
Kinetics and Energetics of Intramolecular Electron Transfer in Single-Point Labeled TUPS-Cytochrome c Derivatives

 , ,
, ,  ,
, 
Abstract
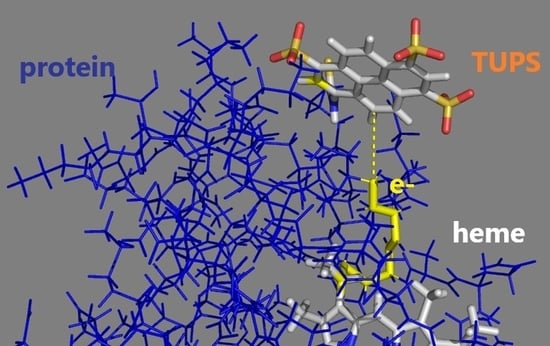
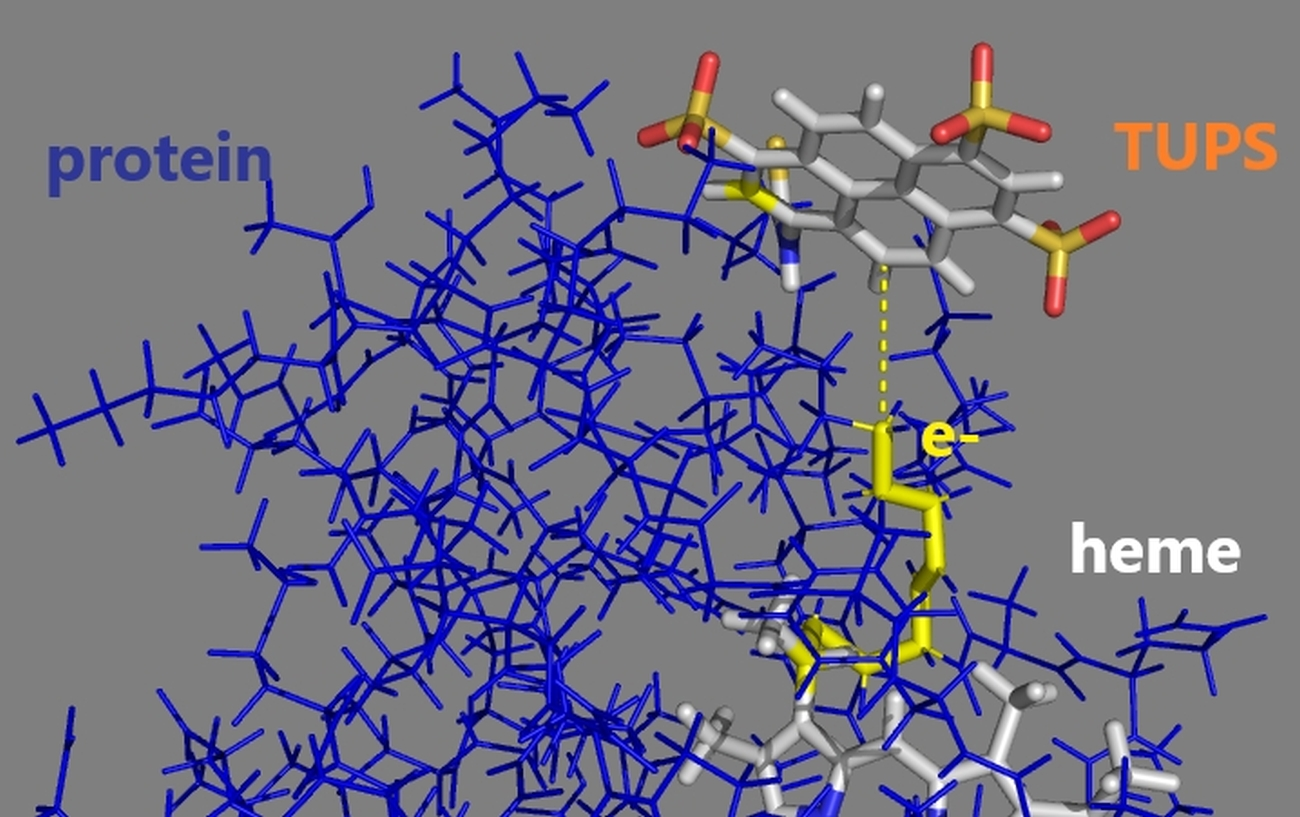
:
1. Introduction
2. Results and Discussion
2.1. Preparation of Cytochrome c-TUPS Derivatives
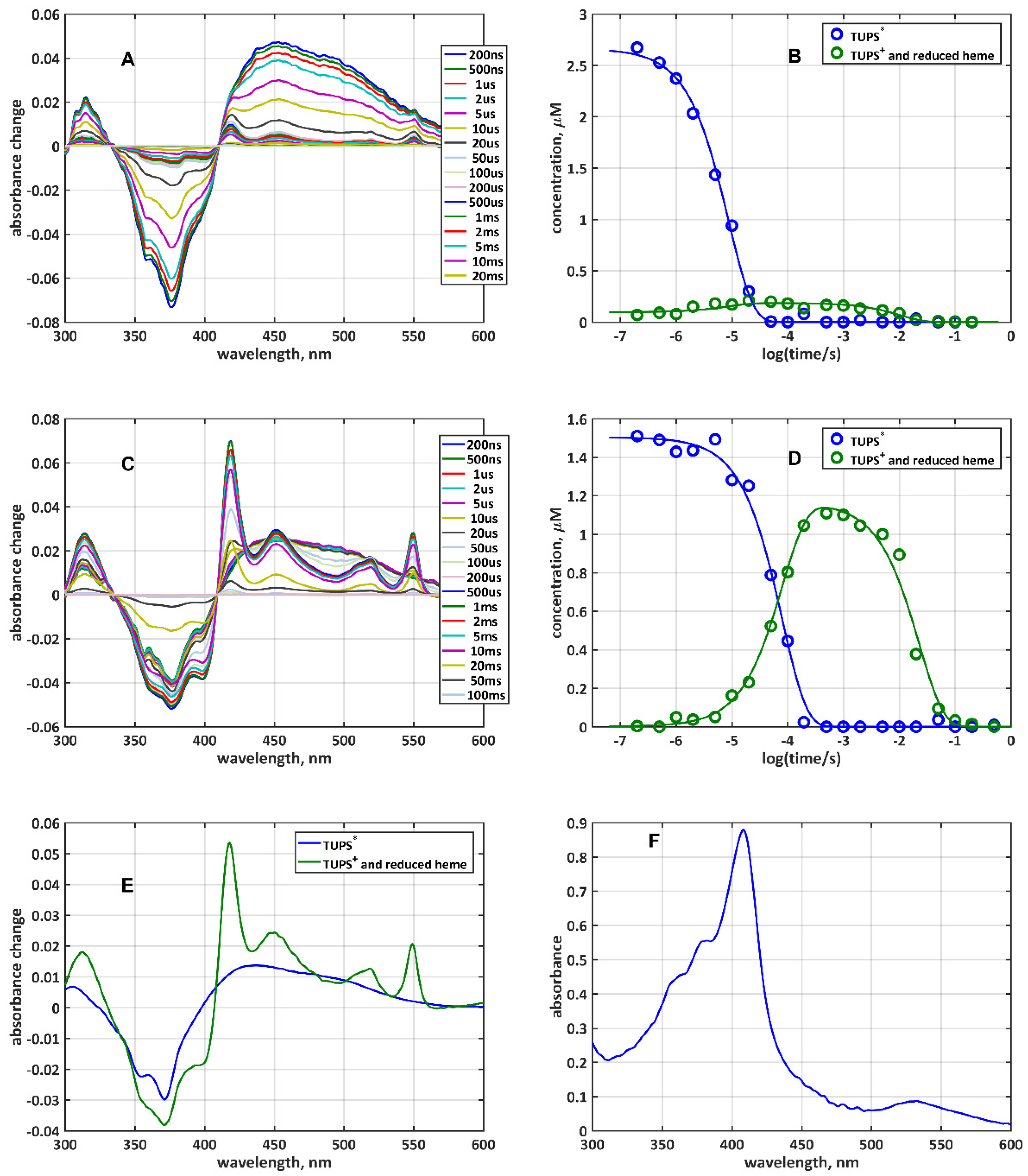
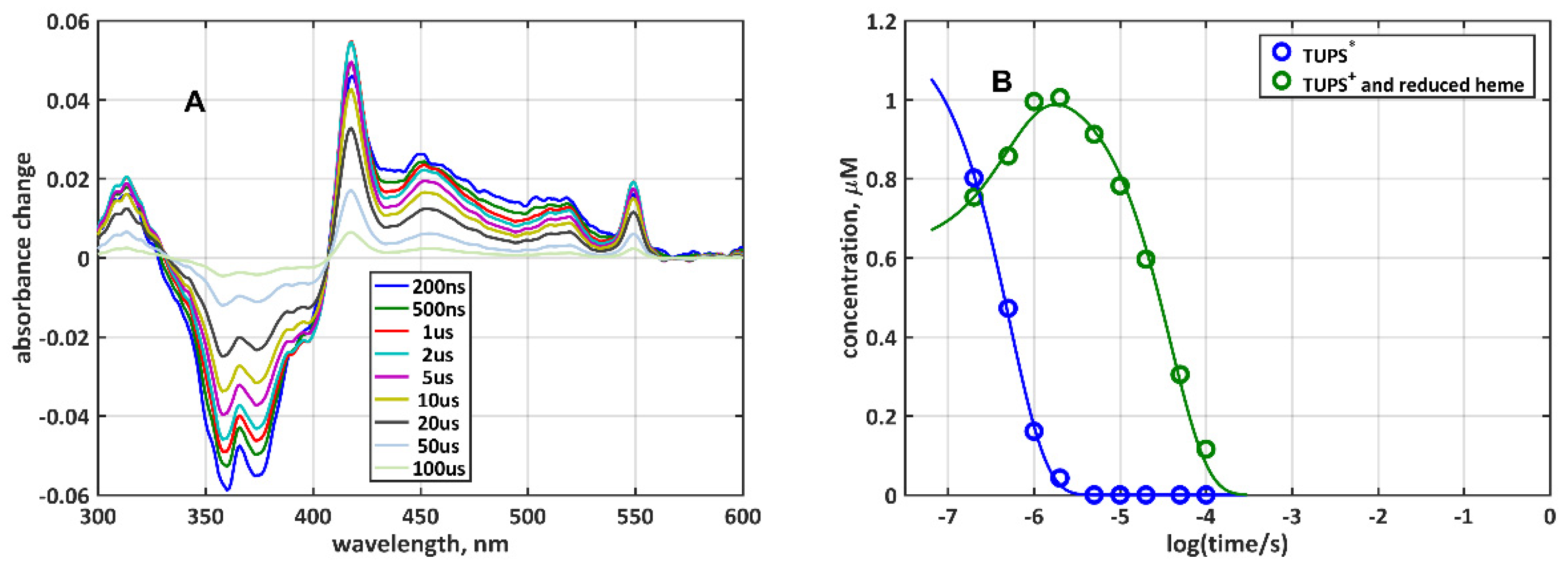
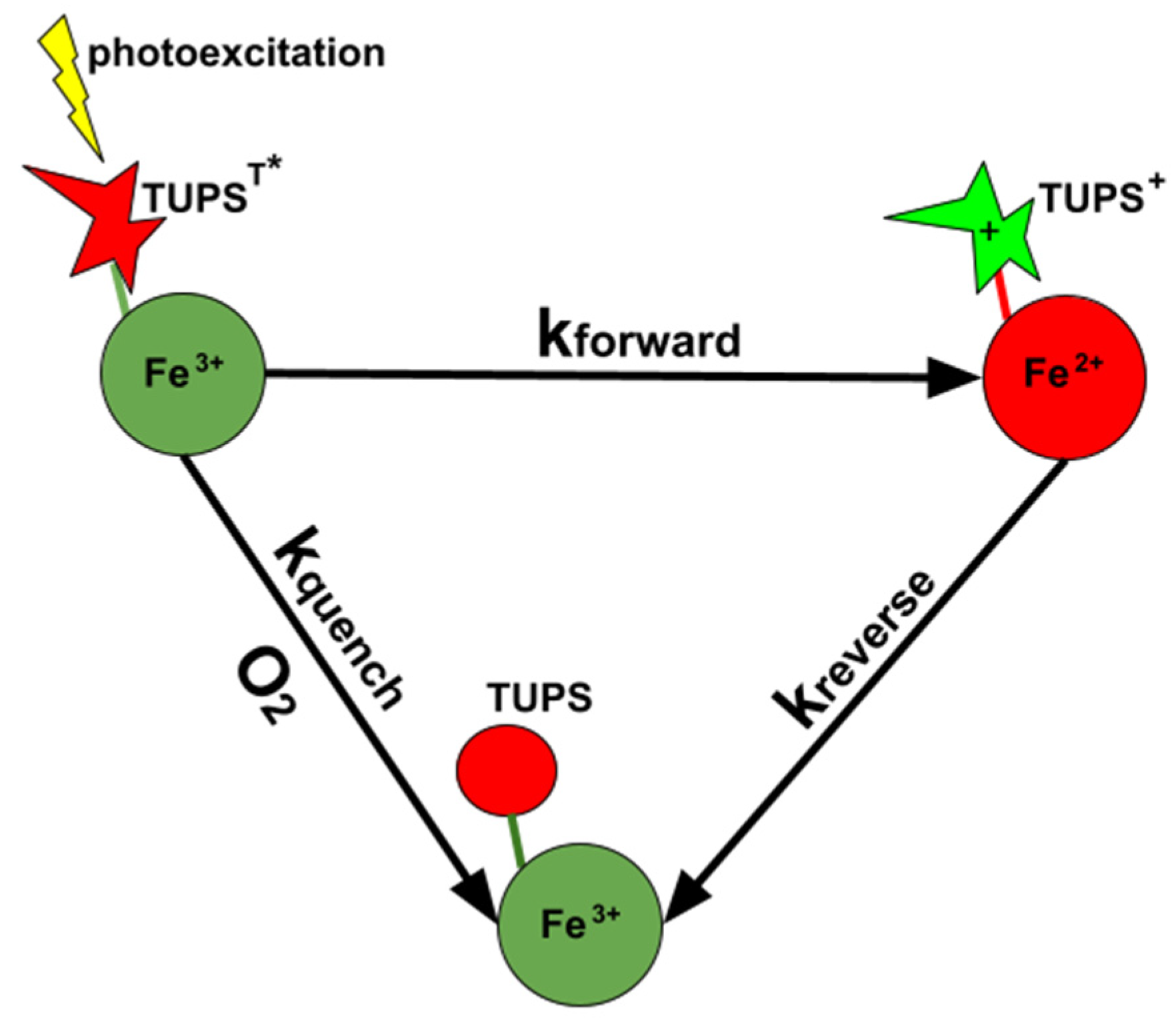
2.2. Quenching of TUPS Triplet Excited State Must Be Taken into Consideration in the Analysis of the Electron Transfer
2.3. The Instantaneous Light-Induced Appearance of the {TUPS+ + hemered} Species: Role of Solvated Electrons

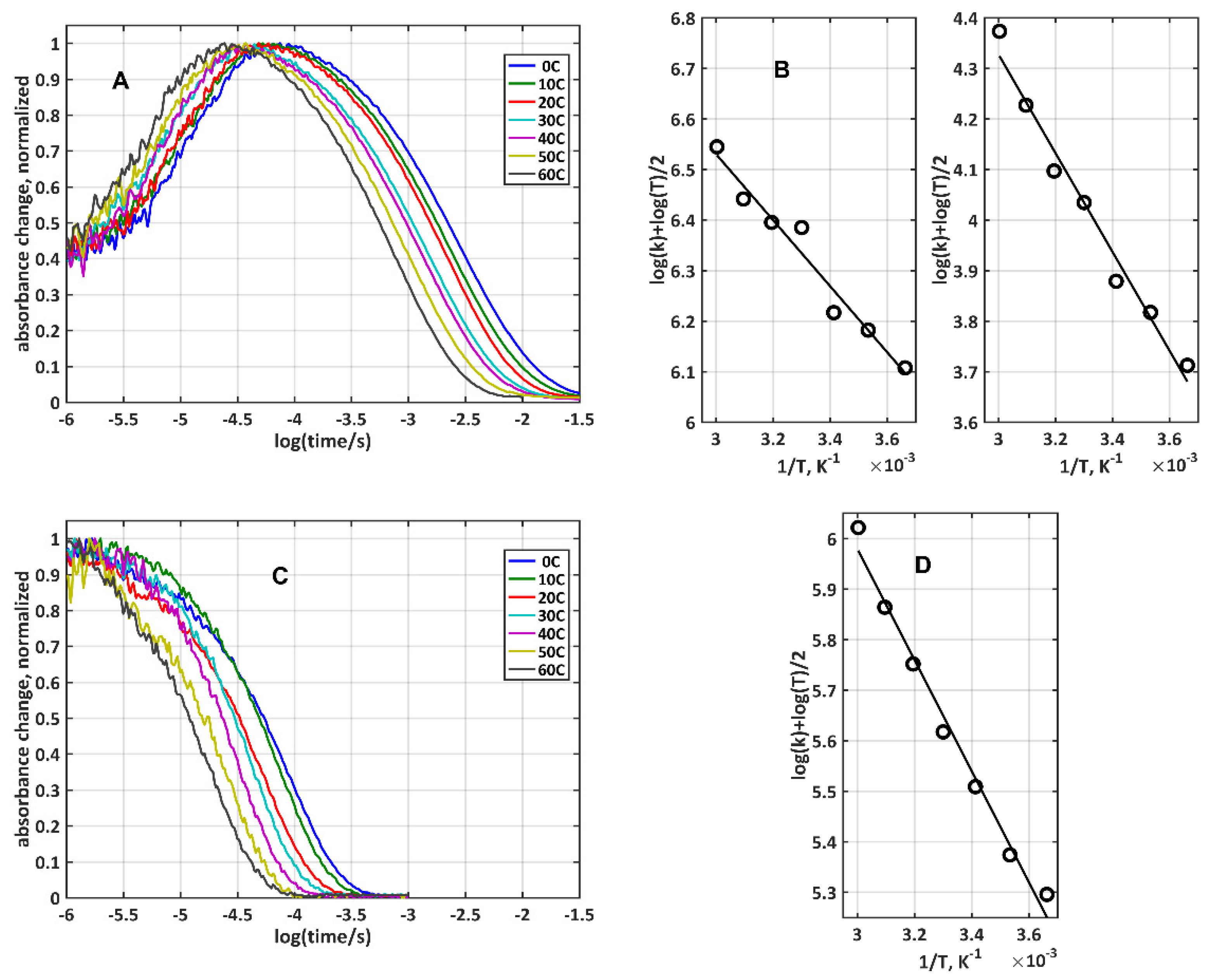
2.4. Determination of the Coupling Terms and Reorganization Energies for Electron Transfer from Temperature Dependent Experiments
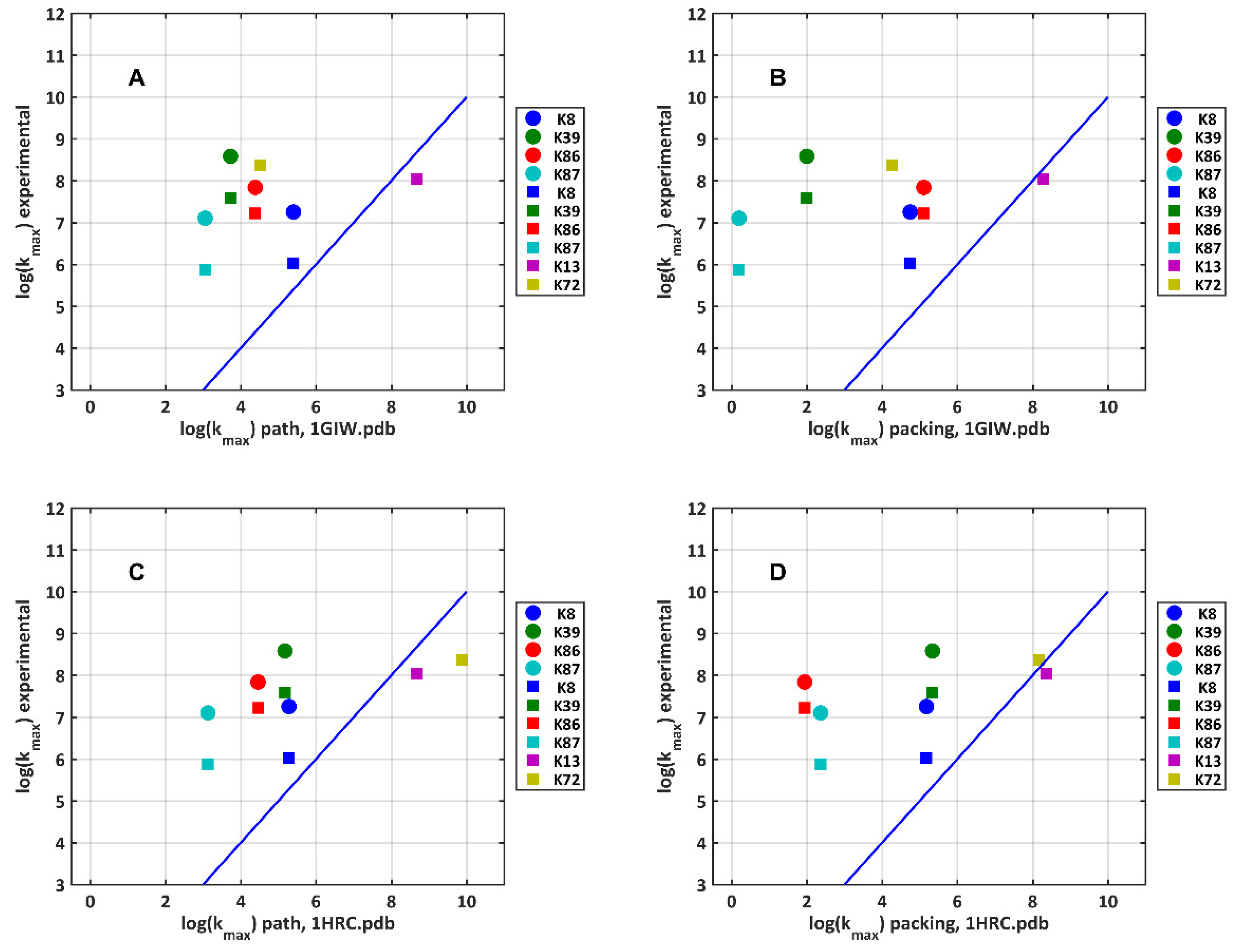
2.5. Electron Transfer Routes between TUPS and the Heme: Maximal Rate from Experiment and Model Calculations
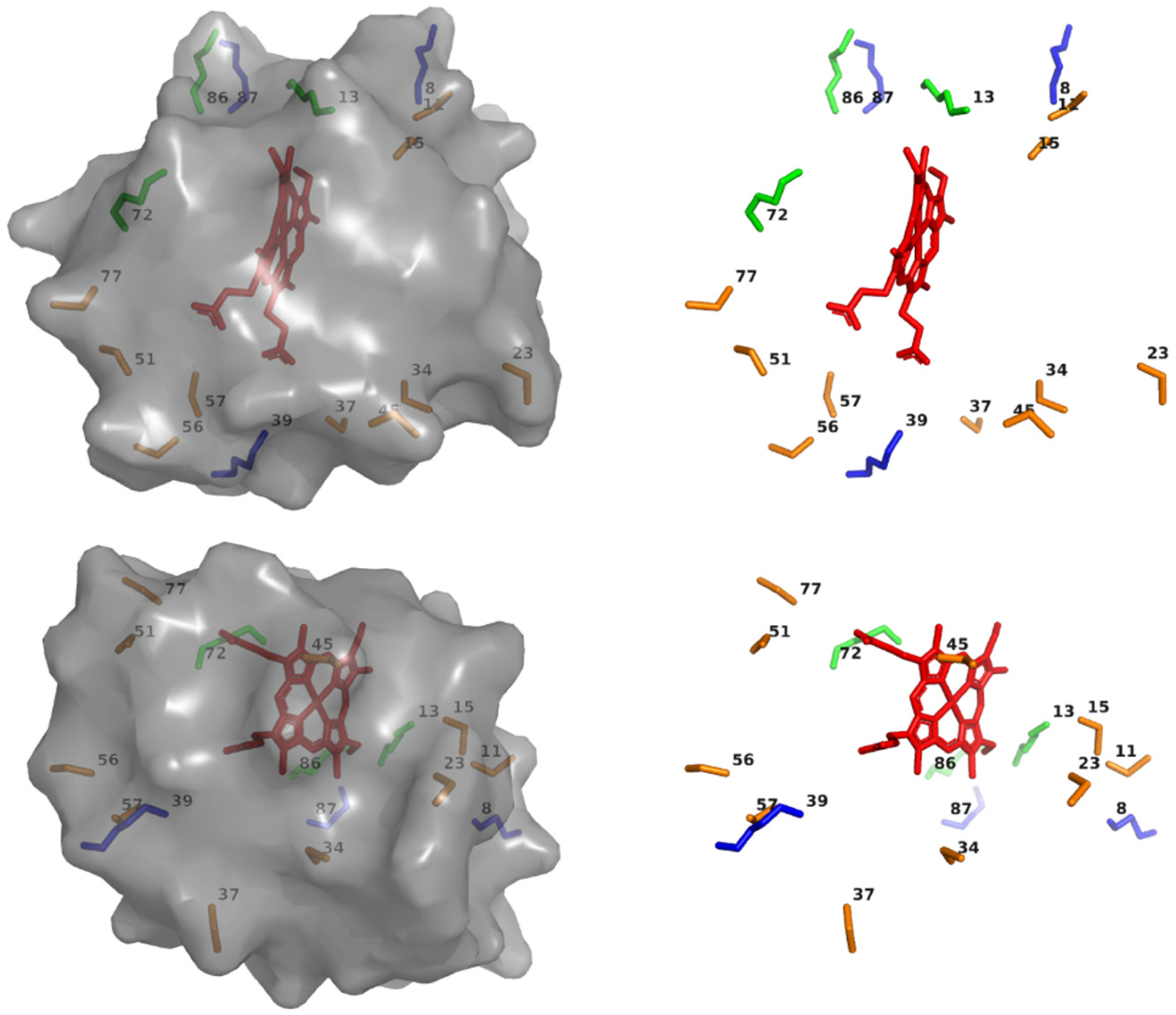
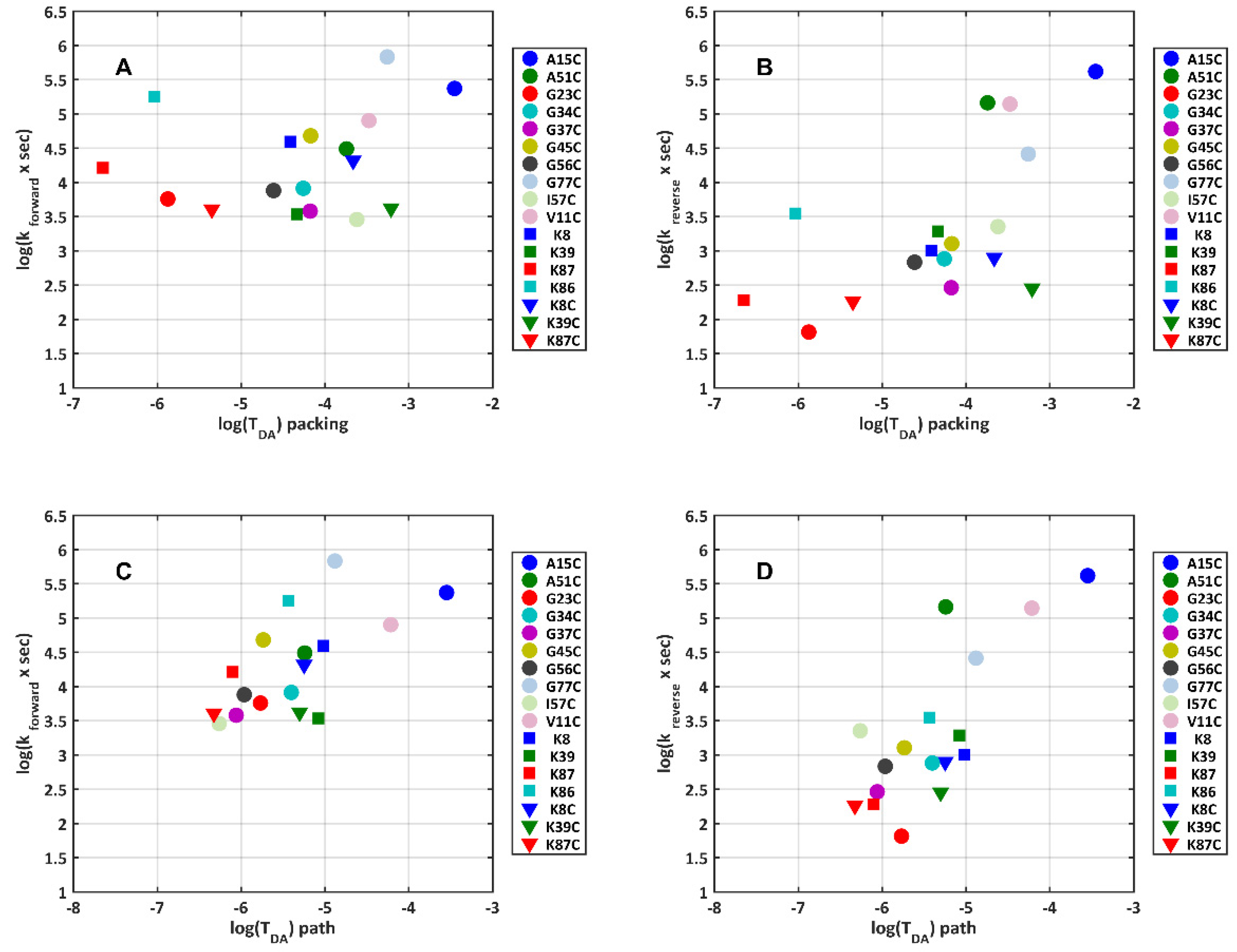
2.6. Electron Transfer Dynamics with Various TUPS-Labeled Cysteines
3. Materials and Methods
3.1. Chemicals
3.2. Construction of Mutant Genes of Cytochrome c
3.3. Expression, Isolation, and Purification of Cytochrome c Mutants
3.4. Preparation of TUPS-Modified Cytochrome c Derivatives
3.5. Kinetic Spectroscopy
3.6. Data Analysis and Modeling
