionally, TNTs contribute to acquiring resistance to cancer therapy, probably via their ability to rescue cells by ameliorating various pathological stresses, such as oxidative stress, reactive oxygen species (ROS), mitochondrial dysfunction, and apoptotic stress. Moreover, mesenchymal stem cells play a crucial role in the rejuvenation of targeted cells with mitochondrial heteroplasmy and oxidative stress by transferring healthy mitochondria through TNTs. Recent research has focussed on uncovering the key regulatory molecules involved in the biogenesis of TNTs. However further work will be required to provide detailed understanding of TNT regulation. In this review, we discuss possible associations with Rho GTPases linked to oxidative stress and apoptotic signals in biogenesis pathways of TNTs and summarize how intercellular trafficking of cargo and organelles, including mitochondria, via TNTs plays a crucial role in disease progression and also in rejuvenation/therapy.
Similar content being viewed by others

Introduction
Cell-to-cell communication plays an important role in maintaining tissue homeostasis. Intercellular communication can be facilitated by many soluble factors such as growth factors, neurotransmitters, cytokines, and extracellular vesicles (EVs), such as exosomes. A study in 2004 [1], first described intercellular transfer of molecular information directly between distal cells forming f-actin containing membrane lipid bilayer encircled ‘tunnel’ structures. Since then, the term “tunnelling nanotube” (TNT) has referred to this membrane f-actin conduit. Originally, the diameter of TNTs was reported to be 50–200 nm [1]. Later studies reported a relatively thicker diameter of around 700–900 nm, using optical resolution limited methods [2]. Cancer cells form networks of TNT-like but relatively thicker membrane protrusions, termed as tumour microtubes (TMs), consisting of both f-actin and tubulin. They are closed-ended and connected via gap junctions at the ends to transfer electrical signals and small molecules [3, 4]. Several studies have also referred to thinner nano-scaled membrane actin closed-ended protrusions as TNTs. Conventionally, f-actin containing open ended nanostructures are termed as TNTs. Recently, correlative FIB-SEM, light- and cryo-electron microscopy of neuronal cells revealed that TNTs of diameter 550 nm are made of 2–11 bundles of thinner channels (iTNTs), where the average diameter of each iTNTs was 123 ± 66 nm [5]. TNTs allow for the intercellular transport of various cargos, including viruses, organelles, RNAs, proteins, and toxic materials such as neurodegenerative protein aggregates [6]. Transfer of mitochondria has been implicated in disease progression and also in regeneration. Several studies have shown that intracellular build-up of prions or prion-like proteins facilitate disease progression by transferring toxic aggregates of these proteins or stressed organelles such as lysosomes and mitochondria from pathological donor cells to healthier acceptor cells [7, 8]. On the other hand, healthy mitochondria from mesenchymal stem cells (MSCs) are transferred to targeted acceptor cells with non-functional mtDNA/mitochondria [6, 9, 10]
In addition to mediating intercellular communication, TNTs rescue cells by relieving diverse cellular stresses caused by pathological conditions, such as oxidative stress, reactive oxygen species (ROS), mitochondrial heteroplasmy and apoptotic stress [11, 12]. Although the molecular drivers for the formation of TNTs under various pathophysiological conditions are unclear, studies over the last two decades indicate that cells form direct long-range connections between neighbouring cells via TNTs to alleviate cellular stress. Cytoskeletal dynamics play a pivotal role in the formation of TNTs and several studies have implicated the localized control of Rho GTPases in TNT-linked actin polymerization pathways [3, 13]. It has become evident that classical Rho GTPases (Rac1, Cdc42, and RhoA) control the complex regulatory balance in cell cycle progression and apoptotic signalling pathways [13, 14]. The capacity of MSCs to donate healthy mitochondria to targeted acceptor cells via TNTs correlates with the activity and expression of the atypical mitochondrial Rho GTPases [15], Miro-1 [9] and Miro-2 [16]. In this review, we summarize the role of TNTs in counteracting oxidative stress, mitochondrial heteroplasmy and apoptosis-related diverse cellular stresses, and the possible association of Rho GTPase-linked apoptotic signalling pathways in cytoskeleton remodelling and plasma membrane surface dynamics in the biogenesis of TNTs.
TNTs in intercellular transport
The original report [1], showed the transfer of endocytic vesicles and organelles as intercellular mediators between pheochromocytoma (PC12) cells. Subsequently, several studies in various cellular systems have shown the presence of TNTs and a range of organelles and cargo transportation via TNTs. These cargos include cytosolic proteins [17], ions [18], and miRNAs [19] that propagate between cells.
Various cellular stresses and pathological conditions promote intercellular transfer of organelles including the endoplasmic reticulum, golgi [12], mitochondria [20], endosomes [21] and lysosomes [7] via TNTs. Transfer of lysosomes from healthy endothelial progenitor cells to stressed human umbilical vein endothelial cells (HUVEC) has been reported, and this transfer helps to maintain lysosomal pH [22]. Oxidative stress-induced transfer of aberrant mitochondria via TNTs helps to propagate pathology from stressed to healthy cells in several diseases [23]. On the other hand, the transfers of healthy mitochondria from MSCs to targeted stressed cells is emerging as a potential therapy in regeneration [9, 10, 24, 25]
TNTs in the spread of disease pathology
Studies in 2005–2010 reported the transfer of prion proteins [26], bacteria [27], and viruses [28] from cell to cell through nanotubes leading to the spread of pathology. Viruses such as human immunodeficiency virus (HIV), and herpesviruses use this intercellular mode of dissemination without exposing themselves to the extracellular environment, thereby escaping the humoral immunity of the host [29, 30]. The first report about the propagation of virus particles from infected to uninfected T cells via TNTs was described for HIV [28]. Later, the involvement of TNTs in the spread of viruses has also been demonstrated for the influenza A virus [31], DNA viruses including alpha herpesvirus [32], bovine herpesvirus 1 [33] and human T-cell leukemia virus type 1 [34].
Initial studies demonstrated in 2009 that prions can hijack TNTs to spread the prion pathology in a cell-to-cell manner [26]. Subsequently, the intercellular propagation of amyloidogenic proteins via TNTs has been widely studied. Several such studies have demonstrated the spread of neurodegenerative proteins such as α-synuclein [35, 36], tau [37, 38], amyloid β [12, 39] and huntingtin [40]. One of the major hallmarks of neurodegenerative diseases is insufficient degradative capacity of lysosomes due to the accumulation of proteotoxic aggregates [41, 42], and lysosomal accumulation generates mitochondrial toxicity and increased oxidative stress. Evidence from several studies indicates that lysosomes can mediate the spread of neurodegenerative protein aggregates via TNTs [7]. It has also been demonstrated that α-synuclein aggregates can be transferred from cell to cell bound to mitochondria travelling within TNTs between neuronal cells [8].
TNTs in cancer malignancy
TNTs that are formed between malignant cells or between malignant cells and other cells in the tumour matrix are known to initiate tumour formation and metastasis [43]. Cell-to-cell transfer of mitochondria via TNTs plays a crucial role in maintaining metabolic homeostasis in cancer cells [44]. Below we discuss several key reports regarding cancer malignancy and TNTs.
