
Crystal Structure of the ORP8 Lipid Transport ORD Domain: Model of Lipid Transport
 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Expression and Purification
2.2. Crystallization and Data Collection
2.3. Lipids and Other Chemicals
2.4. LUV Formation
2.5. Kinetics Assays
2.6. Microscopy
2.7. All-Atom MD Simulations
2.8. Coarse-Grained MD Simulations
3. Results
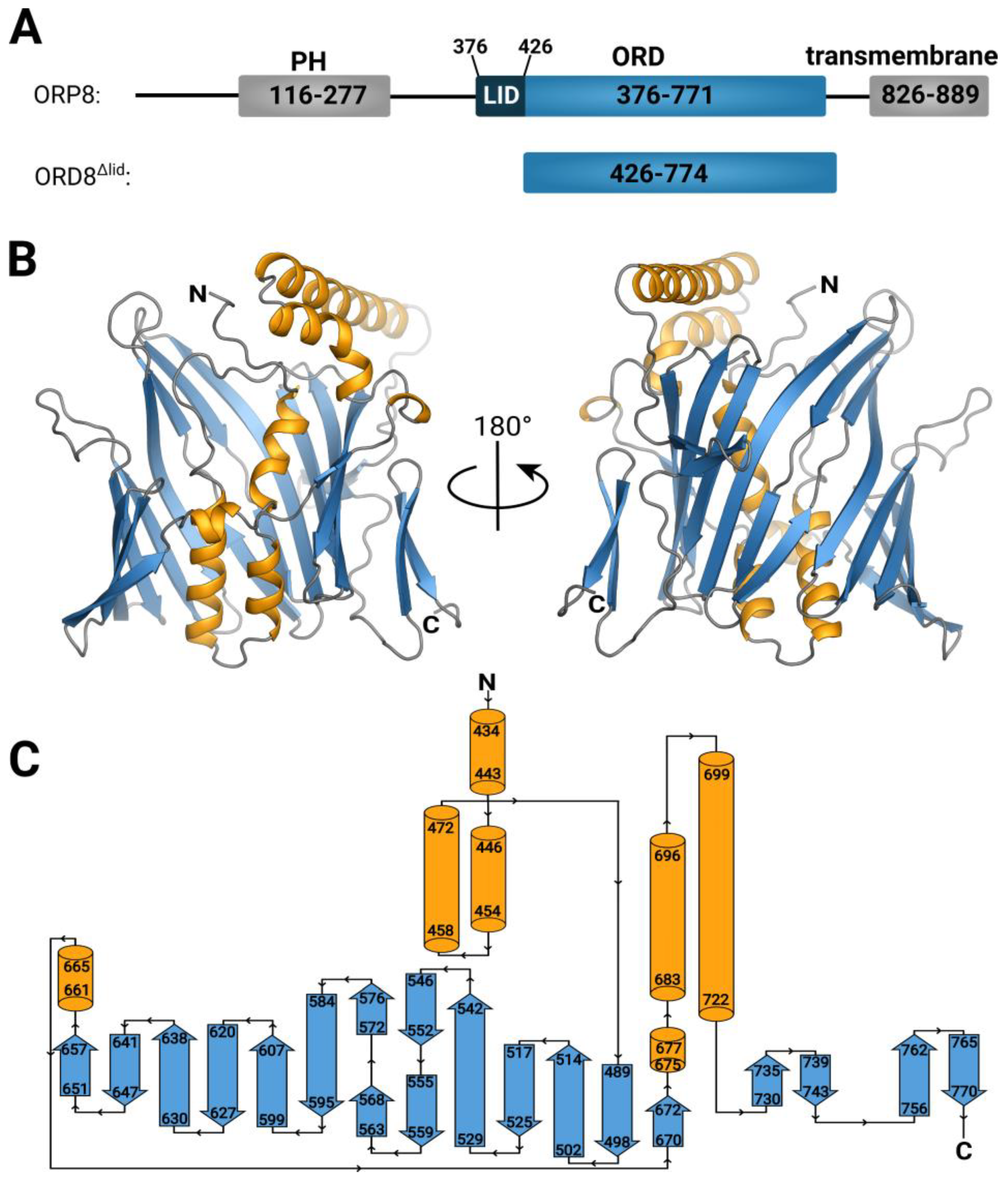
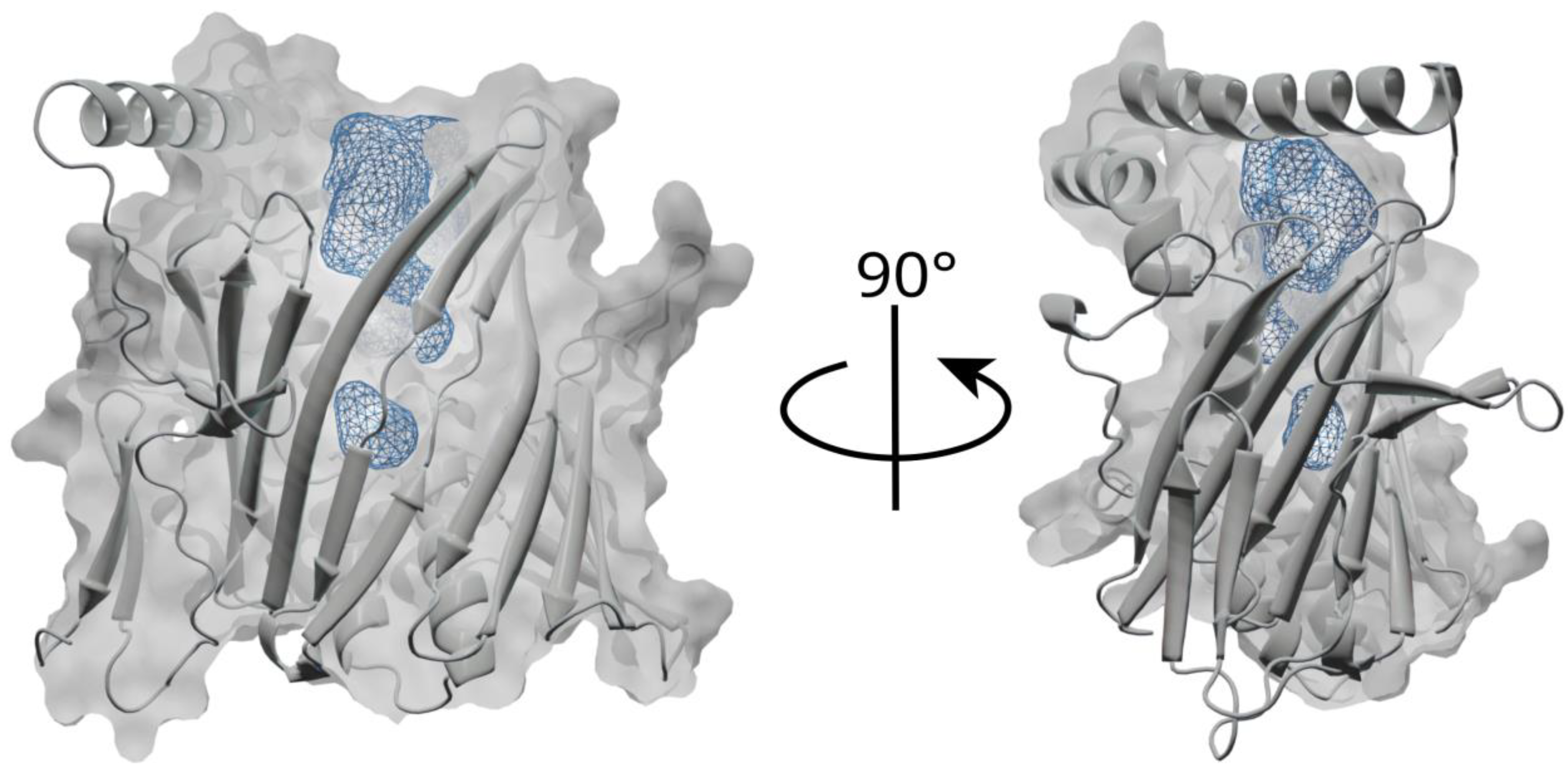
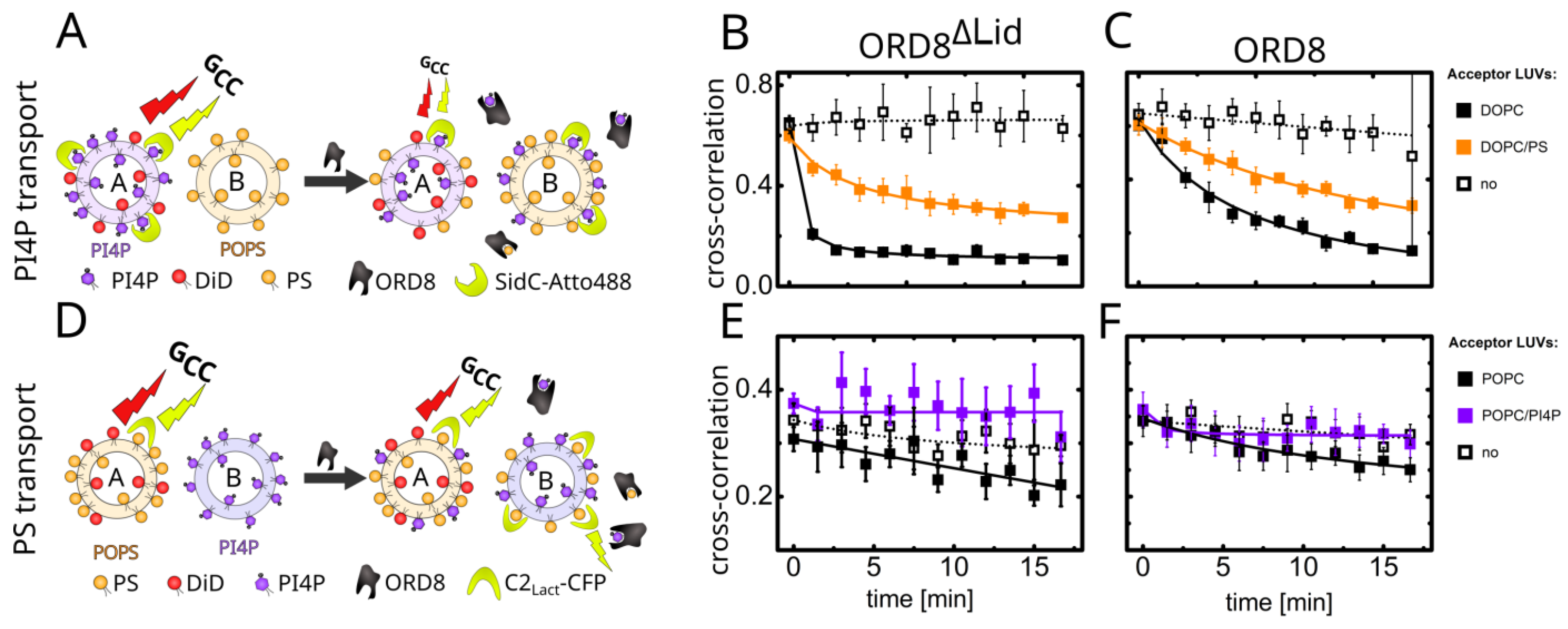
3.1. Overall Structure and Functional Characterization of the ORP8 Lipid Transport Domain
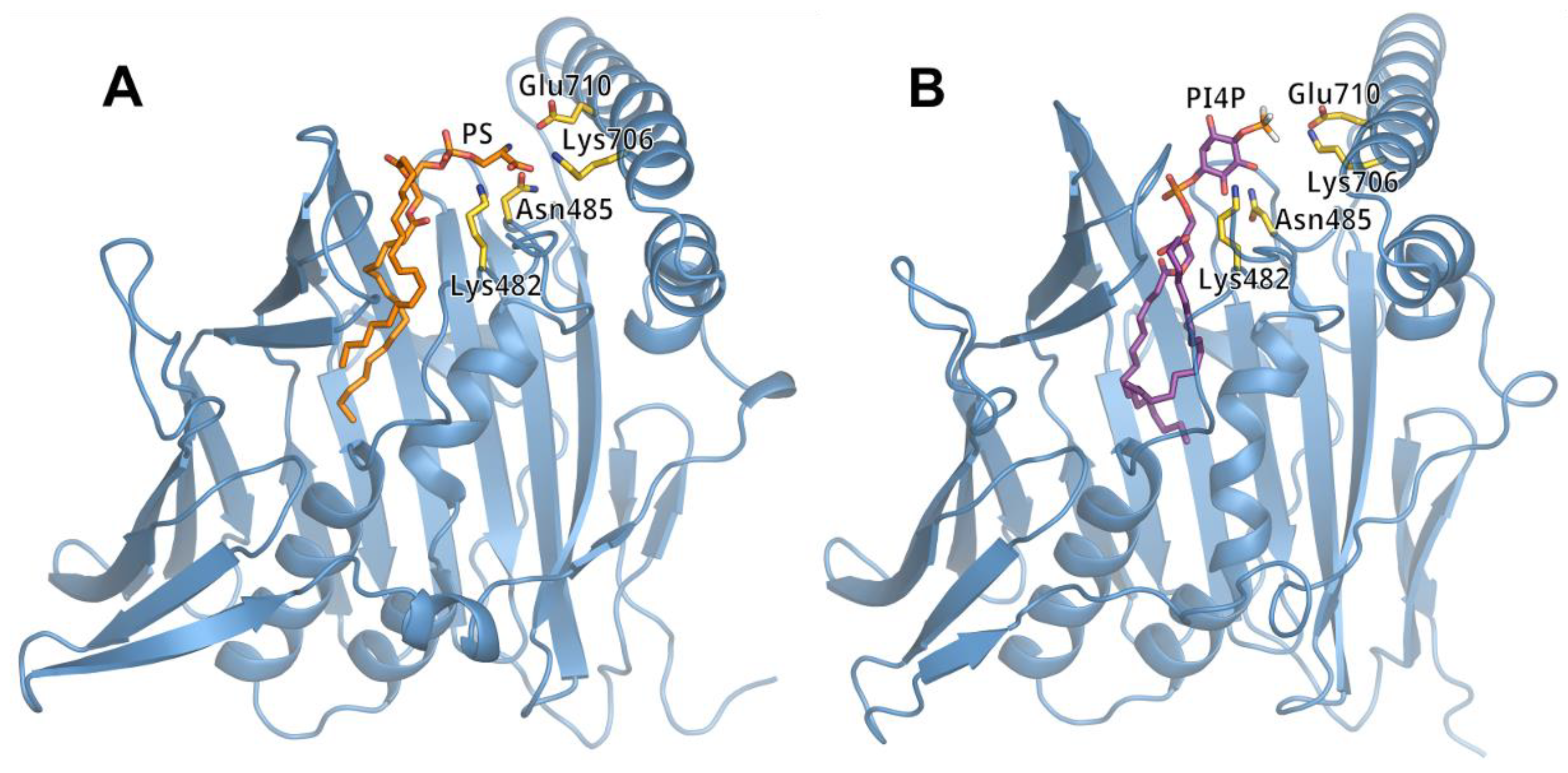
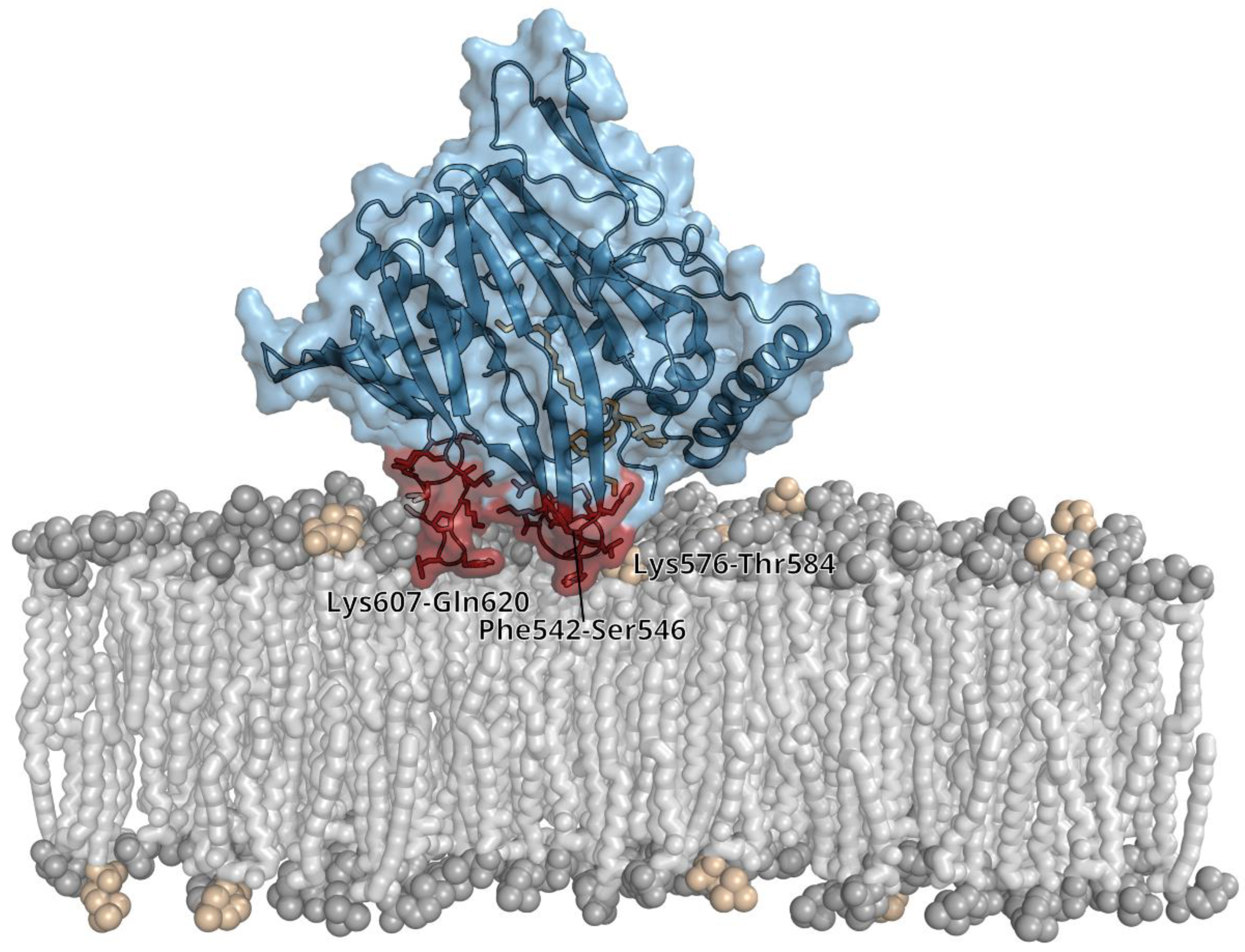
3.2. Lipid Binding Mode of the ORP8 ORD Domain
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reinisch, K.M.; Prinz, W.A. Mechanisms of nonvesicular lipid transport. J. Cell Biol. 2021, 220, e202012058. [Google Scholar] [CrossRef]
- Kentala, H.; Weber-Boyvat, M.; Olkkonen, V.M. OSBP-Related Protein Family: Mediators of Lipid Transport and Signaling at Membrane Contact Sites. Int. Rev. Cell Mol. Biol. 2016, 321, 299–340. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.; Burke, J.E. Novel roles of phosphoinositides in signaling, lipid transport, and disease. Curr. Opin. Cell Biol. 2020, 63, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Prinz, W.A. The Diverse Functions of Oxysterol-Binding Proteins. Annu. Rev. Cell Dev. Biol. 2010, 26, 157–177. [Google Scholar] [CrossRef] [Green Version]