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Moser, C.C.; Keske, J.M.; Warncke, K.; Farid, R.S.; Dutton, P.L. Nature of biological electron transfer. Nature 1992, 355, 796–802. [Google Scholar] [CrossRef]
- Winkler, J.R.; Gray, H.B. Electron tunneling in proteins: Role of the intervening medium. JBIC J. Biol. Inorg. Chem. 1997, 2, 399–404. [Google Scholar] [CrossRef]
- Beratan, D.N.; Onuchic, J.N.; Winkler, J.R.; Gray, H.B. Electron-tunneling pathways in proteins. Science 1992, 258, 1740–1741. [Google Scholar] [CrossRef]
- Page, C.C.; Moser, C.C.; Chen, X.; Dutton, P.L. Natural engineering principles of electron tunnelling in biological oxidation–reduction. Nature 1999, 402, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Gray, H.B.; Winkler, J.R. Photoinduced electron transfer in ruthenium-modified cytochrome c. Pure Appl. Chem. 1992, 64, 1257–1262. [Google Scholar] [CrossRef] [Green Version]
- Gray, H.B.; Winkler, J.R. Electron tunneling through proteins. Q. Rev. Biophys. 2003, 36, 341–372. [Google Scholar] [CrossRef]
- Kotlyar, A.B.; Borovok, N.; Hazani, M. Photoinduced electron transfer in singly labeled thiouredopyrenetrisulfonate cytochrome c derivatives. Biochemistry 1997, 36, 15828–15833. [Google Scholar] [CrossRef] [PubMed]
- Borovok, N.; Kotlyar, A.B.; Pecht, I.; Skov, L.K.; Farver, O. Photoinduced electron transfer in singly labeled thiouredopyrenetrisulfonate azurin derivatives. FEBS Lett. 1999, 457, 277–282. [Google Scholar] [CrossRef] [Green Version]
- Kotlyar, A.B.; Borovok, N.; Khoroshyy, P.; Tenger, K.; Zimányi, L. Redox photochemistry of thiouredopyrenetrisulfonate. Photochem. Photobiol. 2004, 79, 489–493. [Google Scholar] [CrossRef]