Tumour cells network via nano-sized actin membrane open-ended conduits (TNTs proper) or by relatively thicker closed-ended micro-sized tubes (TMs) containing tubulin to transport organelles. The initial study [45], first demonstrated TNT like structures in intact solid tumours dissected from patients with lung adenocarcinoma and pleural mesothelioma malignant tissues. More recently, tumour cell-derived networks of membrane-tubes were observed in animal models of astrocytic brain tumours, including glioblastomas (GBM tumours) [46]. The structures are longer and thicker in diameter, and referred to as TMs. Intercellular transfer of mitochondria from tumour-activated stromal cells (TASC) by means of TNTs, EVs or cannibalism promotes proliferation of patient derived primary cultures of GBM cells in a 3D environment [47]. GBM stem-like cells (GSLCs) used in 2D culture and 3D organoid culture showed mitochondrial transfer via TNTs. These studies proposed a role of TNTs and TMs in the context of malignancy spread in organoid tumour models [48].
Mitochondrial transfer by means of TNTs from non-malignant bone marrow stromal cells to multiple myeloma cells resulted in tumour progression [49]. The same study also showed that shRNA-mediated CD38 knockdown inhibited mitochondrial transfer in vivo. The same knockdown in the in vivo model resulted in attenuation of tumour growth and improved survival rate of animal. In addition, hypoxia elevated the formation of TNTs and malignancy in ovarian and colon cancer [50]. This state of oxygen insufficiency results in increased levels of ROS in tumour cells, which leads to increased metabolic rate, gene expression, mitochondrial peroxidation, cellular stress and apoptotic stress [51, 52]. Cancer cells can counteract ROS induced apoptosis by enzymatic and non-enzymatic antioxidant defences, and it is now well accepted that moderate levels of ROS contributes to tumour progression by promoting several signalling pathways and gene mutations [53]. Several recent studies have shown that ROS promotes formation of TNTs and TNTs contribute in developing malignancy and resistance to cancer therapy [54].
Bcl-2, a highly conserved anti-apoptotic protein plays a central role in acquiring resistance to cancer therapy. A recent study [55] has shown that TNTs contribute to the progression of colorectal cancer by upregulating ERK (extracellular signal regulated kinase) expression in recipient cells by transferring mutant KRAS to these cells. They tend to develop TNTs as a part of their invasion and migration processes, and to transfer miRNAs as regulators of signalling pathways [56,57,58]. All these recent reports and several other studies (summarized in the Table 1) document that TNT formation is directly related to tumour malignancy and plays a significant role in tumour adaptation.
TNTs in drug resistance
Intercellular communications were suggested as a potential target for anti-cancer therapies as early as 2004 [70]. Several recent studies have demonstrated that TNT and TM networks play crucial roles in making these tumours exceptionally resistant to therapy [48]. Mitochondrial transfer from tumour activated stromal cells (TASC) to glioblastoma (GBM) cells was observed via TNTs, and the process provided chemo- and radio-resistance of the GBM [47]. Another study around the same time showed, GBM cells import the DNA repair enzyme O6-methylguanine-DNA methyltransferase via TNTs, thus enhancing resistance to temozolomide [71]. A self-repair mechanism of laser irradiated brain tumour cells was observed, and it involved formation of a network of TNTs and TMs [46]. Furthermore, GBM cells irradiated with α- particles establish a network of TNTs more rapidly compared to control irradiated cells in vitro within 24 h [72].
TNT-mediated cancer drug resistance and rescue from apoptotic cell death is a great challenge in cancer treatment. Acquisition of mitochondria in cancer cells (MCF-7) from endothelial cells through TNTs resulted in doxorubicin resistance in MCF-7 cells [63]. Later, in 2015 [73], it was shown that disruption of TNTs decreased the resistance of B-cell precursor acute lymphoblastic leukemia (BCP-ALL) cell to antileukemic drug prednisolone. A study in pancreatic cancer cells showed, doxorubicin increased the formation of TNTs in vitro in a dose-dependent manner and the biogenesis of TNTs promotes resistance to chemotherapy. The observation of drug resistance was also demonstrated in vivo in tumour specimens from patients diagnosed with pancreatic adenocarcinoma and neuroendocrine carcinoma [74]. The study by Wang et al. [66], showed that mitochondrial exchange through TNTs from Jurkat cells to MSCs by ICAM-1 mediated cell adhesion led to chemoresistance (Ara C and Methotrexate) in Jurkat cells. They also showed inhibition of TNT formation led to reduced chemoresistance in primary T-ALL cells (T cell acute lymphoblastic leukemia). Chemotherapy drugs, cytarabine (Ara-C) and doxorubicin (DNR), activated MSCs to disseminate mitochondria to surrounding ALL cells, and as a result chemoresistance developed [65]. Moreover, transfer of myosin containing cellular vesicles from stromal cells to chronic myeloid leukemia cells resulted in increased resistance of leukemic cells to imatinib which is a tyrosine kinase inhibitor [75].
Mitochondrial transfer from mesenchymal stem cells via TNTs
From a therapeutic point of view, TNTs can play a significant role in stem cell therapy, while the same cellular processes can be detrimental in certain pathological conditions. Several studies have shown that the transfer of mitochondria primarily depends on the communication between MSCs and target cells, and this communication is governed by several mechanisms. They include EVs, gap junctions, cell fusions, and TNTs [76]. Mitochondria provide the capacity for aerobic respiration, play important roles in aging and dysfunction in various heritable and acquired diseases. The human mitochondrial genome has 16,568 bp and encodes for only a small set of mitochondria-specific proteins, rRNAs and tRNAs, while majority of proteins are encoded by the nucleus [77]. The mutation rate in the mtDNA genome is high because it is not protected by histones and has low-efficiency nucleotide repair mechanisms [78].The first report of mitochondrial transfer from MSCs was published in 2006, and showed rescue of aerobic respiration by transferring functioning mitochondria via TNTs to cancer cells devoid of mtDNA [10]. Following this, several studies reported a high propensity of mitochondrial propagation and dynamics through TNTs extended from MSCs to the targeted somatic cells [20, 79,80,81]. Researchers have documented transfer of mitochondria from MSCs to the HUVEC, which are initially subject to ischemia–reperfusion injury [82]. A study in a mouse model of lung injury showed transfer of mitochondria from bone marrow-derived stromal cells to pulmonary alveoli caused alleviation of respiratory damage [83].
Recent research has shown that MSCs from tissue of divers origins, such as bone marrow, Wharton's jelly, adipose, and dental pulp play a role in protecting damaged cells from oxidative stress by donating mitochondria [84]. Studies have also demonstrated that MSCs play a crucial role in reducing mitochondrial ROS levels during repair pathways [9, 85]. However, it is not clear why MSCs exclusively form TNTs to targeted cells and what signal stimulates healthy MSCs to induce TNTs and transfer functional mitochondria. Paracrine factors released from neighbouring stressed cells modulate MSCs to initiate its action of damage repair. One study has shown that phosphatidylserine externalized on the surface of damaged cells (apoptotic epithelial cell) prompted MSCs to form TNTs [80]. In another study, it has been shown that connexin 43 plays a vital role in the regulation of TNT formation [86]. The same study has shown that iPSC derived MSCs transfer mitochondria via TNTs to rescue injured lung epithelial cells in a mouse model as well as in an in vitro model. This “donation” of mitochondria helped in alleviating asthma-related inflammation levels due to hypoxic conditions, and also prevented apoptosis of epithelial cells. One study [87] has shown that transfer of mitochondria via TNTs from MSCs to ocular cells helped in increasing the aerobic capacity and upregulation of mitochondrial genes. The work [88] suggested that both paracrine factors and mitochondrial transfer protect cardiomyocytes against stress, independent of each other.
