1. Introduction
Multidrug resistance (MDR) is a common feature of many cancer types and a major challenge in clinical cancer treatment with chemotherapeutics [
1,
2,
3]. In MDR, cancer cells have an intrinsic or acquired resistance towards a wide variety of anticancer drugs that significantly reduces the effectiveness of chemotherapy and most often also causes cancer recurrence. MDR may emerge from several mechanisms depending on the structure of anticancer agents. One of the most studied mechanisms is the over-expression of ATP-binding cassette (ABC) superfamily, drug effluxing transporters functioning on the plasma membrane, exemplified with historically the most significant efflux transporter, P-glycoprotein (P-gp,
ABCB1) [
2,
4]. Later on, other efflux transporters, such as multidrug resistance protein 1 (MRP1,
ABCC1) and breast cancer resistant protein (BCRP,
ABCG2), also known as mitoxantrone resistant protein (MXR), were also associated with clinical MDR. Despite the extensive research with efflux transporter inhibitors, their success in clinical use has been modest, mainly because of the challenge of targeting these inhibitors only into the cancer cells [
2,
3,
4,
5].
Ganciclovir (GCV), a synthetic analog of 2′-deoxyguanosine, is an antiviral drug discovered in the 1980s that has been used against cytomegalovirus (CMV) infections [
6,
7,
8]. To be active, GCV needs to be phosphorylated firstly to GCV-monophosphate (GCV-MP) by a viral thymidine kinase followed by subsequent phosphorylation to GCV-diphosphate (GCV-DP) and -triphosphate (GCV-TP) by ubiquitous cellular kinases. GCV-TP then inhibits DNA polymerase and thereby prevents viral DNA replication. The apoptosis-resulting effect of GCV has also been utilized in glioma therapy by introducing the herpes simplex virus thymidine kinase (HSV-TK) gene in an adenovirus-vector to remaining dividing cells after surgical removal of a brain tumor [
9,
10]. However, only a modest increase in median survival has been achieved in the clinical trials, most likely because of the poor targeting and delivery of the vector as well as GCV into these dividing cancer cells [
9,
11]. To solve the latter, it has been recommended that a more lipophilic derivative of GCV would be helpful, as GCV itself is very hydrophilic and less well penetrated across the cell membranes [
12]. Moreover, it has been reported that GCV, in its non-phosphorylated form, is an MRP4-substrate, and therefore cancer cells overexpressing MRP4 are highly resistant to HSV-TK/GCV therapy [
13].
In the present study, it was evaluated whether GCV and its more lipophilic novel derivative could serve as an efflux transporter inhibitors that could improve the outcome of another chemotherapeutic agent either in estrogen receptor-positive MCF-7 breast cells or triple-negative MDA-MB-231 breast cells. Thus, in this study, the effects of GCV and its more lipophilic derivative on methotrexate (MTX) cellular accumulation and subsequent apoptosis were explored, since MTX suffers from MRP1, 2, 3, and 4-related chemoresistance [
14,
15] and since it has been previously reported that MCF-7 and MDA-MB-231 cells overexpress MRP1/4 proteins [
16,
17]. Furthermore, the biocompatibility and effects of the novel derivative and GCV, as well as their combinations with MTX were evaluated in human plasma, erythrocytes, and endothelial cells (HUVEC).
3. Discussion
In the present study, the quantitative proteomics revealed that there is no major difference in MRP1 and MRP4 expression levels among estrogen receptor-positive MCF-7 and triple-negative MDA-MB-231 cells (
Figure 1), oppositely to the previous Western blot analyses [
16,
17]. However, a significant 13-fold difference was observed in BCRP expression between these cell lines, MCF-7 cells expressing BCRP to a greater extent than MDA-MB-231 cells. Previously, it has been reported that some sublines of MCF-7, such as MCF-7/AdrVp but not MCF-7 cells, express BCRP based on mRNA detection [
21]. Moreover, it has been proposed that drug-sensitive MCF-7 and MDA-MB231 cells can be transfected with BCRP to resemble the phenotype of MCF-7/AdrVp. All in all, these quantitative proteomic results point out that it is highly important to recognize the expression profiles of efflux transporters in different cancer cell types that are responsible for the clinical MDR in order to find effective chemotherapy that may include a combination of specific efflux inhibitors. Furthermore, the expression of these transporters should be studied with highly sensitive methods, such as the quantitative proteomics method. However, it should be kept in mind that the expression of efflux transporters is inducible, and thus depends highly on the microenvironment of cancer cells.