- Chung, J.; Torta, F.; Masai, K.; Lucast, L.; Czapla, H.; Tanner, L.B.; Narayanaswamy, P.; Wenk, M.R.; Nakatsu, F.; De Camilli, P. Intracellular Transport. PI4P/phosphatidylserine countertransport at ORP5- and ORP8-mediated ER-plasma membrane contacts. Science 2015, 349, 428–432. [Google Scholar] [CrossRef] [Green Version]
- Sohn, M.; Korzeniowski, M.; Zewe, J.P.; Wills, R.C.; Hammond, G.R.; Humpolickova, J.; Vrzal, L.; Chalupska, D.; Veverka, V.; Fairn, G.D.; et al. PI(4,5)P2 controls plasma membrane PI4P and PS levels via ORP5/8 recruitment to ER-PM contact sites. J. Cell Biol. 2018, 217, 1797–1813. [Google Scholar] [CrossRef]
- Ghai, R.; Du, X.; Wang, H.; Dong, J.; Ferguson, C.; Brown, A.J.; Parton, R.G.; Wu, J.W.; Yang, H. ORP5 and ORP8 bind phosphatidylinositol-4, 5-biphosphate (PtdIns(4,5)P (2)) and regulate its level at the plasma membrane. Nat. Commun. 2017, 8, 757. [Google Scholar] [CrossRef] [Green Version]
- Batrouni, A.G.; Baskin, J.M. The chemistry and biology of phosphatidylinositol 4-phosphate at the plasma membrane. Bioorg. Med. Chem. 2021, 40, 116190. [Google Scholar] [CrossRef]
- Hammond, G.R.; Fischer, M.J.; Anderson, K.E.; Holdich, J.; Koteci, A.; Balla, T.; Irvine, R.F. PI4P and PI(4,5)P-2 Are Essential But Independent Lipid Determinants of Membrane Identity. Science 2012, 337, 727–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boura, E.; Nencka, R. Phosphatidylinositol 4-kinases: Function, structure, and inhibition. Exp. Cell Res. 2015, 337, 136–145. [Google Scholar] [CrossRef]
- Zewe, J.P.; Wills, R.C.; Sangappa, S.; Goulden, B.D.; Hammond, G.R. SAC1 degrades its lipid substrate PtdIns4P in the endoplasmic reticulum to maintain a steep chemical gradient with donor membranes. eLife 2018, 7, e35588. [Google Scholar] [CrossRef] [PubMed]