In the last 15 years, several studies have documented transfer of mitochondria from different types of MSCs to aberrant cells via TNTs. In Table 2, we have summarized these studies, most of which have shown the involvement of oxidative stress, mitochondrial stress, ROS and/or apoptotic stress in the biogenesis of TNTs or cell-to-cell transfer via TNTs. Transfer of mtDNA and healthy mitochondria from MSCs via TNTs can be a potential remedy.
However, a deeper understanding is needed to implement the transfer of mitochondria as a therapy, and focus should be given to unravelling various stress signals that could affect transcellular trafficking of mitochondria via TNTs, both in diseases and in rejuvenation [3, 107, 108].
Association of tunnelling nanotubes with oxidative stress, apoptosis, and mitochondrial homeostasis
Mitochondria play an important role in oxidative phosphorylation, aerobic metabolism, calcium signalling, and apoptosis [109]. Mitochondrial dysfunction-related oxidative stress is associated with diseases such as cardiomyopathy, ischemic heart diseases, lung disorders, brain injury, stroke, and neurodegenerative diseases like Alzheimer’s and Parkinson’s disease. Exchange of mtDNA between cells via transfer of mitochondria could modulate respiration and cell cycle arrest. Levels and homoplasmic polymorphism of mtDNA regulate mtDNA-processing enzymes, replication, and transcription of mtDNA and respiratory complexes. Dysfunction of these processes can result in aberrant mitochondria with formation of ROS and also cell cycle arrest due to impaired function of the respiration-linked enzyme dihydroorotate dehydrogenase [110]. Melanoma cancer cells devoid of mtDNA injected in to syngeneic C57BL/6Nsu9-DsRed2 mice expressed with red fluorescent mitochondrial protein can recover to form tumours after import of mtDNA by acquiring whole mitochondria from neighbouring healthy cells [81]. Oxidative stress and ROS promote the biogenesis of TNTs in several pathological conditions [54]. Hydrogen peroxide (H2O2) treatment in the primary hippocampal rat astrocytes and neurons promotes the biogenesis of TNTs, at the same time the induced cellular stress activates tumour suppressor protein p53 [12]. However, later studies were reported that p53 is not the key element for TNT formation, and the effect of H2O2 on TNTs is cell type-specific [111].
The crucial role of intercellular, horizontal transfer of mitochondria demonstrated recently under various pathophysiological conditions, primarily in rescuing tumourigenesis and bioenergetic deficiencies. Tan et al. [108] have shown that the mtDNA-deficient cells acquired functional mitochondrial genome from the surrounding tumour microenvironment or MSCs to regulate many factors related to mitochondrial respiration. In cancer cells, delaying apoptosis resulted in the restoration of cell survival and enhancement of tumourigenicity or metastasis. MSCs from different sources exert different rescue capacities against aerobic respiration ability and postpone apoptosis of the recipient cells [23, 107]. It is possible that paracrine factors related to oxidative stress and/or ROS sent from stressed cells trigger MSCs to make cellular bridges via TNT structures for transferring mitochondria.
The role of TNTs in rescue from apoptotic cell death has also been demonstrated in neuronal cells [25]. This study showed that PC12 cells that were treated with UV light were rescued by non-cancer cells by transfer of mitochondria via TNT-like structures when compared with untreated cells. The UV treated cells that had lost cytochrome C formed TNTs but did not enter the apoptotic cascade. The study suggests that transfer of mitochondria from healthy cells via TNTs reverses the cellular stress in early stage of apoptosis. A recent work [112] has shown that α-synuclein protofibril-induced defects in cellular degradation machineries in microglia enhance cell to cell networks via TNTs to transfer the burden of proteotoxic aggregates to neighbouring cells. The study has also shown that mitochondrial shuffling and sharing of proteotoxic burdens via TNTs alleviate ROS levels and rescue cells from ROS-induced apoptosis.
Rho GTPase related signals counteract apoptosis via tunnelling nanotubes
TNTs mediate direct intercellular transport between neighbouring cells and, structurally, they are open-ended membrane actin conduits. Thus, modulation of membrane and cytoskeleton dynamics may play a major role in the biogenesis of TNTs. Several studies have shown that actin-depolymerizing agents such as cytochalasin B and latrunculin B inhibit TNT formation [1, 113]. The master regulators of the cytoskeleton, Rho family of GTPases (Rac1, Cdc42, and RhoA), are implicated in TNT formation by many studies [13]. The localized control of Rho GTPase regulators, the GTPase activating proteins (GAPs) and guanine nucleotide exchange factors 42 (GEFs) have been proposed to play a role in TNT assembly. One study [114] in immune cells has reported that Cdc42 and Rac1, and their respective effector molecules WASP and WAVE2, are involved in the biogenesis of TNTs by modulating actin polymerization via the Arp2/3 complex. Using FRET-based biosensors, the study has demonstrated that Rac1 stays distributed throughout the TNT structures, while Cdc42 is involved in initiating the biogenesis of TNTs. Transfer of oncogenic KRAS promotes formation of TNTs by regulating the ERK pathway in colorectal cancer. It is thus important to note that Rho GTPase-regulated ERK signalling pathway controls the expression of pro-survival or anti-apoptotic Bcl-2 family of proteins [55].
Two actin regulators downstream of Rho GTPases, βCamKII and cofilin, have recently been demonstrated to play a role in the biogenesis of TNTs. Cross-talk between the signalling cascades of Rho GTPases with the actin regulatory molecules βCamKII, cofilin and Arp2/3 is well documented in the early development of the dendritic spine [115]. Vargas et al. [116], showed that stability of TNTs depends on the activation of the Wnt/Ca2+ signal-dependent modulation of βCamKII in the CAD (mouse catecholaminergic neuronal cell line) cells and primary neurons. The actin-binding ability of the protein is modulated by phosphorylation of βCamKII [117]. Inactivation of cofilin by the RNA-binding protein nucleolin induces TNT biogenesis [118]. In addition, the alphaherpesvirus-induced biogenesis of TNTs depends on the US3 protein kinase-mediated activation of p21-activated kinases (PAKs) apparently by activation of Cdc42/Rac1 and Rho signalling axis, within a poorly understood complex mechanism [32]. PAK kinases are considered primarily the effector of the Rho family GTPases Cdc42 and Rac1. Additionally, studies have shown that PAK1 inhibitor IPA-3 attenuates alpha herpes virus-induced TNT-like membrane actin projections [32, 119]. PAK2 has also been reported in HIV-1 Nef protein-mediated TNT formation [120].
In our recent study, we have observed that Alzheimer’s pathogenesis, the amyloid-β oligomers internalize via PAK1 dependent actin mediated endocytic pathway, and the internalization process promotes formation of TNT-like structures and direct cell-to-cell transfer of oligomers in neuronal cells [39]. The study has also shown colocalization of activated PAK1 with f-actin throughout the TNT network.