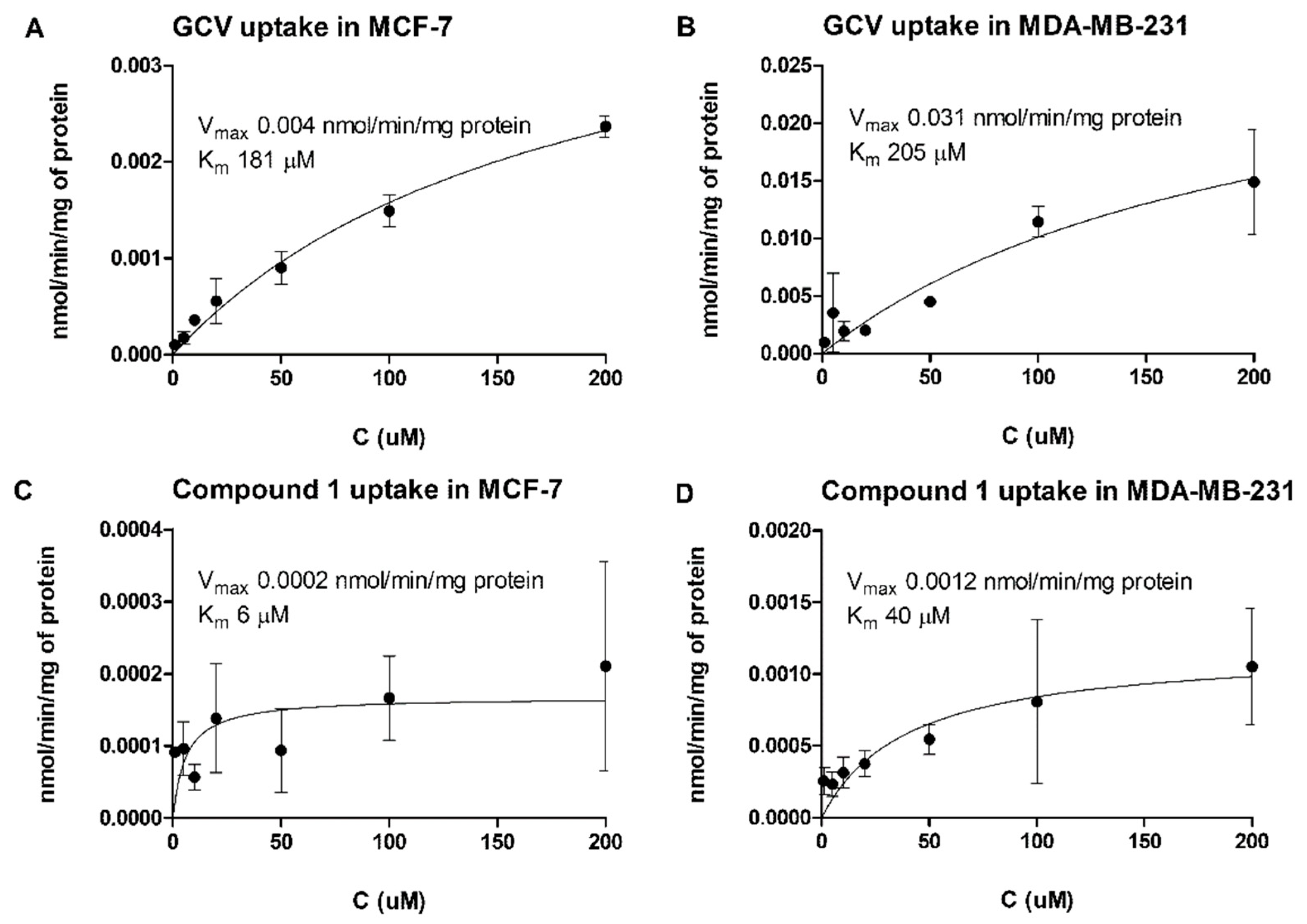
It has been stated that the uptake of GCV into the rat glioma cells (BT4C) is not transporter-mediated, and because of the polar nature of GCV, the cellular (passive) permeation is poor [
12]. Based on this ideology, we designed and synthesized more lipophilic derivative
1 of GCV (
Scheme 1 and
Table 1) and studied the cellular accumulation of GCV and the derivative
1 into MCF-7 and MDA-MB-231 cells. Curiously, the concentration-dependent uptake revealed that GCV was utilizing most likely some saturable transporter for its cellular accumulation. Furthermore, the maximum transport capacity (V
max) of GCV into MDA-MB-231 cells was eight times greater than into MCF-7 cells (
Figure 2A,B), indicating a possible efflux mechanism in MCF-7 cells. Similarly, derivative
1 showed a saturable transport mechanism, which was six times higher into MDA-MB-231 cells than MCF-7, although the cellular uptake in both breast cancer cells was much lower compared to GCV (
Figure 2C,D). Indeed, it has been reported that GCV can use several distinct solute carriers (SLCs), including multidrug and toxin extrusion protein 1 (MATE1,
SLC47A1) and 2-K (MATE2-K,
SLC47A2), organic anion transporters 1 (OAT1,
SLC22A6) and 2 (OAT2,
SLC22A7), organic cation transporter 1 (OCT1,
SLC22A1), as well as equilibrative nucleobase transporter 1 (EEG1, ENBT1,
SLC43A3) [
22,
23,
24,
25]. Thus, based on the presented results, increasing the lipophilicity and size of a GCV-derivative decreased the transporter-mediated cellular uptake and is therefore not a feasible strategy to improve the delivery of GCV into the cancer cells. Instead, GCV derivatives should be designed based on the structure–activity relationships of the above mentioned transporters and the fact these transporters are overexpressed in target cancer cell types.