- Lange, C.; Hunte, C. Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc. Natl. Acad. Sci. USA 2002, 99, 2800–2805. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, H.; Kraut, J. Crystal structure of a complex between electron transfer partners, cytochrome c peroxidase and cytochrome c. Science 1992, 258, 1748–1755. [Google Scholar] [CrossRef]
- Roberts, V.A.; Pique, M.E. Definition of the interaction domain for cytochrome c on cytochrome c oxidase. III. Prediction of the docked complex by a complete, systematic search. J. Biol. Chem. 1999, 274, 38051–38060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez-Paggi, D.; Meister, W.; Kuhlmann, U.; Weidinger, I.; Tenger, K.; Zimányi, L.; Rákhely, G.; Hildebrandt, P.; Murgida, D.H. Disentangling electron tunneling and protein dynamics of cytochrome c through a rationally designed surface mutation. J. Phys. Chem. B 2013, 117, 6061–6068. [Google Scholar] [CrossRef]
- Kotlyar, A.B.; Borovok, N.; Hazani, M.; Szundi, I.; Einarsdóttir, Ó. Photoinduced intracomplex electron transfer between cytochrome c oxidase and TUPS-modified cytochrome c. Eur. J. Biochem. 2000, 267, 5805–5809. [Google Scholar] [CrossRef] [PubMed]
- Szundi, I.; Cappuccio, J.A.; Borovok, N.; Kotlyar, A.B.; Einarsdóttir, Ó. Photoinduced Electron Transfer in the Cytochrome c/Cytochrome c Oxidase Complex Using Thiouredopyrenetrisulfonate-Labeled Cytochrome c. Optical Multichannel Detection. Biochemistry 2001, 40, 2186–2193. [Google Scholar] [CrossRef] [PubMed]