- Eisenreichova, A.; Różycki, B.; Boura, E.; Humpolickova, J. Osh6 Revisited: Control of PS Transport by the Concerted Actions of PI4P and Sac1 Phosphatase. Front. Mol. Biosci. 2021, 8, 747601. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Anand, K.; Chiapparino, A.; Kumar, A.; Poletto, M.; Kaksonen, M.; Gavin, A.-C. Interactome map uncovers phosphatidylserine transport by oxysterol-binding proteins. Nature 2013, 501, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Von Filseck, J.M.; Vanni, S.; Mesmin, B.; Antonny, B.; Drin, G. A phosphatidylinositol-4-phosphate powered exchange mechanism to create a lipid gradient between membranes. Nat. Commun. 2015, 6, 6671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Ma, Q.; Qi, Y.; Dong, J.; Du, X.; Rae, J.; Wang, J.; Wu, W.-F.; Brown, A.J.; Parton, R.G.; et al. ORP2 Delivers Cholesterol to the Plasma Membrane in Exchange for Phosphatidylinositol 4, 5-Bisphosphate (PI(4,5)P2). Mol. Cell 2019, 73, 458–473.e7. [Google Scholar] [CrossRef] [Green Version]
- Balla, T. Phosphoinositides: Tiny Lipids with Giant Impact on Cell Regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef]
- Moser von Filseck, J.; Čopič, A.; Delfosse, V.; Vanni, S.; Jackson, C.L.; Bourguet, W.; Drin, G. Intracellular Transport. Phosphatidylserine transport by ORP/Osh proteins is driven by phosphatidylinositol 4-phosphate. Science 2015, 349, 432–436. [Google Scholar] [CrossRef]
- D’Ambrosio, J.M.; Albanèse, V.; Lipp, N.F.; Fleuriot, L.; Debayle, D.; Drin, G.; Čopič, A. Osh6 requires Ist2 for localization to ER-PM contacts and efficient phosphatidylserine transport in budding yeast. J. Cell Sci. 2020, 133, jcs243733. [Google Scholar] [CrossRef]