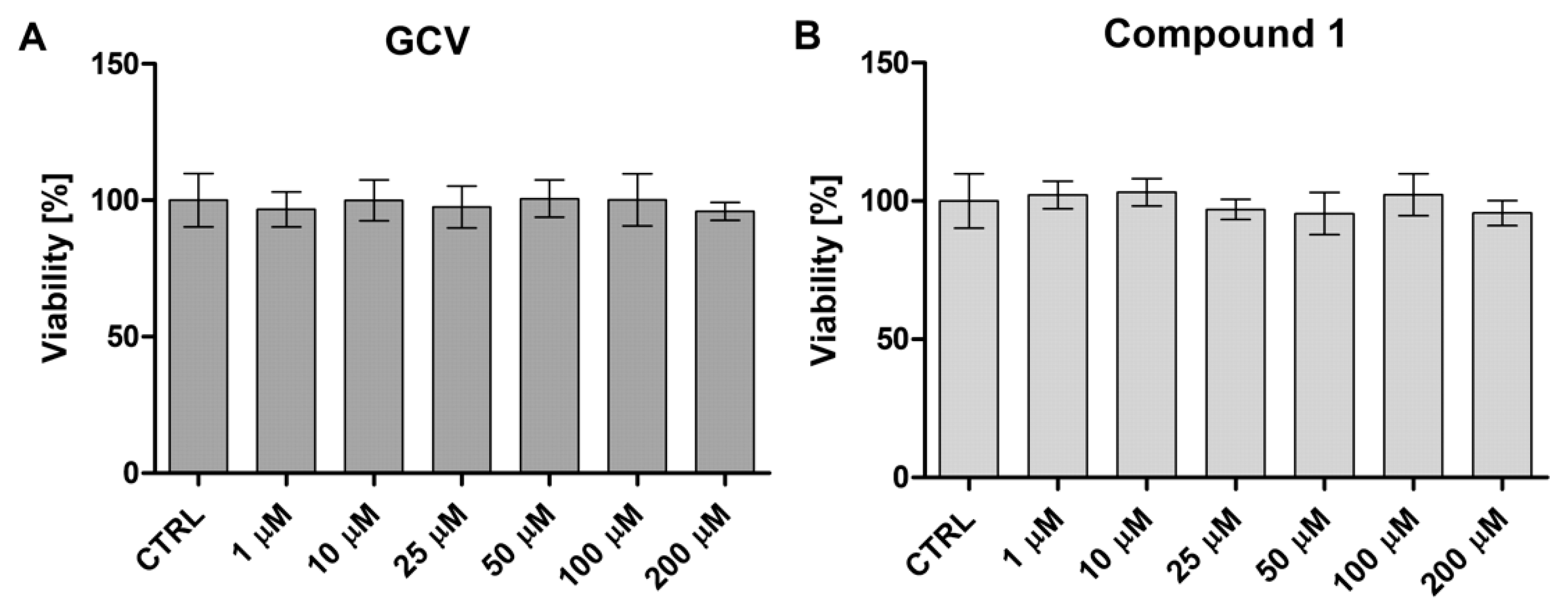
Both GCV as well as its more lipophilic derivative
1 were found to be biocompatible in human plasma and not causing any major effects on plasma haemostasis parameters (
Figure 5). Moreover, neither of the compounds affected the viability of HUVEC (
Figure 6 and
Figure 7). However, both compounds were noticed to increases hemolysis at high concentrations (100 µM) and the hemolytic effects of derivative
1 were greater than those of GCV (
Figure 8 and
Figure 9). Therefore, the combination of MTX (100 µM) and compound
1 (100 µM) was also hemolytic, while GCV together with MTX can be considered a safer combination. GCV has been previously reported to be poorly tolerated in a combination of nucleoside reverse transcriptase inhibitor zidovudine in AIDS patients with cytomegalovirus infections. It has been suggested that MRP4 inhibition by GCV may have increased the cellular levels of zidovudine above cytotoxic levels. Therefore, a combination of GCV, a clinically approved drug, with any other compound can result in effective anticancer treatment, particularly where this primary chemotherapeutic suffers from efflux transporter-related chemoresistance. However, the levels of cytotoxicity in cancer cells vs. healthy cells need to be carefully studied to remain in a safe but effective therapeutic window.
Based on the above-mentioned cellular uptake results, it was explored if GCV or compound
1 can increase the cellular accumulation of the anticancer agent, MTX, since it has been reported that in addition to MRP1–4, MTX can interact also with BCRP [
26,
27]. In MCF-7, both compounds were able to improve the exposure of the cells to MTX, GCV increasing the MTX accumulation by 21-fold while compound
1 increased it by 13-fold (
Figure 3A). It was also demonstrated in the present study that both GCV and its more lipophilic derivative
1 were interacting with BCRP, since a selective BCRP-selective inhibitor, FMC, was able to increase the amount of these compounds in the MCF-7 cells, when incubating FMC together with these compounds (
Figure 3B). However, in this study, the conclusions were based only on the competitive cellular uptake studies, and thus more sophisticated methods, e.g., computational modeling, are needed to clarify the exact interactions between the compounds and BCRP [
28,
29,
30]. Curiously, interactions of GCV and other anticancer nucleosides with BCRP have been demonstrated also in other studies [
31,
32], which supports our conclusions.
