
TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness

 , , , , , and
, , , , , and
Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Virus
2.2. Inhibitors, Antibodies, and Reagents
2.3. Cell Stimulation In Vitro and Viral Infection
2.4. Immunofluorescence Staining and Confocal Microscopy
2.5. Measurement of IL-6 Secretion
2.6. Determination of IL-6, CCL2 /MCP-1, SDF-1, α-SMA, and IP-10 Expression
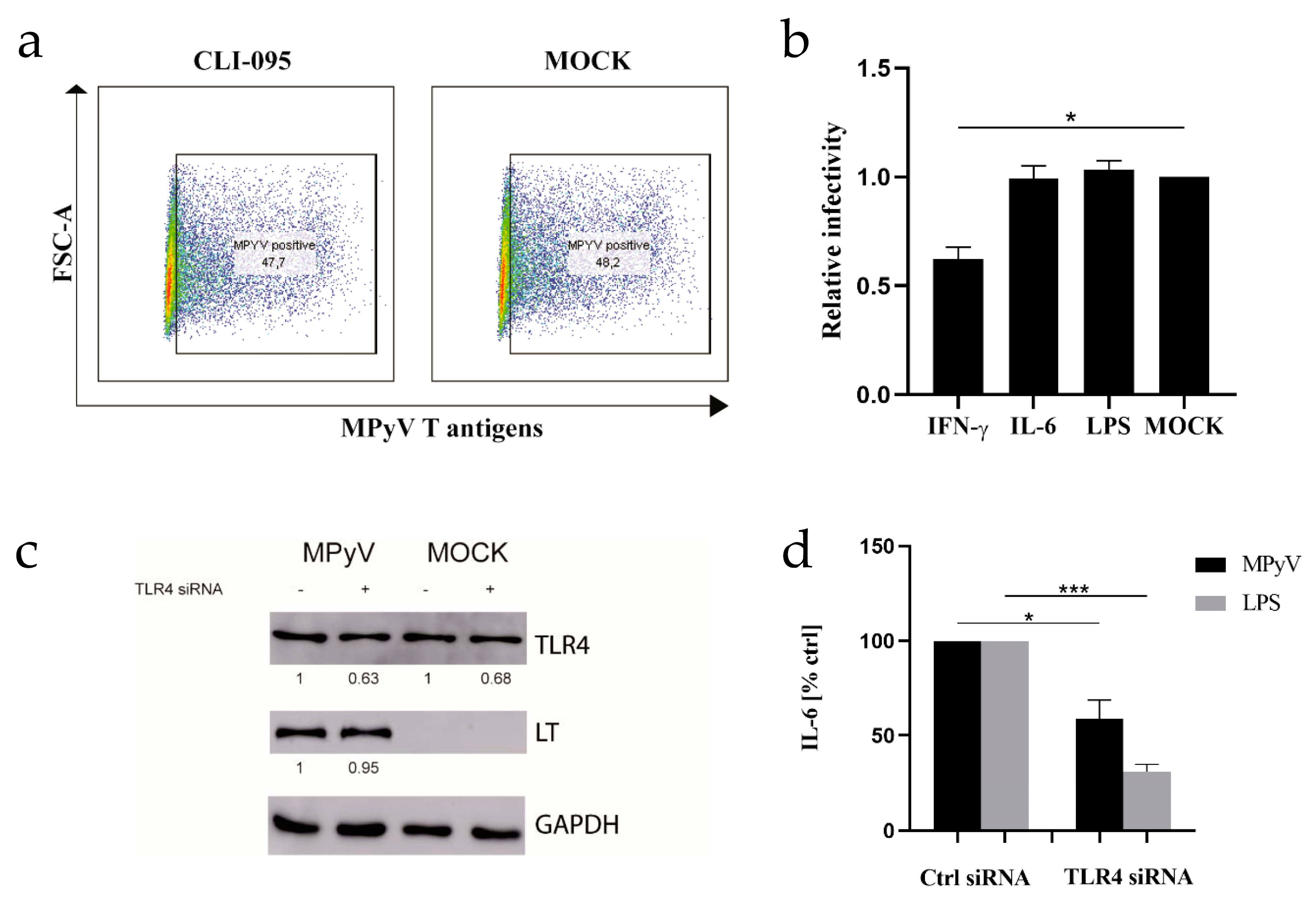
2.7. TLR4 Silencing Using siRNA
2.8. Determination of STAT3 Phosphorylation, Alpha-SMA, ALIX, and TLR4 by Immunoblotting
2.9. Determination of STAT3 Phosphorylation and MPyV Infection of MEFs by Flow Cytometry
2.10. Cell Invasiveness Assay
2.11. Statistical Analysis
3. Results
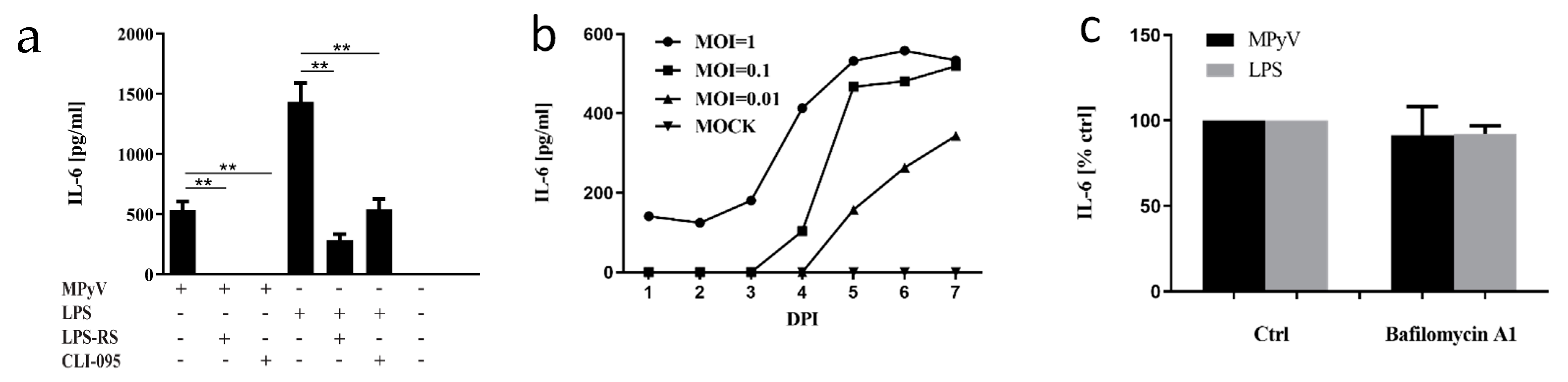
3.1. Mouse Embryonic Fibroblasts and 3T6 Cells Recognize MPyV through TLR4 and Secrete IL-6
3.2. TLR4 Colocalizes with MPyV Capsid Protein VP1 in MEF
3.3. Neither TLR4 Activation Nor Recombinant IL-6 Inhibits MPyV Replication in MEFs and 3T6 Cells
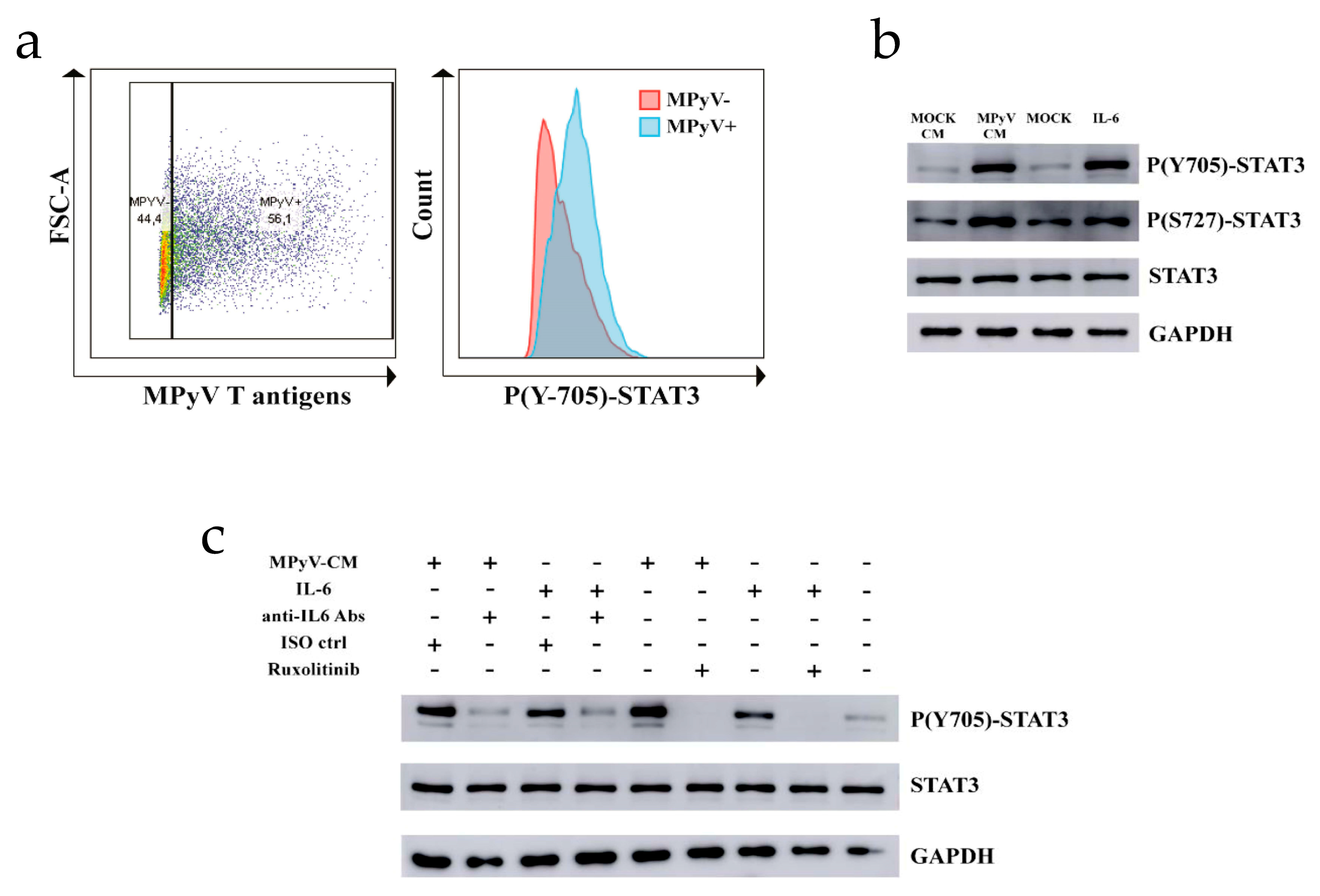
3.4. MPyV Induces STAT3 Phosphorylation via IL-6
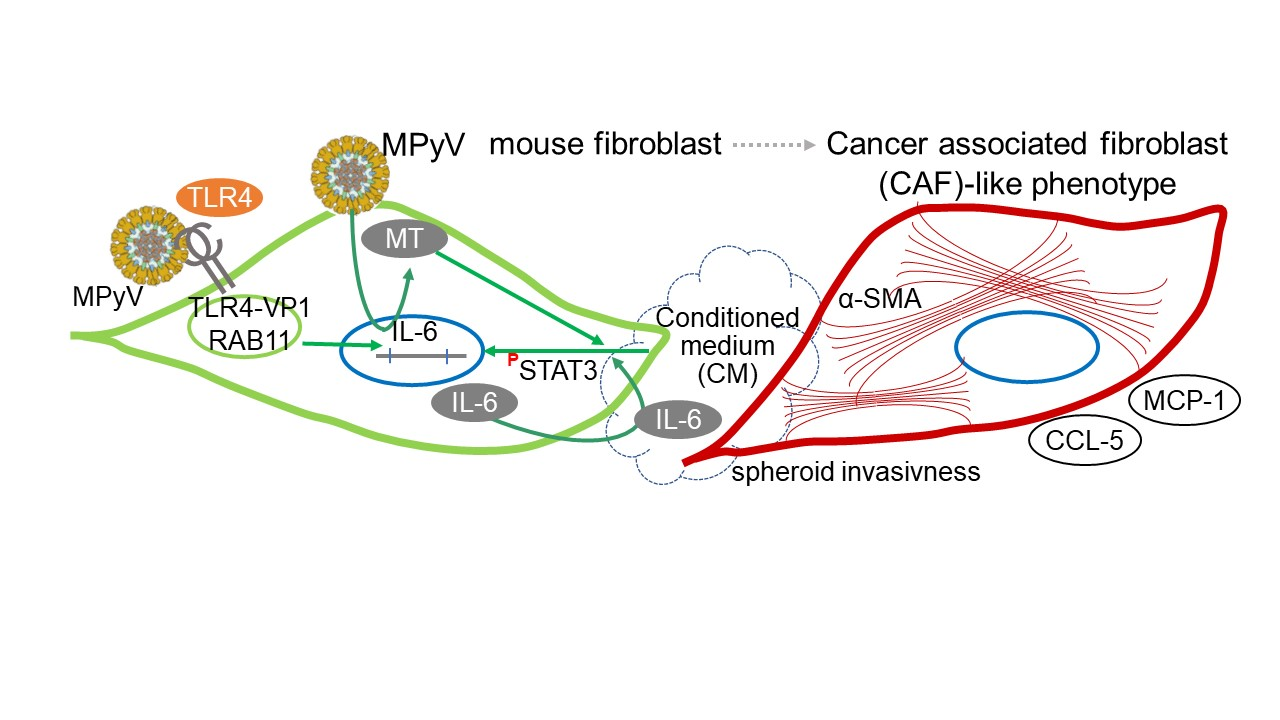
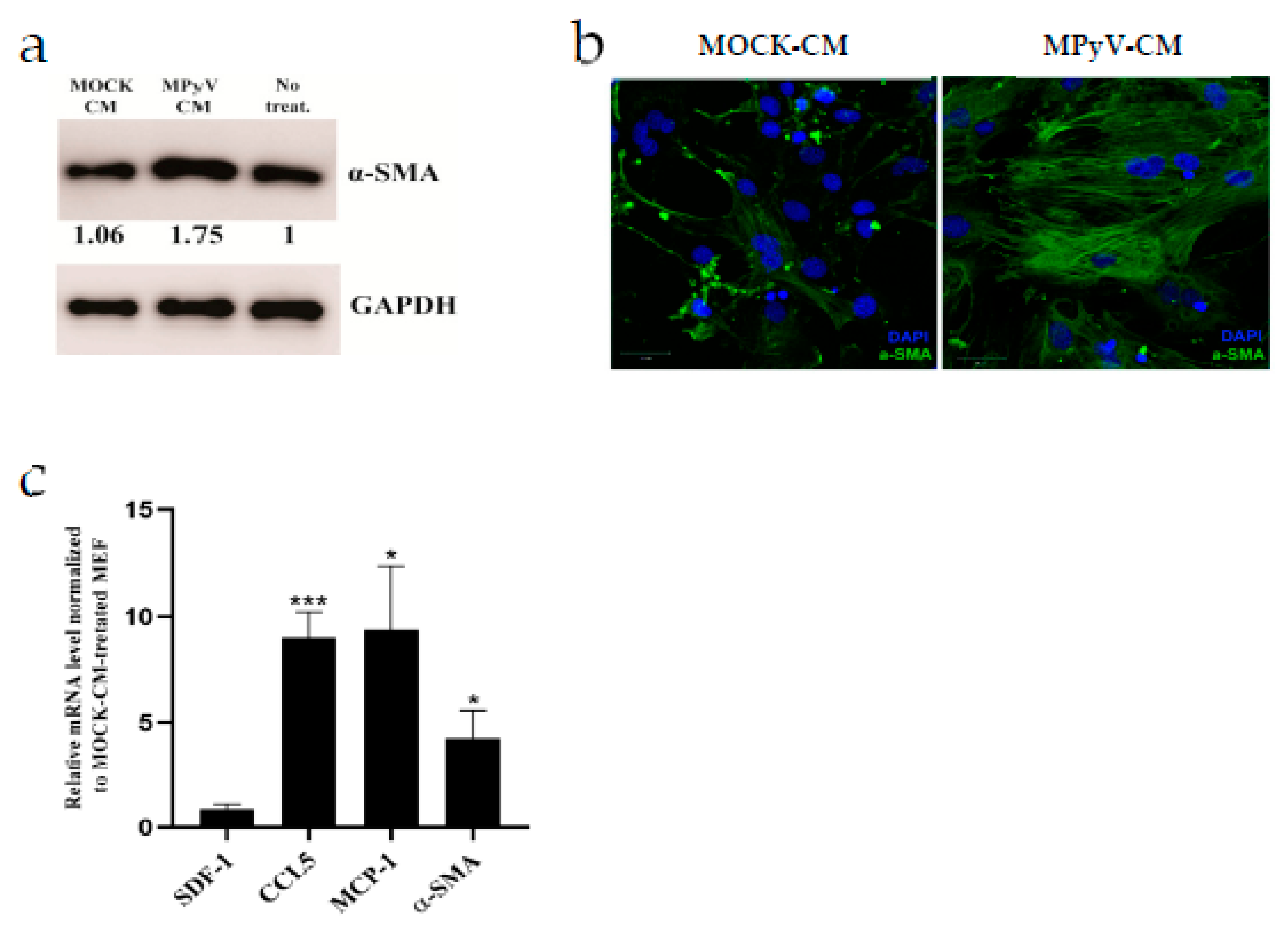
3.5. MPyV Infection Induces Cytokine Environment That Changes MEF Phenotype
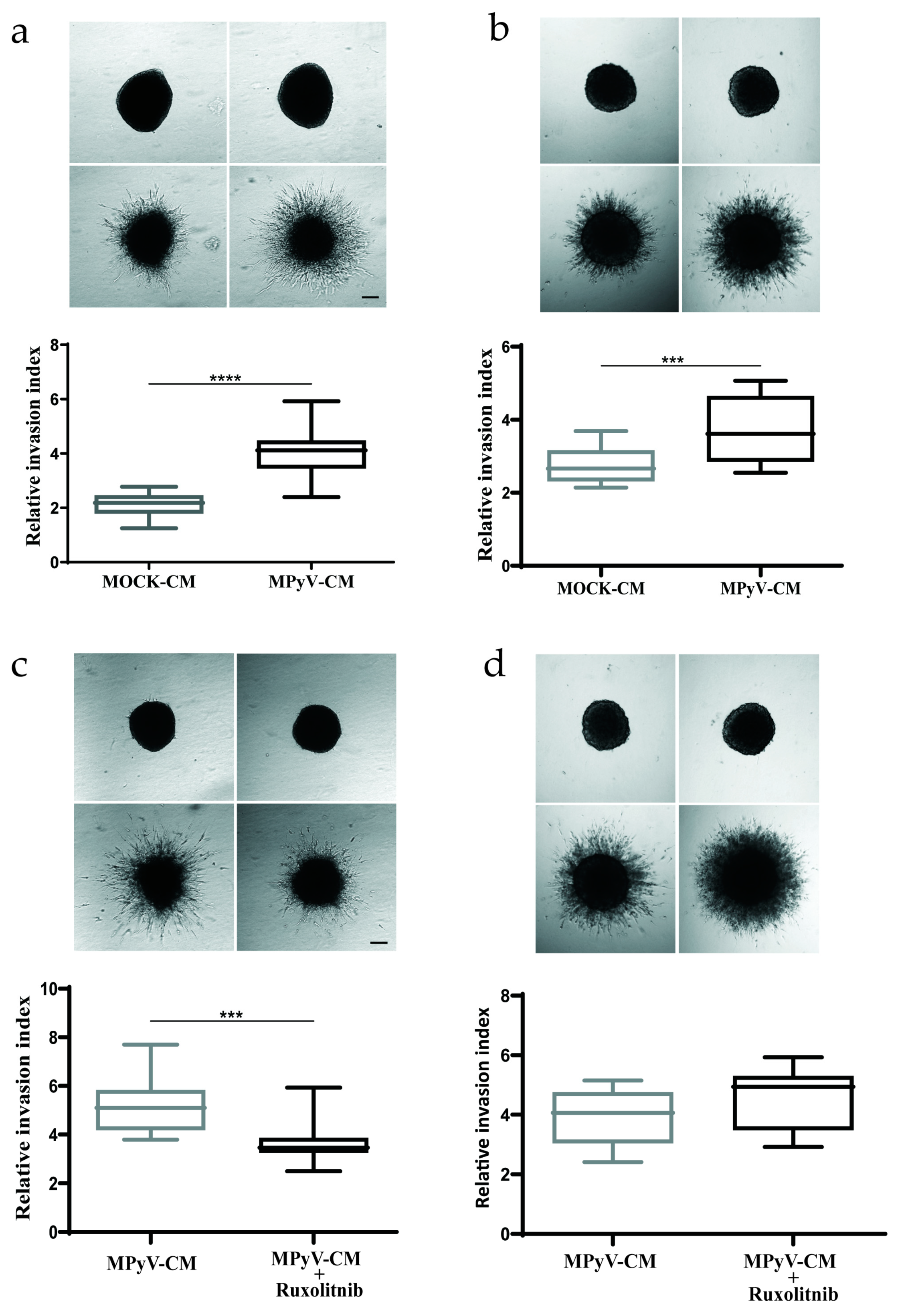
3.6. MPyV Infection in Fibroblasts Establishes Cytokine Environment That Supports Cell Invasiveness