- Wong, A.K.O.; Young, B.P.; Loewen, C.J. Ist2 recruits the lipid transporters Osh6/7 to ER-PM contacts to maintain phospholipid metabolism. J. Cell Biol. 2021, 220, e201910161. [Google Scholar] [CrossRef]
- Fischer, M.A.; Temmerman, K.; Ercan, E.; Nickel, W.; Seedorf, M. Binding of plasma membrane lipids recruits the yeast integral membrane protein Ist2 to the cortical ER. Traffic 2009, 10, 1084–1097. [Google Scholar] [CrossRef]
- Lipp, N.-F.; Gautier, R.; Magdeleine, M.; Renard, M.; Albanèse, V.; Čopič, A.; Drin, G. An electrostatic switching mechanism to control the lipid transfer activity of Osh6p. Nat. Commun. 2019, 10, 3926. [Google Scholar] [CrossRef] [Green Version]
- Koukalova, A.; Eisenreichova, A.; Rozycki, B.; Boura, E.; Humpolickova, J. Coordination of transporter, cargo, and membrane properties during non-vesicular lipid transport. bioRxiv 2023, 28, 546834. [Google Scholar]
- Chalupska, D.; Eisenreichova, A.; Różycki, B.; Rezabkova, L.; Humpolickova, J.; Klima, M.; Boura, E. Structural analysis of phosphatidylinositol 4-kinase IIIbeta (PI4KB)-14-3-3 protein complex reveals internal flexibility and explains 14-3-3 mediated protection from degradation in vitro. J. Struct. Biol. 2017, 200, 36–44. [Google Scholar] [CrossRef]
- Chalupska, D.; Różycki, B.; Humpolickova, J.; Faltova, L.; Klima, M.; Boura, E. Phosphatidylinositol 4-kinase IIIbeta (PI4KB) forms highly flexible heterocomplexes that include ACBD3, 14-3-3, and Rab11 proteins. Sci. Rep. 2019, 9, 567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, U.; Förster, R.; Hellmig, M.; Huschmann, F.U.; Kastner, A.; Malecki, P.; Pühringer, S.; Röwer, M.; Sparta, K.; Steffien, M.; et al. The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 2015, 130, 141. [Google Scholar] [CrossRef]