Moreover, it is highly important also to evaluate how the compounds affect the function and/or expression of BCRP in the future. Notably, the interactions of GCV or compound
1 with BCRP in the present study seemed to be stronger than with MRPs, since MK-571 (MRP inhibitor) did not increase their cellular uptake significantly. Nevertheless, the expression of MRP1 and 4 was lower than, e.g., the one of BCRP in MCF-7 cells, which may affect these conclusions. Therefore, more detailed affinity studies are needed to clarify this comparison between BCRP and MRPs in the future. It needs to be also remembered that we cannot exclude the possibility of GCV or compound
1 affecting MTX cellular accumulation by other means, such as increasing its metabolic stability, which also needs to be studied more thoroughly in the future. For example, MTX is polyglutamated by folylpolyglutamate synthase (FPGS), which increases the retention of MTX in the cells and thus MTX cytotoxicity [
33,
34]. On the other hand, γ-glutamyl hydrolase (γ-GH) removes these polyglutamates and thus predisposes MTX to efflux mechanisms. It is highly likely that the expression or function of these enzymes is affected by GCV and its derivatives.
The greater MTX exposure in the GCV or compound
1 combinations also resulted in lower cell viability in MCF-7 cells compared to the single treatments (MTX, GCV, or compound
1). Accordingly, GCV as well as compound
1 contributed to the significantly lower number of living cells and a higher number of early and late apoptotic cells compared with the single treatment of MTX (
Table 2,
Figure 4). Curiously, there was no significant difference between the combination treatments, although GCV increased MTX exposure to a greater extent than compound
1. Thus, considering the higher MTX accumulation and effects in MCF-7 cells after GCV or compound
1 co-treatments, which both had interactions with BCRP, it can be concluded that increased exposure of MTX is the most obvious explanation for the apoptotic effects seen with combination treatments. Moreover, the transport mechanisms of MTX (influx via reduced folate carrier 1 (RFC1) and efflux via several transporters, including MRPs and BCRP) have been stated to be the probable reasons for MTX resistance [
34,
35]. However, less focus has been paid to the metabolism of MTX. The cellular accumulation in this study was carried out with 30 min incubation, while in the apoptosis study, the cells were incubated for 24 h. Therefore, it is highly likely that in the MTX accumulation study, no metabolism was seen, while in the apoptosis study FPGS and γ-GH had enough time to affect MTX accumulation. Since the metabolism can differ significantly among different cancer cell types, these enzyme expressions and activities should be studied more thoroughly in the future.
4. Materials and Methods
4.1. Chemicals
All reagents and solvents used in these studies were commercial and high purity of analytical grade or ultra-gradient LC-MS-grade purchased from MilliporeSigma (St. Louis, MO, USA), J.T. Baker (Deventer, The Netherlands), Merck (Darmstadt, Germany), Riedel-de Haën (Seelze, Germany) or Thermo Fisher Scientific (Waltham, MA, USA), unless otherwise stated. Water was purified using a Milli-Q Gradient system (Millipore, Milford, MA, USA). The following reagents were used for basic coagulology screening, Bio-Ksel System APTTs reagent and calcium chloride (Bio-Ksel, Grudziądz, Poland), Bio-Ksel PT plus reagent (thromboplastin and solvent, Bio-Ksel, Grudziądz, Poland), and thrombin (3.0 UNIH/mL, Bio-Ksel, Grudziądz, Poland). Coefficients of variability were calculated using Bio-Ksel normal plasma (Bio-Ksel, Poland), and water for injection (Polpharma, Gdańsk, Poland). Triton X-100 used in the erythrotoxicity test was obtained from Polish Chemical Reagents (Gliwice, Poland).
4.2. Biological Material
The studies on the biological material were approved by the Bioethics Committee of the Medical University of Lodz (RNN/109/16/KE; 19 April 2016 and RNN/104/20/KE; 2 April 2020). RBCs for erythrotoxicity studies were separated from the plasma by centrifugation (3000× g, 10 min) at 20 °C and washed three times with 0.9% saline. Plasma was stored in small aliquots at −30 °C. Before the experiments, the material was thawed for 15 min at 37 °C.
4.3. Cell Cultures
MCF-7 human breast adenocarcinoma cells (HTB-22; RRID: CVCL_0031) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA), MDA-MB-231 human breast adenocarcinoma cells (RRID: CVCL_0062) from Sigma-Aldrich (European Collection of Authenticated Cell Cultures (ECACC, Public Health England, Salisbury, UK, Cat. No 92020424), and human umbilical vein endothelial cells (HUVEC; RRID CVCL_2959) from Lonza (Basel, Switzerland, Cat. No. CC-2517). Both human breast cancer cell lines were cultured in standard conditions (37 °C, 5% CO2) using Dulbecco’s modified Eagle medium (DMEM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA) supplemented with L-glutamine (2 mM, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), heat-inactivated fetal bovine serum (10%, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), penicillin (50 U/mL, Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and streptomycin (50 μg/mL, Gibco, Thermo Fisher Scientific, Waltham, MA, USA). Once the cells reached 80% confluence in culture bottles (75 cm2), the cells were washed twice with DPBS solution (Gibco, Thermo Fisher Scientific, Waltham, MA, USA), and harvested using 5 mL of accutase (Sigma Aldrich, St. Louis, MO, USA). HUVEC were subcultured according to the manufacturers’ (Lonza, Basel, Switzerland) guidelines. Cell culturing ingredients for HUVEC included medium EGM-2 − medium + bullet kit (Lonza, Clonetics, Basel, Switzerland), accutase solution (Sigma Aldrich, St. Louis, MO, USA), and HEPES buffered saline solution (Lonza, Basel, Switzerland).