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, W.; Langhoff, E. Polyomavirus in human cancer development. Adv. Exp. Med. Biol. 2006, 577, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Gheit, T.; Dutta, S.; Oliver, J.; Robitaille, A.; Hampras, S.; Combes, J.D.; McKay-Chopin, S.; Le Calvez-Kelm, F.; Fenske, N.; Cherpelis, B.; et al. Isolation and characterization of a novel putative human polyomavirus. Virology 2017, 506, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Cook, L. Polyomaviruses. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Yang, R.; Payne, A.S.; Schowalter, R.M.; Spurgeon, M.E.; Lambert, P.F.; Xu, X.; Buck, C.B.; You, J. Identifying the Target Cells and Mechanisms of Merkel Cell Polyomavirus Infection. Cell Host. Microbe 2016, 19, 775–787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, T.L. Polyoma virus: Old findings and new challenges. Virology 2001, 289, 167–173. [Google Scholar] [CrossRef] [Green Version]
- Fluck, M.M.; Schaffhausen, B.S. Lessons in Signaling and Tumorigenesis from Polyomavirus Middle T Antigen. Microbiol. Mol. Biol. Rev. 2009, 73, 542–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, K.A.; Villarreal, L.P. Natural Biology of Polyomavirus Middle T Antigen. Microbiol. Mol. Biol. Rev. 2001, 65, 288–318. [Google Scholar] [CrossRef] [Green Version]