- Schkolnik, G.; Utesch, T.; Salewski, J.; Tenger, K.; Millo, D.; Kranich, A.; Zebger, I.; Schulz, C.; Zimányi, L.; Rákhely, G.; et al. Mapping local electric fields in proteins at biomimetic interfaces. Chem. Commun. 2012, 48, 70–72. [Google Scholar] [CrossRef] [Green Version]
- Ranieri, A.; Borsari, M.; Casalini, S.; Rocco, G.D.; Sola, M.; Bortolotti, C.A.; Battistuzzi, G. How to Turn an Electron Transfer Protein into a Redox Enzyme for Biosensing. Molecules 2021, 26, 4950. [Google Scholar] [CrossRef]
- Tenger, K.; Khoroshyy, P.; Leitgeb, B.; Rákhely, G.; Borovok, N.; Kotlyar, A.; Dolgikh, D.A.; Zimányi, L. Complex kinetics of the electron transfer between the photoactive redox label TUPS and the heme of cytochrome c. J. Chem. Inf. Model. 2005, 45, 1520–1526. [Google Scholar] [CrossRef]
- Kotlyar, A.B.; Borovok, N. Intramolecular oxidation of cytochrome c by covalently attached sulfoaromatic molecules. Biochim. Biophys. Acta-Bioenerg. 1997, 1321, 221–228. [Google Scholar] [CrossRef] [Green Version]
- Kotlyar, A.B.; Borovok, N.; Hazani, M. Use of thiouredopyrenetrisulfonate photochemistry for driving electron transfer reactions in aqueous solutions. Biochemistry 1997, 36, 15823–15827. [Google Scholar] [CrossRef]
- Kotlyar, A.B.; Borovok, N.; Raviv, S.; Zimányi, L.; Gutman, M. Fast Redox Perturbation of Aqueous Solution by Photoexcitation of Pyranine. Photochem. Photobiol. 1996, 63, 448–454. [Google Scholar] [CrossRef]