4.4. Protein Expression of Efflux Transporters in MCF-7 and MDA-MB-231 Cells
The absolute expression of the efflux transporters BCRP, MRP1, MRP4, and P-gp was quantified in the crude membrane fractions of MCF-7 and MDA-MB-231 cells by the LC-MS/MS method following a multiplexed multiple reaction monitoring (MRM) analysis mode according to the protocol described by Uchida et al. [
36] with minor modifications. First, the crude membrane fractions were isolated from three distinct sets of cell culture plates using Membrane Protein Extraction Kit (BioVision Incorporated, Milpitas, CA, USA) according to the manufacturer’s instructions. The protein content for each fraction was measured by Bio-Rad Protein Assay, based on the Bradford dye-binding method (EnVision, Perkin Elmer, Inc., Waltham, MA, USA). A total amount of 50 µg protein from each fraction was solubilized/denatured in 7 M Guanidine hydrochloride, 0.5 M Tris-HCl and 10 mM EDTA-Na. The proteins were then reduced by dithiothreitol (1:50,
w/
w) and
S-carboxymethylated by iodoacetamide (1:20,
w/
w) (Sigma-Aldrich, St. Louis, MO, USA). The alkylated proteins were precipitated by methanol/chloroform/water (4:1:3) and centrifuged at 18,000×
g for 5 min at 4 °C. The pellet was resuspended in 6 M urea and mixed for 10 min at room temperature before the dilution with 0.1 M Tris-HCl (pH 8.5) to a final concentration of 1.2 M urea, and dissolved completely by intermittent sonication (Branson 3510, Danbury, CT, USA). The dissolved proteins were first digested with LysC (1/100,
w/
w) (MilliporeSigma, St. Louis, MO, USA) and 0.05% ProteaseMax (Promega Biotech AB, Nacka, Sweden) for 3 h at room temperature. Then, the samples were spiked with 10 µL (30 fmol) of the labeled peptides for absolute quantification (JPT Peptide Technologies GmbH, Berlin, Germany) (
Table 1). The samples were incubated with (1/100,
w/
w) TPCK-Trypsin (Promega Biotech AB, Nacka, Sweden) for 18 h at 37 °C. The tryptic digestion was then quenched by adding 40 µL of 5% formic acid. The samples were then centrifuged at 18,000×
g for 5 min at 4 °C, and the supernatants were transferred to vials for the analysis.
The digested peptides were analyzed using an ultra-performance liquid chromatography system coupled with a triple quadrupole mass spectrometer with a heated electrospray ionization source in the positive mode (UPLC 1290 and MSD 6495, Agilent Technologies, Santa Clara, CA, USA). A total amount of 20 µL of the digested peptides (10 µg) was separated using AdvanceBio Peptide Map 2.1 × 250 mm, 2.7 μm column (Agilent Technologies, Santa Clara, CA, USA) and LC eluents of 0.1% formic acid in water (A) and acetonitrile (B). The peptides were eluted following a constant flow rate of 0.3 mL/min and a gradient of 2–7% B for 2 min, followed by 7–30% B for 48 min, 30–45% B for 3 min, and 45–80% B for 2.5 min before re-equilibrating the column again for 4.5 min. The proteins were quantified based on the ratio between the light and heavy standard peptides, as described previously (
Table 3) [
37]. Data were acquired using Agilent MassHunter Workstation Acquisition (Agilent Technologies, Data Acquisition for Triple Quadrupole, version B.03.01) and processed by using Skyline software, MacCossLab, University of Washington, Seattle, WA, USA (version 20.1). The results were normalized to a housekeeping protein Na
+/K
+ATPase and expressed as fmol/µg of the total amount of protein in the samples.
4.5. General Synthetic Procedures
All reagents used in the synthesis of compound 1 were obtained from MilliporeSigma (St. Louis, MO, USA) or ThermoFisher Scientific (Waltham, MA, USA). Thin-layer chromatography with aluminum sheets coated with silica gel 60 F245 (0.24 mm) were used to monitor the reactions under UV and with a suitable coloring agent. Purifications were performed by flash chromatography on silica gel 60 (0.063–0.200 mm mesh). Nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 500 spectrometer (Bruker Biospin, Fällanden, Switzerland) operating at 500.13 MHz (1H) and 125.75 (13C), by using tetramethylsilane as an internal standard. Not all pH-dependent protons of the compounds were observed. ESI-MS spectra were recorded by an Agilent 1200 Infinity LC system coupled with an Agilent 6410 triple quadrupole mass spectrometer with an electrospray ionization source (Agilent Technologies, Palo Alto, CA, USA). Over 95% purity was confirmed by the UPLC system (Agilent Technologies 1290 Infinity II system; Agilent Technologies Inc., Wilmington, DE, USA), which comprised a high-speed pump, a multi-sampler, a multi-column thermostat (MCT), a diode array detector (DAD), and an Agilent ZORBAX Eclipse Plus C18 analytical column (2.1 × 50 mm, 1.8 µm) (Agilent Technologies Inc., Wilmington, DE, USA) eluting with water containing 0.1% formic acid (pH ca. 3.0) and acetonitrile containing 0.1% formic acid at the flow rate of 1.0 mL/min at room temperature.