Conversely, the Cdc42/IRSp53/VASP system plays a role in the filopodia-promoting network, being negatively correlated with formation of TNTs in neuronal cells [121]. Recently, another study has reported that Arp2/3 negatively regulates biogenesis of TNTs in CAD cells [5]. Another recent study [112] has shown that inhibition of ROCK (using chemical inhibitor Y-27632), a downstream signalling molecule of Rho/Rac/Cdc42, promotes biogenesis of TNTs. The study has indicated that ROCK inhibition promotes TNT formation via Myosin II regulated f-actin modulation. Altogether, these studies suggest a complex regulatory mechanism of Rho GTPases in TNT biogenesis. A further recent report [118], has also shown that M-sec regulated exocyst complex needs to function together with actin polymerization by inhibiting activity of cofilin in the biogenesis of TNTs in multiple mammalian cellular models. The study suggests that in addition to actin polymerization, M-Sec-dependent plasma membrane (PM) re-modelling is a necessary step in formation of TNTs.
The rescue capacity of MSCs mediated via TNTs correlates with the Miro-1 expression, as shown for the transfer capacity of mitochondria from MSCs to stressed alveolar epithelial cells via TNTs [9]. Miro-1 and -2 belong to a class of novel Rho-GTPase, amino acid sequence revealed GTPases domain homolog to the classical Rho-GTPases in the N-terminal part of the protein [16]. Interestingly, Miro proteins lack the membrane-binding motif CAAX in their C-terminal domains, unlike small GTPases but contain a second GTP-binding domain without homology to typical Rho-GTPases [15]. Studies have shown that overexpression of Miro-1 protein leads to an increase in the mitochondrial transfer capacity and, hence, there is a decrease in the apoptosis level and mitochondrial ROS production, and alleviation of respiratory dysfunction [122]. A recent study has shown that the monooxygenase domain of MICAL2PV, a spliced isoform product of the neuronal guidance gene MICAL2, interacts with Miro-2, inhibiting TNT formation by depolymerization of f-actin. MICAL2PV plays crucial role in cell survival and down-regulation of MICAL2PV, and protect lung cancer cells treated with chemotherapeutic drugs [123].
Rho GTPases in cell surface dynamics and TNT biogenesis
Several cytoskeleton remodelling signals are correlated with cell surface dynamics and PM remodelling [124]. Small GTPases Arf and Rab regulate exocytosis of specific vesicles to discrete sites of the PM. Rho GTPases and their regulatory factors contribute to the process by modulating the tethering and subsequent fusion of exocytic vesicles. One study [125], showed that formation of TNTs is regulated by the exocyst complex protein M-Sec in HeLa cells, which is involved in exosome fusion and membrane expansion. The exocyst complex contributes to PM recruitment of the actin remodelling proteins Ral-GTPase and filamin to promote TNTs. The regulatory molecules associated with the recycling of endocytic vesicles and vesicle trafficking, which regulates PM surface dynamics, have also been implicated by several studies in the biogenesis of TNTs. Rab class of small GTPases, Rab8a, Rab11a, and Rab35 are implicated in TNT formation by regulating membrane recycling in neuronal and cancer cells [126, 127]. Rab35-GTP, ACAP2, ARF6-GDP, and EHD1 promote TNT formation in a cascade-like manner in neuronal cells. It may therefore be that modulation of cytoskeleton remodelling via actin polymerization signalling cascades is linked to cell membrane surface dynamics to induce formation of membrane actin-derived TNTs.
Rho GTPases in cell cycle progression, apoptosis, and TNT biogenesis
RhoA, Rac1, and Cdc42 are the most studied typical Rho GTPases, not only involved in the regulation of distinct actin cytoskeleton and PM structures, they are also interlinked via complex molecular signalling events to regulate cell cycle progressions and apoptosis [128]. Rac1-regulated oxidase was reported to modulate acute cellular necrosis, apoptosis, and acute inflammatory response in hepatic ischemia. Rac1-induced production of ROS by an NADPH oxidase was also reported in both phagocytic and non-phagocytic cells [129]. Rac1 can also activate signalling downstream of NFκB, PAK, and ERK by ROS-mediated pathways in neuronal cells to counteract apoptosis. Neuronal cells have limited regenerative capability, and continuous ‘fitness’ of these cells is vital; these cells possess intrinsic competence to attenuate apoptosis [130]. Instead, apoptosis due to elevated stress/ROS levels in neuronal cells may induce formation of TNTs to ameliorate cellular stress [54]. In cancer cells, Rac1-mediated MAPK/ERK and Akt signalling involves the upregulation of the pro-survival or anti-apoptotic Bcl-2 family of proteins [131]. The pro-survival signalling of MAPK/ERK involving formation of TNTs occurs in various cancer cells [132], and TNTs promote cell proliferation and cancer malignancy levels [48]. In addition, TNTs are involved in transfer of apoptosis regulators from healthy cells to diseased cells. Several studies have also shown that the pro-apoptotic Fas ligand is transferred via TNTs to T lymphocytes to induce cell death [133, 134].
Conclusions
The discovery of TNTs in 2004, opened up a novel mechanism of long-range intercellular communication. TNTs are actin-membrane conduits, thereby, actin regulation together with dynamic PM modulatory cellular events play major roles in their biogenesis. The complex functions of Rho GTPase signalling cascades have been implicated by several studies in TNT biogenesis. However, some contradictions exist in the literature and there may be some variability in TNT regulation in different cell types. Moreover, discrepancies also exist in the definition of supercellularity of TNT structures in different studies. It is challenging to resolve TNTs and TMs in ex vivo organoid models or in vivo animal models. Detection methods using advanced imaging tools or exclusive markers need to be explored to make advancement in the field.
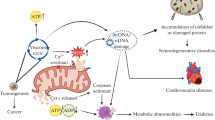
Rho GTPase signalling cascades, that are not only related to the regulation of distinct actin cytoskeleton and PM dynamics, downstream of their linear axis are interlinked via complex molecular signalling events to regulate cell cycle progression and apoptosis (Fig. 1) [128, 131]. Direct cell-to-cell transfer of organelles or cargo via TNTs has emerged as an important mechanism for maintaining cellular homeostasis, and this process has been implicated in disease spread and disease resistance [1]. The widespread association of oxidative stress, apoptosis, mitochondrial homeostasis, and mitochondrial heteroplasmy with the biogenesis of TNTs has been established by several studies [1, 113]. Cell types that possess an inherent mechanism to resist apoptosis, such as neuronal cells and cancer cells, promote the biogenesis of TNTs possibly to maintain cell survival under pathological stress. Some studies for example [10], have shown that ROS and apoptotic stress promotes the biogenesis of TNTs, however, the molecular events associated with apoptosis signalling or oxidative stresses are not the primary regulatory elements. Biogenesis of TNTs increases the survival of cancer cells treated with chemotherapy, radiotherapy, UV radiation, and laser-induced phototoxicity. MSCs rescue cells from apoptotic death triggered by oxidative stress or mitochondrial heteroplasmy. Therefore, MSC-mediated transfer of mitochondria could have therapeutic potential, for example, by promoting wound healing in response to mitochondrial import [135]. On the other hand, transfer of healthy mitochondria rescues ROS-induced apoptosis in cancer cells and promotes cancer malignancies. It is unclear to what extent damage to mitochondria triggers the formation of TNTs. Do damaged recipient cells actively form TNTs to healthy neighbouring cells? If not then, what signal triggers healthy cells to make direct connections via TNTs to transfer mitochondria. Several articles have shown that the atypical Rho GTPases, Miro 1 and Miro 2, play significant roles in cell to cell transfer of mitochondria from MSCs. Classical Rho GTPases are implicated in other cell types, such as neuronal cells, immune cells and in the transfer of virus spreading. Structurally and functionally these two types of Rho GTPases are distinct, although they do share several homologous domains and may have overlapping functions in TNT signalling pathways. Thus, future studies are required to investigate the emerging role of Rho GTPase signalling cascades in TNT biogenesis and in the formation of supercellular structures with potential importance in maintaining tissue homeostasis and pathophysiological conditions.