- Zhou, A.Y.; Ichaso, N.; Adamarek, A.; Zila, V.; Forstova, J.; Dibb, N.J.; Dilworth, S.M. Polyomavirus Middle T-Antigen Is a Transmembrane Protein That Binds Signaling Proteins in Discrete Subcellular Membrane Sites. J. Virol. 2011, 85, 3046–3054. [Google Scholar] [CrossRef] [Green Version]
- Courtneidge, S.A. Activation of the pp60c-src kinase by middle T antigen binding or by dephosphorylation. EMBO J. 1985, 4, 1471–1477. [Google Scholar] [CrossRef]
- Dunant, N.M.; Senften, M.; Ballmer-Hofer, K. Polyomavirus middle-T antigen associates with the kinase domain of Src-related tyrosine kinases. J. Virol. 1996, 70, 1323–1330. [Google Scholar] [CrossRef] [Green Version]
- Utermark, T.; Schaffhausen, B.S.; Roberts, T.M.; Zhao, J.J. The p110α Isoform of Phosphatidylinositol 3-Kinase Is Essential for Polyomavirus Middle T Antigen-Mediated Transformation. J. Virol. 2007, 81, 7069–7076. [Google Scholar] [CrossRef] [Green Version]
- Su, W.; Liu, W.; Schaffhausen, B.S.; Roberts, T.M. Association of Polyomavirus Middle Tumor Antigen with Phospholipase C-γ1. J. Biol. Chem. 1995, 270, 12331–12334. [Google Scholar] [CrossRef] [Green Version]
- Meili, R.; Cron, P.; Hemmings, B.A.; Ballmer-Hofer, K. Protein kinase B/Akt is activated by polyomavirus middle-T antigen via a phosphatidylinositol 3-kinase-dependent mechanism. Oncogene 1998, 16, 903–907. [Google Scholar] [CrossRef] [Green Version]