4.6. N-(9-(((1,3-Dihydroxypropan-2-yl)oxy)methyl)-6-oxo-6,9-dihydro-1H-purin-2-yl)benzamide (1)
Ganciclovir (GCV; 0.10 g, 0.39 mmol) and benzoyl chloride (0.27 mL, 2.35 mmol) were stirred in anhydrous pyridine under Ar-atm at RT overnight. The solvent was removed and the residue was redissolved in CH2Cl2 (50 mL), washed with cold 3 M HCl (2 × 50 mL), saturated NaHCO3 (50 mL), and brine (50 mL), dried over Na2SO4, filtered, and evaporated to dryness under high vacuum. The intermediate was used for the next step without further purification.
The intermediate from the previous step was reacted with NaOMe (0.13 g, 2.34 mmol) in anhydrous MeOH under Ar-atm at RT overnight. The solvent was removed and the residue was redissolved in DCM and purified by a flash column chromatography eluting with MeOH:CH2Cl2 (0–20%) to yield an off-white solid, which was triturated with Et2O to give white solid 0.7 g (47% over 2 steps). 1H NMR (600 MHz, MeOD-d6) δ 8.23 (s, 1H, purine CH), 7.91–7.87 (m, 2H, benzene ring), 7.56–7.49 (m, 1H, benzene ring), 7.47–7.43 (m, 2H, benzene ring), 5.76 (s, 2H, NHCH2O), 4.08 (s, 2H, OH), 3.74 (tt, J = 6.3, 4.3 Hz, 1H, OCH(CH2OH)2), 3.51 (dd, J = 11.9, 4.2 Hz, 2H, OCH(CH2OH)2), 3.43 (dd, J = 11.9, 6.3 Hz, 2H, OCH(CH2OH)2). 13C NMR (126 MHz, DMSO-d6) δ 170.28 (amide CO), 163.57 (purine ring amide CO), 155.60 (NC(=)N), 149.52 (HNC=N), 139.67 (N=CN), 132.44 (benzene ring C), 132.28 (benzene ring C), 129.23 (benzene ring C), 128.48 (benzene ring C), 128.42 (benzene ring C), 128.24 (benzene ring C), 119.90 (C=C(N)CO), 80.40 (O-CH2-(CH2OH)2), 71.95 (NCH2O), 60.92 (2C, (O-CH2-(CH2OH)2)). MS (ESI+) for C16H18N5O5 (M+H)+: Calcd 360.35, Found 360.03. UPLC (254 nm) purity: 96.5%.
4.7. Cellular Uptake of GCV and Compound 1
For the cell uptake experiments, MCF-7 and MDA-MB-231 cells (passages 8–15) were seeded at the density of 1 × 105 cells/well onto 24-well plates a day before the experiments. Cellular uptake of compounds was studied by incubating the cells at 37 °C for 30 min (uptake was linear with all compounds up to 30 min) with compounds at the concentration of 1–200 μM in pre-warmed HBSS buffer (250 μL). Subsequently, the cells were washed three times with ice-cold HBSS and lysed with 250 μL of NaOH (0.1 M) for 60 min. The lysates were diluted with acetonitrile (ACN) including the selected internal standard (labetalol) with a ratio of 1:3 and centrifuged at 10,000× g for 10 min. The samples were analyzed by liquid chromatography-tandem mass spectrometric (LC-MS/MS) methods with an Agilent 1200 Series Rapid Resolution LC System (Agilent Technologies, Palo Alto, CA, USA) together with an Agilent 6410 Triple Quadrupole Mass Spectrometer equipped with an electrospray ionization source by using a Zorbax Eclipse XDB-C18 column (50 mm × 4.6 mm, 1.8 μm, Agilent Technologies, Santa Clara, CA, USA).
The chromatographic separation of analytes was achieved with a gradient elution of water containing 0.1% (v/v) formic acid (A) and acetonitrile containing 0.1% (v/v) formic acid (B) at the flow rate of 0.50 mL/min, running with 15–40% B gradient for 0–0.5 min, 40–90% B gradient from 0.5 to 2 min, running isocratically 90% B from 2 to 5 min, B gradient return from 90% to 15% from 5 to 5.1 min, and finally the column was equilibrated with 15% B from 5.1 to 8 min. The column temperature was 40 °C and the injection volume was 5 μL. The following mass spectrometry conditions were used: electrospray ionization, positive ion mode; drying gas (nitrogen) temperature, 300 °C; drying gas flow rate, 6 L/min; nebulizer pressure, 25 psi; and capillary voltage, 4000 V. Analyte detection was performed using multiple reaction monitoring, the transitions being 256→152; 359.9→259 and 105; and 329→294 and 162 for GCV, compound 1, and the internal standard (labetalol), respectively. Fragmentor voltages were 100 V for GCV and compound 1, and 70 V for the internal standard. The collision energies were 10 V for GCV, 10 V, and 30V for compound 1, and 10 V for the internal standard. Agilent MassHunter Workstation Acquisition software (Data Acquisition for Triple Quadrupole Mass Spectrometer, version B.03.01) was used for data acquisition, and Quantitative Analysis (B.04.00) software was used for the data processing and analysis. The lower limit of quantification (LLOQ) for the samples was 1.0 nM for compound 1 and 10 nM for GCV and the methods were linear selective, accurate (100 ± 5% of nominal concentration), and precise (RSD < 15%) over the range 1.0–2500 nM and 12.5–2500 nM, respectively. The concentrations of analytes in cell lysates were calculated from the standard curve that was prepared by spiking known amounts of compounds to ACN, including the selected internal standard and normalized with protein concentration. The protein concentrations on each plate were determined as a mean of three samples by Bio-Rad Protein Assay, based on the Bradford dye-binding method, using bovine serum albumin (BSA) as a standard protein and measuring the absorbance (595 nm) with a multiplate reader (EnVision, Perkin Elmer, Inc., Waltham, MA, USA).