Schematic summary of TNT studies indicating the involvement of Rho GTPase signalling cascades in the biogenesis of TNTs by modulating actin cytoskeleton proteins, PM dynamics and potentially alleviating cellular or apoptotic stress
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- BMSC:
-
Bone marrow stromal cells
- CAD:
-
Catecholaminergic neuronal cells
- EVs:
-
Extracellular vesicles
- ERK:
-
Extracellular signal-regulated kinase
- GBM:
-
Glioblastoma Multiforme
- HIV:
-
Human immunodeficiency virus
- HUVEC:
-
Human umbilical vein endothelial cells
- iPSC:
-
Induced pluripotent stem cells
- MIRO:
-
Mitochondrial Rho GTPase
- MSCs:
-
Mesenchymal stem cells
- mtDNA:
-
Mitochondrial DNA
- PAK1:
-
P21-activated kinase 1
- PC12:
-
Pheochromocytoma cells
- Rho:
-
Ras homologous protein
- ROS:
-
Reactive oxygen species
- TASC:
-
Tumour activated stromal cells
- TNTs:
-
Tunnelling nanotubes
- TMs:
-
Tumour microtubes
- UV:
-
Ultraviolet
References
Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH (2004) Nanotubular highways for intercellular organelle transport. Science 303(5660):1007–1010
Gerdes HH, Rustom A, Wang X (2013) Tunneling nanotubes, an emerging intercellular communication route in development. Mech Dev 130(6–8):381–387
Ljubojevic N, Henderson JM, Zurzolo C (2021) The ways of actin: why tunneling nanotubes are unique cell protrusions. Trends Cell Biol 31(2):130–142
Roehlecke C, Schmidt MHH (2020) Tunneling nanotubes and tumor microtubes in cancer. Cancers (Basel) 12(4):857
Sartori-Rupp A, Cordero Cervantes D, Pepe A, Gousset K, Delage E, Corroyer-Dulmont S et al (2019) Correlative cryo-electron microscopy reveals the structure of TNTs in neuronal cells. Nat Commun 10(1):342
Mittal R, Karhu E, Wang JS, Delgado S, Zukerman R, Mittal J et al (2019) Cell communication by tunneling nanotubes: implications in disease and therapeutic applications. J Cell Physiol 234(2):1130–1146
Victoria GS, Zurzolo C (2017) The spread of prion-like proteins by lysosomes and tunneling nanotubes: implications for neurodegenerative diseases. J Cell Biol 216(9):2633–2644
Valdinocci D, Kovarova J, Neuzil J, Pountney DL (2021) Alpha-synuclein aggregates associated with mitochondria in tunnelling nanotubes. Neurotox Res 39(2):429–443
Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M et al (2014) Miro1 regulates intercellular mitochondrial transport and enhances mesenchymal stem cell rescue efficacy. EMBO J 33(9):994–1010
Spees JL, Olson SD, Whitney MJ, Prockop DJ (2006) Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci USA 103(5):1283–1288
Zhu D, Tan KS, Zhang X, Sun AY, Sun GY, Lee JC (2005) Hydrogen peroxide alters membrane and cytoskeleton properties and increases intercellular connections in astrocytes. J Cell Sci 118(Pt 16):3695–3703
Wang Y, Cui J, Sun X, Zhang Y (2011) Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ 18(4):732–742
Zhang S, Kazanietz MG, Cooke M (2020) Rho GTPases and the emerging role of tunneling nanotubes in physiology and disease. Am J Physiol Cell Physiol 319(5):C877–C884
Olson MF, Ashworth A, Hall A (1995) An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science 269(5228):1270–1272
Fransson A, Ruusala A, Aspenstrom P (2003) Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278(8):6495–6502
Nahacka Z, Zobalova R, Dubisova M, Rohlena J, Neuzil J (2021) Miro proteins connect mitochondrial function and intercellular transport. Crit Rev Biochem Mol Biol 56(4):401–425
Biran A, Perelmutter M, Gal H, Burton DG, Ovadya Y, Vadai E et al (2015) Senescent cells communicate via intercellular protein transfer. Genes Dev 29(8):791–802
Watkins SC, Salter RD (2005) Functional connectivity between immune cells mediated by tunneling nanotubules. Immunity 23(3):309–318
Thayanithy V, Dickson EL, Steer C, Subramanian S, Lou E (2014) Tumor-stromal cross talk: direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl Res 164(5):359–365
Koyanagi M, Brandes RP, Haendeler J, Zeiher AM, Dimmeler S (2005) Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ Res 96(10):1039–1041
Onfelt B, Nedvetzki S, Benninger RK, Purbhoo MA, Sowinski S, Hume AN et al (2006) Structurally distinct membrane nanotubes between human macrophages support long-distance vesicular traffic or surfing of bacteria. J Immunol 177(12):8476–8483
Yasuda K, Khandare A, Burianovskyy L, Maruyama S, Zhang F, Nasjletti A et al (2011) Tunneling nanotubes mediate rescue of prematurely senescent endothelial cells by endothelial progenitors: exchange of lysosomal pool. Aging (Albany NY) 3(6):597–608
Valdinocci D, Simoes RF, Kovarova J, Cunha-Oliveira T, Neuzil J, Pountney DL (2019) Intracellular and intercellular mitochondrial dynamics in Parkinson’s disease. Front Neurosci 13:930
Rogers RS, Bhattacharya J (2013) When cells become organelle donors. Physiology (Bethesda) 28(6):414–422
Wang X, Gerdes HH (2015) Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ 22(7):1181–1191
Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT et al (2009) Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11(3):328–336
Dubey GP, Ben-Yehuda S (2011) Intercellular nanotubes mediate bacterial communication. Cell 144(4):590–600
Sowinski S, Jolly C, Berninghausen O, Purbhoo MA, Chauveau A, Kohler K et al (2008) Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat Cell Biol 10(2):211–219
Okafo G, Prevedel L, Eugenin E (2017) Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci Rep 7(1):16660
Jansens RJJ, Tishchenko A, Favoreel HW (2020) Bridging the gap: virus long-distance spread via tunneling nanotubes. J Virol 94(8):e02120-19
Roberts KL, Manicassamy B, Lamb RA (2015) Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol 89(3):1537–1549
Van den Broeke C, Radu M, Deruelle M, Nauwynck H, Hofmann C, Jaffer ZM et al (2009) Alphaherpesvirus US3-mediated reorganization of the actin cytoskeleton is mediated by group A p21-activated kinases. Proc Natl Acad Sci USA 106(21):8707–8712
Panasiuk M, Rychlowski M, Derewonko N, Bienkowska-Szewczyk K (2018) Tunneling nanotubes as a novel route of cell-to-cell spread of herpesviruses. J Virol 92(10):e00090-18
Omsland M, Pise-Masison C, Fujikawa D, Galli V, Fenizia C, Parks RW et al (2018) Inhibition of tunneling nanotube (TNT) formation and human T-cell leukemia virus type 1 (HTLV-1) transmission by cytarabine. Sci Rep 8(1):11118