- Kabsch, W. Xds. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef] [Green Version]
- Afonine, P.V.; Poon, B.K.; Read, R.J.; Sobolev, O.V.; Terwilliger, T.C.; Urzhumtsev, A.; Adams, P.D. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018, 74, 531–544. [Google Scholar] [CrossRef] [Green Version]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.; Hintze, B.; Hung, L.W.; Jain, S.; McCoy, A.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D-Struct. Biol. 2019, 75, 861–877. [Google Scholar] [CrossRef] [Green Version]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [Green Version]
- Boura, E.; Hurley, J.H. Structural basis for membrane targeting by the MVB12-associated beta-prism domain of the human ESCRT-I MVB12 subunit. Proc. Natl. Acad. Sci. USA 2012, 109, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM All-Atom Additive Force Field for Lipids: Validation on Six Lipid Types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone phi, psi and Side-Chain chi(1) and chi(2) Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Ingólfsson, H.I.; Cheng, X.; Lee, J.; Marrink, S.J.; Im, W. CHARMM-GUI Martini Maker for Coarse-Grained Simulations with the Martini Force Field. J. Chem. Theory Comput. 2015, 11, 4486–4494. [Google Scholar] [CrossRef]
- Souza, P.C.T.; Alessandri, R.; Barnoud, J.; Thallmair, S.; Faustino, I.; Grünewald, F.; Patmanidis, I.; Abdizadeh, H.; Bruininks, B.M.H.; Wassenaar, T.A.; et al. Martini 3: A general purpose force field for coarse-grained molecular dynamics. Nat. Methods 2021, 18, 382–388. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [Green Version]
- Quigley, D.; Probert, M.I.J. Langevin dynamics in constant pressure extended systems. J. Chem. Phys. 2004, 120, 11432–11441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Ormö, M.; Cubitt, A.B.; Kallio, K.; Gross, L.A.; Tsien, R.Y.; Remington, S.J. Crystal Structure of the Aequorea victoria Green Fluorescent Protein. Science 1996, 273, 1392–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, J.; Tan, L.; Im, Y.J. Structure of human ORP3 ORD reveals conservation of a key function and ligand specificity in OSBP-related proteins. PLoS ONE 2021, 16, e0248781. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, Z.; Ye, S.; Zhang, R. The crystal structure of ORP3 reveals the conservative PI4P binding pattern. Biochem. Biophys. Res. Commun. 2020, 529, 1005–1010. [Google Scholar] [CrossRef]
- Chwastyk, M.; Jaskolski, M.; Cieplak, M. The volume of cavities in proteins and virus capsids. Proteins Struct. Funct. Bioinform. 2016, 84, 1275–1286. [Google Scholar] [CrossRef]
- Li, F.-L.; Fu, V.; Liu, G.; Tang, T.; Konradi, A.W.; Peng, X.; Kemper, E.; Cravatt, B.F.; Franklin, J.M.; Wu, Z.; et al. Hippo pathway regulation by phosphatidylinositol transfer protein and phosphoinositides. Nat. Chem. Biol. 2022, 18, 1076–1086. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Lin, D.; Kusov, Y.; Nian, Y.; Ma, Q.; Wang, J.; Von Brunn, A.; Leyssen, P.; Lanko, K.; Neyts, J.; et al. alpha-Ketoamides as Broad-Spectrum Inhibitors of Coronavirus and Enterovirus Replication: Structure-Based Design, Synthesis, and Activity Assessment. J. Med. Chem. 2020, 63, 4562–4578. [Google Scholar] [CrossRef] [Green Version]