- Mori, Y.; Shinoda, H.; Nakano, T.; Kitagawa, T. Formation and Decay Behaviors of Laser-Induced Transient Species from Pyrene Derivatives 1. Spectral Discrimination and Decay Mechanisms in Aqueous Solution. J. Phys. Chem. A 2002, 106, 11743–11749. [Google Scholar] [CrossRef]
- Devault, D. Quantum mechanical tunnelling in biological systems. Q. Rev. Biophys. 1980, 13, 387–564. [Google Scholar] [CrossRef] [PubMed]
- Marcus, R.A.; Sutin, N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta-Rev. Bioenerg. 1985, 811, 265–322. [Google Scholar] [CrossRef]
- Miyashita, O.; Okamura, M.Y.; Onuchic, J.N. Interprotein electron transfer from cytochrome c2 to photosynthetic reaction center: Tunneling across an aqueous interface. Proc. Natl. Acad. Sci. USA 2005, 102, 3558–3563. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Paggi, D.; Castro, M.A.; Tórtora, V.; Castro, L.; Radi, R.; Murgida, D.H. Electrostatically Driven Second-Sphere Ligand Switch between High and Low Reorganization Energy Forms of Native Cytochrome c. J. Am. Chem. Soc. 2013, 135, 4389–4397. [Google Scholar] [CrossRef]
- Beratan, D.N.; Onuchic, J.N.; Hopfield, J.J. Electron tunneling through covalent and noncovalent pathways in proteins. J. Chem. Phys. 1998, 86, 4488. [Google Scholar] [CrossRef]
- Zimányi, L.; Sipos, Á.; Sarlós, F.; Nagypál, R.; Groma, G.I. Machine-learning model selection and parameter estimation from kinetic data of complex first-order reaction systems. PLoS ONE 2021, 16, e0255675. [Google Scholar] [CrossRef] [PubMed]