Abounit S, Bousset L, Loria F, Zhu S, de Chaumont F, Pieri L et al (2016) Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J 35(19):2120–2138
Dieriks BV, Park TI, Fourie C, Faull RL, Dragunow M, Curtis MA (2017) Alpha-synuclein transfer through tunneling nanotubes occurs in SH-SY5Y cells and primary brain pericytes from Parkinson’s disease patients. Sci Rep 7:42984
Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A et al (2009) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11(7):909–913
Tardivel M, Begard S, Bousset L, Dujardin S, Coens A, Melki R et al (2016) Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 4(1):117
Dilna A, Deepak KV, Damodaran N, Kielkopf CS, Kagedal K, Ollinger K et al (2021) Amyloid-beta induced membrane damage instigates tunneling nanotube-like conduits by p21-activated kinase dependent actin remodulation. Biochim Biophys Acta Mol Basis Dis 1867(12):166246
Costanzo M, Abounit S, Marzo L, Danckaert A, Chamoun Z, Roux P et al (2013) Transfer of polyglutamine aggregates in neuronal cells occurs in tunneling nanotubes. J Cell Sci 126(Pt 16):3678–3685
Domert J, Rao SB, Agholme L, Brorsson AC, Marcusson J, Hallbeck M et al (2014) Spreading of amyloid-beta peptides via neuritic cell-to-cell transfer is dependent on insufficient cellular clearance. Neurobiol Dis 65:82–92
Nath S, Agholme L, Kurudenkandy FR, Granseth B, Marcusson J, Hallbeck M (2012) Spreading of neurodegenerative pathology via neuron-to-neuron transmission of beta-amyloid. J Neurosci 32(26):8767–8777
Sahu P, Jena SR, Samanta L (2018) Tunneling nanotubes: a versatile target for cancer therapy. Curr Cancer Drug Targets 18(6):514–521
Hekmatshoar Y, Nakhle J, Galloni M, Vignais ML (2018) The role of metabolism and tunneling nanotube-mediated intercellular mitochondria exchange in cancer drug resistance. Biochem J 475(14):2305–2328
Lou E, Fujisawa S, Morozov A, Barlas A, Romin Y, Dogan Y et al (2012) Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS One 7(3):e33093
Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, Blaes J et al (2015) Brain tumour cells interconnect to a functional and resistant network. Nature 528(7580):93–98
Salaud C, Alvarez-Arenas A, Geraldo F, Belmonte-Beitia J, Calvo GF, Gratas C et al (2020) Mitochondria transfer from tumor-activated stromal cells (TASC) to primary glioblastoma cells. Biochem Biophys Res Commun 533(1):139–147
Pinto G, Brou C, Zurzolo C (2020) Tunneling nanotubes: the fuel of tumor progression? Trends Cancer 6(10):874–888
Marlein CR, Piddock RE, Mistry JJ, Zaitseva L, Hellmich C, Horton RH et al (2019) CD38-driven mitochondrial trafficking promotes bioenergetic plasticity in multiple myeloma. Cancer Res 79(9):2285–2297
Lou E, Zhai E, Sarkari A, Desir S, Wong P, Iizuka Y et al (2018) Cellular and molecular networking within the ecosystem of cancer cell communication via tunneling nanotubes. Front Cell Dev Biol 6:95
Mattson MP, Culmsee C, Yu ZF (2000) Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 301(1):173–187
Ham PB 3rd, Raju R (2017) Mitochondrial function in hypoxic ischemic injury and influence of aging. Prog Neurobiol 157:92–116
Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G et al (2020) ROS in cancer therapy: the bright side of the moon. Exp Mol Med 52(2):192–203
Rustom A (2016) The missing link: does tunnelling nanotube-based supercellularity provide a new understanding of chronic and lifestyle diseases? Open Biol 6(6):160057
Desir S, Wong P, Turbyville T, Chen D, Shetty M, Clark C et al (2019) Intercellular transfer of oncogenic KRAS via tunneling nanotubes introduces intracellular mutational heterogeneity in colon cancer cells. Cancers (Basel) 11(7):892
Pinto G, Saenz-de-Santa-Maria I, Chastagner P, Perthame E, Delmas C, Toulas C et al (2021) Patient-derived glioblastoma stem cells transfer mitochondria through tunneling nanotubes in tumor organoids. Biochem J 478(1):21–39
Ma L, Weinberg RA (2008) Micromanagers of malignancy: role of microRNAs in regulating metastasis. Trends Genet 24(9):448–456
Ma L, Teruya-Feldstein J, Weinberg RA (2007) Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449(7163):682–688
Boukelmoune N, Chiu GS, Kavelaars A, Heijnen CJ (2018) Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol Commun 6(1):139
Diaz-Carballo D, Klein J, Acikelli AH, Wilk C, Saka S, Jastrow H et al (2017) Cytotoxic stress induces transfer of mitochondria-associated human endogenous retroviral RNA and proteins between cancer cells. Oncotarget 8(56):95945–95964
Marlein CR, Zaitseva L, Piddock RE, Robinson SD, Edwards DR, Shafat MS et al (2017) NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 130(14):1649–1660
Ippolito L, Morandi A, Taddei ML, Parri M, Comito G, Iscaro A et al (2019) Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 38(27):5339–5355
Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N et al (2013) Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11:94
Lu J, Zheng X, Li F, Yu Y, Chen Z, Liu Z et al (2017) Tunneling nanotubes promote intercellular mitochondria transfer followed by increased invasiveness in bladder cancer cells. Oncotarget 8(9):15539–15552
Burt R, Dey A, Aref S, Aguiar M, Akarca A, Bailey K et al (2019) Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134(17):1415–1429
Wang J, Liu X, Qiu Y, Shi Y, Cai J, Wang B et al (2018) Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J Hematol Oncol 11(1):11
Lin HY, Liou CW, Chen SD, Hsu TY, Chuang JH, Wang PW et al (2015) Mitochondrial transfer from Wharton’s jelly-derived mesenchymal stem cells to mitochondria-defective cells recaptures impaired mitochondrial function. Mitochondrion 22:31–44
Walters HE, Cox LS (2021) Intercellular transfer of mitochondria between senescent cells through cytoskeleton-supported intercellular bridges requires mTOR and CDC42 signalling. Oxid Med Cell Longev 2021:6697861
Desir S, Dickson EL, Vogel RI, Thayanithy V, Wong P, Teoh D et al (2016) Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 7(28):43150–43161
Vidulescu C, Clejan S, O’Connor KC (2004) Vesicle traffic through intercellular bridges in DU 145 human prostate cancer cells. J Cell Mol Med 8(3):388–396
Valdebenito S, Audia A, Bhat KPL, Okafo G, Eugenin EA (2020) Tunneling nanotubes mediate adaptation of glioblastoma cells to temozolomide and ionizing radiation treatment. iScience 23(9):101450
Matejka N, Reindl J (2020) Influence of alpha-particle radiation on intercellular communication networks of tunneling nanotubes in U87 glioblastoma cells. Front Oncol 10:1691