- Mejdrová, I.; Chalupská, D.; Placková, P.; Muller, C.; Sála, M.; Klíma, M.; Baumlová, A.; Hrebabecký, H.; Procházková, E.; Dejmek, M.; et al. Rational Design of Novel Highly Potent and Selective Phosphatidylinositol 4-Kinase IIIbeta (PI4KB) Inhibitors as Broad-Spectrum Antiviral Agents and Tools for Chemical Biology. J. Med. Chem. 2017, 60, 100–118. [Google Scholar] [CrossRef]
- Otava, T.; Sala, M.; Li, F.; Fanfrlik, J.; Devkota, K.; Perveen, S.; Chau, I.; Pakarian, P.; Hobza, P.; Vedadi, M.; et al. The Structure-Based Design of SARS-CoV-2 nsp14 Methyltransferase Ligands Yields Nanomolar Inhibitors. ACS Infect. Dis. 2021, 7, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Nencka, R.; Silhan, J.; Klima, M.; Otava, T.; Kocek, H.; Krafcikova, P.; Boura, E. Coronaviral RNA-methyltransferases: Function, structure and inhibition. Nucleic Acids Res. 2022, 50, 635–650. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal | hORP8/ORD | |
|---|---|---|
| PDB accession code | 8P7A | |
| Data collection and processing | ||
| Diffraction source | BESSY 14.2 | |
| Detector | Dectris Pilatus 2M | |
| Wavelength (Å) | 0.9184 | |
| Space group | P 21 21 2 | |
| Cell dimensions | a, b, c (Å) | 105.6 190.2 58.7 |
| α, β, γ (°) | 90.0 90.0 90.0 | |
| Resolution range (Å) | 46.17–2.56 (2.65–2.56) | |
| No. of total reflections | 504,392 (47,110) | |
| Multiplicity | 12.9 (12.3) | |