- Bortolotti, C.A.; Borsari, M.; Sola, M.; Chertkova, R.; Dolgikh, D.A.; Kotlyar, A.B.; Facci, P. Orientation-Dependent Kinetics of Heterogeneous Electron Transfer for Cytochrome c Immobilized on Gold: Electrochemical Determination and Theoretical Prediction. J. Phys. Chem. C 2007, 111, 12100–12105. [Google Scholar] [CrossRef]
- Pepelina, T.Y.; Chertkova, R.V.; Dolgikh, D.A.; Kirpichnikov, M.P. The role of individual lysine residues of horse cytochrome c in the formation of reactive complexes with components of the respiratory chain. Russ. J. Bioorg. Chem. 2010, 36, 90–96. [Google Scholar] [CrossRef]
- Chertkova, R.V.; Brazhe, N.A.; Bryantseva, T.V.; Nekrasov, A.N.; Dolgikh, D.A.; Yusipovich, A.I.; Sosnovtseva, O.; Maksimov, G.V.; Rubin, A.B.; Kirpichnikov, M.P. New insight into the mechanism of mitochondrial cytochrome c function. PLoS ONE 2017, 12, e0178280. [Google Scholar] [CrossRef] [PubMed]
- Chertkova, R.V.; Sharonov, G.V.; Feofanov, A.V.; Bocharova, O.V.; Latypov, R.F.; Chernyak, B.V.; Arseniev, A.S.; Dolgikh, D.A.; Kirpichnikov, M.P. Proapoptotic activity of cytochrome c in living cells: Effect of K72 substitutions and species differences. Mol. Cell. Biochem. 2008, 314, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Pepelina, T.Y.; Chertkova, R.V.; Ostroverkhova, T.V.; Dolgikh, D.A.; Kirpichnikov, M.P.; Grivennikova, V.G.; Vinogradov, A.D. Site-directed mutagenesis of cytochrome c: Reactions with respiratory chain components and superoxide radical. Biochemistry 2009, 74, 625–632. [Google Scholar] [CrossRef]