4.8. Cellular Uptake of MTX
Cellular uptake of MTX was studied as described above for GCV and compound
1 at the concentration of 1–200 μM in MCF-7 cells (30 min incubation). The cell lysates were prepared accordingly and analyzed for MTX with LC-MS/MS (Agilent 1200 Series Rapid Resolution LC System together with an Agilent 6410 Triple Quadrupole Mass Spectrometer equipped with an electrospray ionization source) with a Zorbax Eclipse XDB-C18 column (50 mm × 4.6 mm, 1.8 μm, Agilent Technologies, Santa Clara, CA, USA) with a modified method that has been described previously [
38]. The chromatographic separation of analytes was achieved with a gradient elution of water containing 0.1% (
v/
v) formic acid (A) and acetonitrile containing 0.1% (
v/
v) formic acid (B) at the flow rate of 0.30 mL/min, running with 5% B for 1 min, 5–90% B gradient from 1 to 5 min, 90–5% from 5 to 5.1 min, and finally the column was equilibrated from 5.1 to 8 min. The column temperature was 40 °C and the injection volume was 2 μL. The following mass spectrometry conditions were used: electrospray ionization, positive ion mode; drying gas (nitrogen) temperature, 300 °C; drying gas flow rate, 6 L/min; nebulizer pressure, 20 psi; and capillary voltage, 3000 V. Analyte detection was performed using multiple reaction monitoring, the transitions being 455
→ 308 and 329
→ 294, 162 for MTX and the internal standard (labetalol), respectively. Fragmentor voltages were 200 V for MTX and 70 V for the internal standard. The collision energies were 20 V and 10 V for MTX and the internal standard, respectively. Agilent MassHunter Workstation Acquisition software (Data Acquisition for Triple Quadrupole Mass Spectrometer, version B.03.01) was used for data acquisition, and Quantitative Analysis (B.04.00) software was used for the data processing and analysis. The lower limit of quantification (LLOQ) for the samples was 1.0 nM and the method was linear selective, accurate (100 ± 5% of nominal concentration), and precise (RSD < 15%) over the range 2.5–2500 nM. The concentrations of MTX in cell lysates were calculated from the standard curve that was prepared by spiking known amounts of compounds to ACN, including the selected internal standard and normalized with protein concentration. The protein concentrations on each plate were determined as a mean of three samples by Bio-Rad Protein Assay described above.
The cellular accumulation of MTX in the presence of GCV, compound 1, P-gp/BCRP inhibitor, elacridar or multi-MRP-inhibitor MK-571 was carried out as described above. The cells were pre-incubated with 100 µM GCV, compound 1, elacridar or MK-571 for 10 min and the incubation mixture was removed before adding the studied prodrug and LAT1 inhibitor on the cells. The uptake of 1–200 μM MTX (30 min) with 100 µM GCV, compound 1, elacridar, or MK-571 was then carried out as the normal uptake. The concentrations of MTX were analyzed by the LC-MS/MS method described above and calculated from the spiked standard curve and normalized with the protein concentrations.
4.9. Accumulation of GCV/Compound 1 in the Presence of Efflux Inhibitors
The uptake of GCV and compound 1 in the presence of BCRP-selective inhibitor, 5 µM Fumitremorgin C (FMC), and multi-MRP-inhibitor, 100 µM MK-571, was studied as described above, by pre-incubating MCF-7 cells with FMC or MK-571 in HBSS buffer 10 min before adding the GCV or compound 1 solutions (20 µM) together with FMC or MK-571 on the cells and incubating them for 30 min. The amounts of GCV or its derivative 1 were analyzed by the LC-MS/MS method described above and calculated from the spiked standard curve and normalized with the protein concentrations.
4.10. Cell Viability
The viability of MCF-7 cells (cell passages 8–11) as well as HUVEC (passages 3–4) was determined in the presence of examined compounds (MTX, GCV and compound
1) by using the WST-1 assay (Takara, Takara Bio Europe, Saint-Germain-en-Laye, France), which is based on the reaction of cleavage of tetrazolium salts by mitochondrial dehydrogenase in viable cells [
39]. The cells were seeded at the density of 10,000 (MCF-7 and MDA-MB-231) or 7500 (HUVEC) per well on 96-well plates and maintained for 24 h to obtain 70% confluency, followed by treatment with the studied compounds: MTX (1–100 µM), GCV (1–100 µM) and compound
1 (1–100 µM) in MCF-7 and MDA-MB-231 cells, or GCV (0.1–200 μM) and compound
1 (0.1–200 μM) in HUVEC for 24 h (37 °C, 5% CO
2). Control samples with pure medium and medium containing 10 µL of solvent (methanol+water) were performed to obtain 100% viability and the effects of solvent on cells, respectively. After 24-h incubation, the treating solutions were discarded, the cells were washed with culture medium (100 μL) and incubated with WST-1 reagent dissolved in the cell culture medium. The plates were incubated at 37 °C with 5% CO
2 for between 1 (MCF-7 and MDA-MB-231) and 2 (HUVEC) hours and the absorbance was read at 450 nm using a microplate reader (iMARK, Bio-Rad, Bio-Rad Laboratories Inc., US). The results are expressed as a percentage of the control samples treated with pure medium, which constituted 100% viability. The effects of examined compounds on HUVEC were also evaluated by using an inverted microscope with phase-contrast (magnification 100×) (software OptaView 7, Opta-Tech, Warsaw, Poland).