Polak R, de Rooij B, Pieters R, den Boer ML (2015) B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 126(21):2404–2414
Desir S, O’Hare P, Vogel RI, Sperduto W, Sarkari A, Dickson EL et al (2018) Chemotherapy-induced tunneling nanotubes mediate intercellular drug efflux in pancreatic cancer. Sci Rep 8(1):9484
Kolba MD, Dudka W, Zaręba-Kozioł M, Kominek A, Ronchi P, Turos L et al (2019) Tunneling nanotube-mediated intercellular vesicle and protein transfer in the stroma-provided imatinib resistance in chronic myeloid leukemia cells. Cell Death Dis 10(11):817
Torralba D, Baixauli F, Sanchez-Madrid F (2016) Mitochondria know no boundaries: mechanisms and functions of intercellular mitochondrial transfer. Front Cell Dev Biol 4:107
Brandon MC, Lott MT, Nguyen KC, Spolim S, Navathe SB, Baldi P et al (2005) MITOMAP: a human mitochondrial genome database—2004 update. Nucleic Acids Res 33(Database issue):D611–D613
Mason PA, Matheson EC, Hall AG, Lightowlers RN (2003) Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res 31(3):1052–1058
Shanmughapriya S, Langford D, Natarajaseenivasan K (2020) Inter and intracellular mitochondrial trafficking in health and disease. Ageing Res Rev 62:101128
Liu K, Ji K, Guo L, Wu W, Lu H, Shan P et al (2014) Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92:10–18
Dong LF, Kovarova J, Bajzikova M, Bezawork-Geleta A, Svec D, Endaya B et al (2017) Horizontal transfer of whole mitochondria restores tumorigenic potential in mitochondrial DNA-deficient cancer cells. Elife 6:e22187
Sarmah D, Kaur H, Saraf J, Pravalika K, Goswami A, Kalia K et al (2018) Getting closer to an effective intervention of ischemic stroke: the big promise of stem cell. Transl Stroke Res 9(4):356–374
Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K et al (2012) Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18(5):759–765
Paliwal S, Chaudhuri R, Agrawal A, Mohanty S (2018) Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 25(1):31
Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C et al (2016) Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535(7613):551–555
Yao Y, Fan XL, Jiang D, Zhang Y, Li X, Xu ZB et al (2018) Connexin 43-mediated mitochondrial transfer of iPSC-MSCs alleviates asthma inflammation. Stem Cell Rep 11(5):1120–1135
Jiang D, Chen FX, Zhou H, Lu YY, Tan H, Yu SJ et al (2020) Bioenergetic crosstalk between mesenchymal stem cells and various ocular cells through the intercellular trafficking of mitochondria. Theranostics 10(16):7260–7272
Mahrouf-Yorgov M, Augeul L, Da Silva CC, Jourdan M, Rigolet M, Manin S et al (2017) Mesenchymal stem cells sense mitochondria released from damaged cells as danger signals to activate their rescue properties. Cell Death Differ 24(7):1224–1238
Li CJ, Chen PK, Sun LY, Pang CY (2017) Enhancement of mitochondrial transfer by antioxidants in human mesenchymal stem cells. Oxid Med Cell Longev 2017:8510805
Han H, Hu J, Yan Q, Zhu J, Zhu Z, Chen Y et al (2016) Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol Med Rep 13(2):1517–1524
Lin TK, Chen SD, Chuang YC, Lan MY, Chuang JH, Wang PW et al (2019) Mitochondrial transfer of Wharton’s jelly mesenchymal stem cells eliminates mutation burden and rescues mitochondrial bioenergetics in rotenone-stressed MELAS fibroblasts. Oxid Med Cell Longev 2019:9537504
Chuang YC, Liou CW, Chen SD, Wang PW, Chuang JH, Tiao MM et al (2017) Mitochondrial transfer from Wharton’s jelly mesenchymal stem cell to MERRF cybrid reduces oxidative stress and improves mitochondrial bioenergetics. Oxid Med Cell Longev 2017:5691215
Jiang D, Gao F, Zhang Y, Wong DS, Li Q, Tse HF et al (2016) Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis 7(11):e2467
Yang Y, Ye G, Zhang YL, He HW, Yu BQ, Hong YM et al (2020) Transfer of mitochondria from mesenchymal stem cells derived from induced pluripotent stem cells attenuates hypoxia-ischemia-induced mitochondrial dysfunction in PC12 cells. Neural Regen Res 15(3):464–472
Plotnikov EY, Khryapenkova TG, Galkina SI, Sukhikh GT, Zorov DB (2010) Cytoplasm and organelle transfer between mesenchymal multipotent stromal cells and renal tubular cells in co-culture. Exp Cell Res 316(15):2447–2455
Vallabhaneni KC, Haller H, Dumler I (2012) Vascular smooth muscle cells initiate proliferation of mesenchymal stem cells by mitochondrial transfer via tunneling nanotubes. Stem Cells Dev 21(17):3104–3113
Hu J, Deng G, Tian Y, Pu Y, Cao P, Yuan W (2015) An in vitro investigation into the role of bone marrowderived mesenchymal stem cells in the control of disc degeneration. Mol Med Rep 12(4):5701–5708
Babenko VA, Silachev DN, Zorova LD, Pevzner IB, Khutornenko AA, Plotnikov EY et al (2015) Improving the post-stroke therapeutic potency of mesenchymal multipotent stromal cells by cocultivation with cortical neurons: the role of crosstalk between cells. Stem Cells Transl Med 4(9):1011–1020
Babenko VA, Silachev DN, Popkov VA, Zorova LD, Pevzner IB, Plotnikov EY et al (2018) Miro1 enhances mitochondria transfer from multipotent mesenchymal stem cells (MMSC) to neural cells and improves the efficacy of cell recovery. Molecules 23(3):687
Li H, Wang C, He T, Zhao T, Chen YY, Shen YL et al (2019) Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via Gap Junction. Theranostics 9(7):2017–2035
Jackson MV, Krasnodembskaya AD (2017) Analysis of mitochondrial transfer in direct co-cultures of human monocyte-derived macrophages (MDM) and mesenchymal stem cells (MSC). Bio Protoc 7(9):e2255
Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A et al (2016) Mitochondrial transfer via tunneling nanotubes is an important mechanism by which mesenchymal stem cells enhance macrophage phagocytosis in the in vitro and in vivo models of ARDS. Stem Cells 34(8):2210–2223
Cselenyak A, Pankotai E, Horvath EM, Kiss L, Lacza Z (2010) Mesenchymal stem cells rescue cardiomyoblasts from cell death in an in vitro ischemia model via direct cell-to-cell connections. BMC Cell Biol 11:29
Yang H, Borg TK, Ma Z, Xu M, Wetzel G, Saraf LV et al (2016) Biochip-based study of unidirectional mitochondrial transfer from stem cells to myocytes via tunneling nanotubes. Biofabrication 8(1):015012
Zhang Y, Yu Z, Jiang D, Liang X, Liao S, Zhang Z et al (2016) iPSC-MSCs with High intrinsic MIRO1 and sensitivity to TNF-alpha yield efficacious mitochondrial transfer to rescue anthracycline-induced cardiomyopathy. Stem Cell Rep 7(4):749–763
Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y et al (2014) Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Respir Cell Mol Biol 51(3):455–465
Paliwal S, Chaudhuri R, Agrawal A, Mohanty S (2019) Correction to: Human tissue-specific MSCs demonstrate differential mitochondria transfer abilities that may determine their regenerative abilities. Stem Cell Res Ther 10(1):215
Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J et al (2015) Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21(1):81–94
Wallace DC (1994) Mitochondrial DNA sequence variation in human evolution and disease. Proc Natl Acad Sci USA 91(19):8739–8746
Bajzikova M, Kovarova J, Coelho AR, Boukalova S, Oh S, Rohlenova K et al (2019) Reactivation of dihydroorotate dehydrogenase-driven pyrimidine biosynthesis restores tumor growth of respiration-deficient cancer cells. Cell Metab 29(2):399-416 e10
Andresen V, Wang X, Ghimire S, Omsland M, Gjertsen BT, Gerdes HH (2013) Tunneling nanotube (TNT) formation is independent of p53 expression. Cell Death Differ 20(8):1124
Scheiblich H, Dansokho C, Mercan D, Schmidt SV, Bousset L, Wischhof L et al (2021) Microglia jointly degrade fibrillar alpha-synuclein cargo by distribution through tunneling nanotubes. Cell 184(20):5089-5106 e21
Austefjord MW, Gerdes HH, Wang X (2014) Tunneling nanotubes: diversity in morphology and structure. Commun Integr Biol 7(1):e27934
Hanna SJ, McCoy-Simandle K, Miskolci V, Guo P, Cammer M, Hodgson L et al (2017) The role of Rho-GTPases and actin polymerization during macrophage tunneling nanotube biogenesis. Sci Rep 7(1):8547
Rangamani P, Levy MG, Khan S, Oster G (2016) Paradoxical signaling regulates structural plasticity in dendritic spines. Proc Natl Acad Sci USA 113(36):E5298–E5307
Vargas JY, Loria F, Wu YJ, Cordova G, Nonaka T, Bellow S et al (2019) The Wnt/Ca(2+) pathway is involved in interneuronal communication mediated by tunneling nanotubes. EMBO J 38(23):e101230
Shen K, Meyer T (1999) Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science 284(5411):162–166
Dagar S, Pushpa K, Pathak D, Samaddar S, Saxena A, Banerjee S et al (2021) Nucleolin regulates 14-3-3zeta mRNA and promotes cofilin phosphorylation to induce tunneling nanotube formation. FASEB J 35(1):e21199
Jacob T, Broeke CVD, Waesberghe CV, Troys LV, Favoreel HW (2015) Pseudorabies virus US3 triggers RhoA phosphorylation to reorganize the actin cytoskeleton. J Gen Virol 96(8):2328–2335
Mukerji J, Olivieri KC, Misra V, Agopian KA, Gabuzda D (2012) Proteomic analysis of HIV-1 Nef cellular binding partners reveals a role for exocyst complex proteins in mediating enhancement of intercellular nanotube formation. Retrovirology 9:33
Delage E, Cervantes DC, Penard E, Schmitt C, Syan S, Disanza A et al (2016) Differential identity of filopodia and tunneling nanotubes revealed by the opposite functions of actin regulatory complexes. Sci Rep 6:39632
Las G, Shirihai OS (2014) Miro1: new wheels for transferring mitochondria. EMBO J 33(9):939–941
Wang F, Chen X, Cheng H, Song L, Liu J, Caplan S et al (2021) MICAL2PV suppresses the formation of tunneling nanotubes and modulates mitochondrial trafficking. EMBO Rep 22(7):e52006
Ridley AJ (2006) Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol 16(10):522–529
Hase K, Kimura S, Takatsu H, Ohmae M, Kawano S, Kitamura H et al (2009) M-Sec promotes membrane nanotube formation by interacting with Ral and the exocyst complex. Nat Cell Biol 11(12):1427–1432
Bhat S, Ljubojevic N, Zhu S, Fukuda M, Echard A, Zurzolo C (2020) Rab35 and its effectors promote formation of tunneling nanotubes in neuronal cells. Sci Rep 10(1):16803
Burtey A, Wagner M, Hodneland E, Skaftnesmo KO, Schoelermann J, Mondragon IR et al (2015) Intercellular transfer of transferrin receptor by a contact-, Rab8-dependent mechanism involving tunneling nanotubes. FASEB J 29(11):4695–4712
Aznar S, Lacal JC (2001) Rho signals to cell growth and apoptosis. Cancer Lett 165(1):1–10
Ozaki M, Deshpande SS, Angkeow P, Bellan J, Lowenstein CJ, Dinauer MC et al (2000) Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. FASEB J 14(2):418–429
Lin L, Zhang M, Stoilov P, Chen L, Zheng S (2020) Developmental attenuation of neuronal apoptosis by neural-specific splicing of Bak1 microexon. Neuron 107(6):1180-1196 e8
Stankiewicz TR, Linseman DA (2014) Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front Cell Neurosci 8:314
Cole JM, Dahl R, Cowden Dahl KD (2021) MAPK signaling is required for generation of tunneling nanotube-like structures in ovarian cancer cells. Cancers (Basel) 13(2):274
Arkwright PD, Luchetti F, Tour J, Roberts C, Ayub R, Morales AP et al (2010) Fas stimulation of T lymphocytes promotes rapid intercellular exchange of death signals via membrane nanotubes. Cell Res 20(1):72–88
Luchetti F, Canonico B, Arcangeletti M, Guescini M, Cesarini E, Stocchi V et al (2012) Fas signalling promotes intercellular communication in T cells. PLoS One 7(4):e35766
Levoux J, Prola A, Lafuste P, Gervais M, Chevallier N, Koumaiha Z et al (2021) Platelets facilitate the wound-healing capability of mesenchymal stem cells by mitochondrial transfer and metabolic reprogramming. Cell Metab 33(3):688–690
Funding
Open access funding provided by Manipal Academy of Higher Education, Manipal. A.R thanks Manipal Academy of higher education for Dr. TMA pai fellowship. S.N thanks the Intramural fund of Manipal Academy of Higher Education, Manipal, India and the Indian Council of Medical Research of India (5/4-5/Ad-hoc/Neuro/216/2020-NCD-I) for financial support. D.L.P and J.N thank Griffith University for financial support.
Author information
Authors and Affiliations
Contributions
SN conceived the initial idea for the review; AR, PR and SN researched the literatures and wrote the manuscript; AR and PR drew the figure and collected the tables; JN and DLP edited, revised and added their ideas in the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no competing interests associated with the manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Raghavan, A., Rao, P., Neuzil, J. et al. Oxidative stress and Rho GTPases in the biogenesis of tunnelling nanotubes: implications in disease and therapy. Cell. Mol. Life Sci. 79, 36 (2022). https://doi.org/10.1007/s00018-021-04040-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00018-021-04040-0