| No. of unique reflections | uncorrected | 38,978 (3814) |
| anisotropy-corrected | 21,645 (133) | |
| Completeness (%) | uncorrected | 99.17 (98.90) |
| anisotropy-corrected | 52.87 (3.39) | |
| Mean I/σ(I) | 9.52 (0.51) | |
| Wilson B factor (Å2) | 34.28 | |
| R-merge | 0.2847 (4.747) | |
| R-meas | 0.2964 (4.952) | |
| CC1/2 (%) | 99.8 (49.5) | |
| CC* (%) | 100.0 (81.4) | |
| Structure solution and refinement | ||
| R-work (%) | 21.90 (42.87) | |
| R-free (%) | 25.08 (42.08) | |
| CC-work (%) | 73.4 (67.1) | |
| CC-free (%) | 80.3 (88.7) | |
| R.m.s. deviations | bonds (Å) | 0.003 |
| angles (°) | 0.59 | |
| Average B factor (Å2) | 35.30 | |
| Rotamer outliers (%) | 0.00 | |
| Clashscore | 1.00 | |
| Ramachandran (%) | favoured | 99.13 |
| allowed | 0.87 | |
| outliers | 0.00 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eisenreichova, A.; Klima, M.; Anila, M.M.; Koukalova, A.; Humpolickova, J.; Różycki, B.; Boura, E. Crystal Structure of the ORP8 Lipid Transport ORD Domain: Model of Lipid Transport. Cells 2023, 12, 1974. https://doi.org/10.3390/cells12151974
Eisenreichova A, Klima M, Anila MM, Koukalova A, Humpolickova J, Różycki B, Boura E. Crystal Structure of the ORP8 Lipid Transport ORD Domain: Model of Lipid Transport. Cells. 2023; 12(15):1974. https://doi.org/10.3390/cells12151974
Chicago/Turabian StyleEisenreichova, Andrea, Martin Klima, Midhun Mohan Anila, Alena Koukalova, Jana Humpolickova, Bartosz Różycki, and Evzen Boura. 2023. "Crystal Structure of the ORP8 Lipid Transport ORD Domain: Model of Lipid Transport" Cells 12, no. 15: 1974. https://doi.org/10.3390/cells12151974