4.11. Apoptosis Analysis
Apoptosis was examined using FITC Annexin V Apoptosis Detection Kit with propidium iodide (PI) (Biolegend, London, UK). MCF-7 cells were seeded at the density of 50,000 per well on 24-well plates, followed by 24-h incubation at standard conditions (37 °C, 5% CO2). Then, the medium was replaced by 250 μL of fresh medium (control wells) or medium with tested compounds or their combinations (100 µM MTX, GCV, compound 1 or 100 µM MTX+50 µM GCV or compound 1, 100 µM MTX+ 100 µM GCV or compound 1). Further, 10 µM etoposide was used as a positive control. The cells were incubated for an additional 24 h, followed by harvesting with accutase (Sigma Aldrich, St. Louis, MO, USA), collecting to Eppendorf tubes, and centrifuging (220× g, 5 min). The cell pellets were suspended in cold (4 ℃) cell staining buffer (BioLegend, London, UK) and washed once with this solution (220× g, 5 min). Then, the cells were resuspended in 100 μL of binding buffer (BioLegend, London, UK), and solutions of PI (10 μL) and FITC Annexin (5 μL) were added. The samples were vigorously vortexed and incubated for 20 min at room temperature in the dark. The analysis was conducted on the cytometer (CytoFlex, blue laser, 480 nm, Beckman Coulter, Brea, CA, US), and the results were analyzed using Kaluza 2.1 Beckman Coulter software. The analysis was performed according to the principle that annexin V (−) and PI (−) cells constitute living cells (E−−), annexin V (+) and PI (−) are early apoptotic cells (E−+), annexin V (+) and PI (+) are late apoptotic cells (E++), and annexin V (−) and PI (+) are necrotic cells (E−+). Moreover, 10,000 cells of each sample were analyzed, and the experiments were conducted in triplicate.
4.12. Basic Coagulation Tests (PT, INR, APTT, TT)
Coagulation parameters, including prothrombin time (PT), international normalized ratio (INR), partially activated thromboplastin time (APTT), and thrombin time (TT), were determined in the presence of the studied compounds (GCV and compound 1) according to the routine diagnostic procedure using a coagulometer (CoagChrom-3003 Bio-Ksel, Grudziądz, Poland) as described previously [
40,
41]. Control samples with distilled water and methanol mixture (1:1) were conducted in all tests. The methods were validated using normal plasma (Bio-Ksel, Grudziądz, Poland). Coefficients of variability for the tests were calculated; W
(PT) = 1.98%, W
(APTT) = 1.49%, W
(TT) = 1.15%. The reference values for each test equal PT: 9.7–14.6 s; APTT: 26.7–40.0 s; TT: 13.8–18.0 s for 3.0 UNIH/mL of thrombin.
4.13. Effects on Erythrocytes
The protocol for hemolysis assay has been previously described [
38]. Briefly, red blood cell (RBC) suspension (2%) was prepared in 0.9% saline and incubated at 37 °C for one and 24 h with the tested compounds (GCV or compound
1) at various concentrations ranging from 1 to 100 µM, or MTX (100 µM) or MTX + GCV or compound
1 (50 or 100 µM). The samples were then centrifuged (1000×
g, 10 min), and the absorbance of the supernatant was measured at 550 nm (Cecil 2021, Cecil Instruments Ltd., Cambridge, UK). The degree of hemolysis was expressed as a percentage of released hemoglobin. Further, 2.0%
v/v Triton X-100 was used as a positive control, which constituted 100% of hemolysis, whereas a sample of solvent (methanol + water) solution represented spontaneous hemolysis of RBCs (control). The coefficient of variability was counted: W
(1h) = 8.21%,
n = 6; W
(24h) = 7.58%,
n = 6.
For morphological evaluation, a 2% erythrocyte suspension was incubated with various concentrations of GCV or compound 1 for 1 and 24 h at 37 °C. Afterwards, the RBC morphology was evaluated using a phase-contrast Opta-Tech inverted microscope, at 400× magnification, equipped with software (OptaView 7, Opta-Tech, Poland) for image analysis.
4.14. Data Analysis
Physicochemical properties of the studied compounds were analyzed by ChemDraw Professional (PerkinElmer Informatics, Inc., Waltham, MA, USA, version 16.0.1.4 (77)) and MarvinSketch (ChemAxon, Ltd., Budapest, Hungary, Version 19.24.0) All statistical analyses, including the Michaelis–Menten kinetics of cellular uptake studies, proteomics, and IC50 values (the concentration of tested compound inhibiting cell growth by 50%) of the cell viabilities, and plasma biocompatibility (APTT, PT, TT) analyses were performed using GraphPad Prism v. 5.03 software (GraphPad Software, San Diego, CA, USA). Statistical differences between groups were tested using one-way ANOVA, followed by a Tukey’s multiple comparison test, and presented as mean ± SD, with significant difference denoted by * p < 0.05, *** p < 0.001. All the data are presented as mean ± SD (standard deviation) and the size of n varied among the study type (n = 4–9).


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}