- Freund, R.; Sotnikov, A.; Bronson, R.T.; Benjamin, T.L. Polyoma virus middle T is essential for virus replication and persistence as well as for tumor induction in mice. Virology 1992, 191, 716–723. [Google Scholar] [CrossRef]
- Swanson, P.A.; Lukacher, A.E.; Szomolanyi-Tsuda, E. Immunity to polyomavirus infection: The polyomavirus-mouse model. Semin. Cancer Biol. 2009, 19, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Palm, N.W.; Medzhitov, R. Pattern recognition receptors and control of adaptive immunity. Immunol. Rev. 2009, 227, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velupillai, P.; Sung, C.K.; Andrews, E.; Moran, J.; Beier, D.; Kagan, J.; Benjamin, T. Polymorphisms in Toll-Like Receptor 4 Underlie Susceptibility to Tumor Induction by the Mouse Polyomavirus. J. Virol. 2012, 86, 11541–11547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velupillai, P.; Garcea, R.L.; Benjamin, T.L. Polyoma virus-like particles elicit polarized cytokine responses in APCs from tumor-susceptible and -resistant mice. J. Immunol. 2006, 176, 1148–1153. [Google Scholar] [CrossRef]
- Kondo, T.; Ohshima, T. The dynamics of inflammatory cytokines in the healing process of mouse skin wound: A preliminary study for possible wound age determination. Int. J. Leg. Med. 1996, 108, 231–236. [Google Scholar] [CrossRef]