- Schägger, H.; von Jagow, G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 1987, 166, 368–379. [Google Scholar] [CrossRef]
- Babul, J.; Stellwagen, E. Participation of the Protein Ligands in the Folding of Cytochrome c. Biochemistry 1972, 11, 1195–1200. [Google Scholar] [CrossRef]
- Khoroshyy, P.; Dér, A.; Zimányi, L. Effect of Hofmeister cosolutes on the photocycle of photoactive yellow protein at moderately alkaline pH. J. Photochem. Photobiol. B Biol. 2013, 120, 111–119. [Google Scholar] [CrossRef]
- Krekic, S.; Nagy, D.; Taneva, S.G.; Fábián, L.; Zimányi, L.; Dér, A. Spectrokinetic characterization of photoactive yellow protein films for integrated optical applications. Eur. Biophys. J. 2019, 48, 465–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heyes, D.J.; Quinn, A.M.; Cullis, P.M.; Lee, M.; Munro, A.W.; Scrutton, N.S. Internal electron transfer in multi-site redox enzymes is accessed by laser excitation of thiouredopyrene-3,6,8-trisulfonate (TUPS). Chem. Commun. (Camb) 2009, 7, 1124–1126. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1st Solution | 2nd Solution | |||||
|---|---|---|---|---|---|---|
| λ, eV | HDA, eV | λ, eV | HDA, eV | ΔG, eV | ||
| K8 | forward | 2.18 | 3.96 × 10−5 | 0.57 | 2.84 × 10−5 | −1.12 |
| reverse | 1.86 | 9.19 × 10−6 | 0.23 | 5.48 × 10−6 | −0.66 | |
| K39 | forward | 2.54 | 1.90 × 10−4 | 0.49 | 1.26 × 10−4 | −1.12 |
| reverse | 2.17 | 5.78 × 10−5 | 0.20 | 3.19 × 10−5 | −0.66 | |