- Abend, J.R.; Imperiale, M.J. Transforming growth factor-beta-mediated regulation of BK virus gene expression. Virology 2008, 378, 6–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wollebo, H.S.; Safak, M.; del Valle, L.; Khalili, K.; White, M.K. Role for tumor necrosis factor-α in JC virus reactivation and progressive multifocal leukoencephalopathy. J. Neuroimmunol. 2011, 233, 46–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, F.D.; Barnaba, V. Persisting viruses and chronic inflammation: Understanding their relation to autoimmunity. Immunol. Rev. 1998, 164, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Yao, M.; Brummer, G.; Acevedo, D.; Cheng, N. Cytokine Regulation of Metastasis and Tumorigenicity. Adv. Cancer Res. 2016, 132, 265–367. [Google Scholar] [CrossRef]
- Multhoff, G.; Molls, M.; Radons, J. Chronic Inflammation in Cancer Development. Front. Immunol. 2012, 2, 98. [Google Scholar] [CrossRef] [Green Version]
- Bromberg, J.; Wang, T.C. Inflammation and Cancer: IL-6 and STAT3 Complete the Link. Cancer Cell 2009, 15, 79–80. [Google Scholar] [CrossRef] [Green Version]
- Jobe, N.P.; Rösel, D.; Dvořánková, B.; Kodet, O.; Lacina, L.; Mateu, R.; Smetana, K.; Brábek, J. Simultaneous blocking of IL-6 and IL-8 is sufficient to fully inhibit CAF-induced human melanoma cell invasiveness. Histochem. Cell Biol. 2016, 146, 205–217. [Google Scholar] [CrossRef]
- Cheteh, E.H.; Sarne, V.; Ceder, S.; Bianchi, J.; Augsten, M.; Rundqvist, H.; Egevad, L.; Östman, A.; Wiman, K.G. Interleukin-6 derived from cancer-associated fibroblasts attenuates the p53 response to doxorubicin in prostate cancer cells. Cell Death Discov. 2020, 6, 1–14. [Google Scholar] [CrossRef]