| K86 | forward | 2.27 | 7.85 × 10−5 | 0.55 | 5.52 × 10−5 | −1.12 |
| reverse | 1.96 | 3.72 × 10−5 | 0.22 | 2.16 × 10−5 | −0.66 | |
| K87 | forward | 2.20 | 3.34 × 10−5 | 0.57 | 2.38 × 10−5 | −1.12 |
| reverse | 1.87 | 7.76 × 10−6 | 0.23 | 4.61 × 10−6 | −0.66 | |
| K13 | reverse | 1.97 | 9.54 × 10−5 | 0.22 | 5.52 × 10−5 | −0.66 |
| K72 | reverse | 1.86 | 1.36 × 10−4 | 0.23 | 8.10 × 10−5 | −0.66 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khoroshyy, P.; Tenger, K.; Chertkova, R.V.; Bocharova, O.V.; Kirpichnikov, M.P.; Borovok, N.; Groma, G.I.; Dolgikh, D.A.; Kotlyar, A.B.; Zimányi, L. Kinetics and Energetics of Intramolecular Electron Transfer in Single-Point Labeled TUPS-Cytochrome c Derivatives. Molecules 2021, 26, 6976. https://doi.org/10.3390/molecules26226976
Khoroshyy P, Tenger K, Chertkova RV, Bocharova OV, Kirpichnikov MP, Borovok N, Groma GI, Dolgikh DA, Kotlyar AB, Zimányi L. Kinetics and Energetics of Intramolecular Electron Transfer in Single-Point Labeled TUPS-Cytochrome c Derivatives. Molecules. 2021; 26(22):6976. https://doi.org/10.3390/molecules26226976
Chicago/Turabian StyleKhoroshyy, Petro, Katalin Tenger, Rita V. Chertkova, Olga V. Bocharova, Mikhail P. Kirpichnikov, Natalia Borovok, Géza I. Groma, Dmitry A. Dolgikh, Alexander B. Kotlyar, and László Zimányi. 2021. "Kinetics and Energetics of Intramolecular Electron Transfer in Single-Point Labeled TUPS-Cytochrome c Derivatives" Molecules 26, no. 22: 6976. https://doi.org/10.3390/molecules26226976