- Giraudo, E.; Arese, M.; Toniatti, C.; Strasly, M.; Primo, L.; Mantovani, A.; Ciliberto, G.; Bussolini, F. IL-6 is an in vitro and in vivo autocrine growth factor for middle T antigen-transformed endothelial cells. J. Immunol. 1996, 157, 2618–2623. [Google Scholar]
- Bussolino, F.; De Rossi, M.; Sica, A.; Colotta, F.; Wang, J.M.; Bocchietto, E.; Padura, I.M.; Bosia, A.; Dejana, E.; Mantovani, A. Murine endothelioma cell lines transformed by polyoma middle T oncogene as target for and producers of cytokines. J. Immunol. 1991, 147, 2122–2129. [Google Scholar]
- Horníková, L.; Fraiberk, M.; Man, P.; Janovec, V.; Forstová, J. VP1, the major capsid protein of the mouse polyomavirus, binds microtubules, promotes their acetylation and blocks the host cell cycle. FEBS J. 2017, 284, 301–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liebl, D.; Difato, F.; Horníková, L.; Mannová, P.; Stokrová, J.; Forstová, J. Mouse polyomavirus enters early endosomes, requires their acidic pH for productive infection, and meets transferrin cargo in Rab11-positive endosomes. J. Virol. 2006, 80, 4610–4622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zila, V.; Difato, F.; Klimova, L.; Huerfano, S.; Forstova, J. Involvement of microtubular network and its motors in productive endocytic trafficking of mouse polyomavirus. PLoS ONE 2014, 9, e96922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manders, E.M.; Stap, J.; Brakenhoff, G.J.; van Driel, R.; Aten, J.A. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 1992, 103 Pt 3, 857–862. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Bolte, S.; Cordelières, F.P. A guided tour into subcellular colocalization analysis in light microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Lucifora, J.; Bonnin, M.; Aillot, L.; Fusil, F.; Maadadi, S.; Dimier, L.; Michelet, M.; Floriot, O.; Ollivier, A.; Rivoire, M.; et al. Direct antiviral properties of TLR ligands against HBV replication in immune-competent hepatocytes. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abend, J.R.; Low, J.A.; Imperiale, M.J. Inhibitory Effect of Gamma Interferon on BK Virus Gene Expression and Replication. J. Virol. 2007, 81, 272–279. [Google Scholar] [CrossRef] [Green Version]
- Wilson, J.J.; Lin, E.; Pack, C.D.; Frost, E.L.; Hadley, A.; Swimm, A.I.; Wang, J.; Dong, Y.; Breeden, C.P.; Kalman, D.; et al. Gamma interferon controls mouse polyomavirus infection in vivo. J. Virol. 2011, 85, 10126–10134. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jiang, B.; Huo, Y.; Primo, L.; Dahl, J.S.; Benjamin, T.L.; Luo, J. Shp2 suppresses PyMT-induced transformation in mouse fibroblasts by inhibiting Stat3 activity. Virology 2011, 409, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; van Boxel-Dezaire, A.H.H.; Cheon, H.; Yang, J.; Stark, G.R. STAT3 activation in response to IL-6 is prolonged by the binding of IL-6 receptor to EGF receptor. Proc. Natl. Acad. Sci. USA 2013, 110, 16975–16980. [Google Scholar] [CrossRef] [Green Version]
- Im, K.; Baek, J.; Kwon, W.S.; Rha, S.Y.; Hwang, K.W.; Kim, U.; Min, H. The Comparison of Exosome and Exosomal Cytokines between Young and Old Individuals with or without Gastric Cancer. Int. J. Gerontol. 2018, 12, 233–238. [Google Scholar] [CrossRef]
- Gao, K.; Jin, J.; Huang, C.; Li, J.; Luo, H.; Li, L.; Huang, Y.; Jiang, Y. Exosomes Derived From Septic Mouse Serum Modulate Immune Responses via Exosome-Associated Cytokines. Front. Immunol. 2019, 10, 1560. [Google Scholar] [CrossRef] [Green Version]
- Öhlund, D.; Handly-Santana, A.; Biffi, G.; Elyada, E.; Almeida, A.S.; Ponz-Sarvise, M.; Corbo, V.; Oni, T.E.; Hearn, S.A.; Lee, E.J.; et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J. Exp. Med. 2017, 214, 579–596. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erez, N.; Truitt, M.; Olson, P.; Hanahan, D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-κB-Dependent Manner. Cancer Cell 2010, 17, 135–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, B.; Henriques, A.C.; Silva, P.M.A.; Bousbaa, H. Three-Dimensional Spheroids as In Vitro Preclinical Models for Cancer Research. Pharmaceutics 2020, 12, 1186. [Google Scholar] [CrossRef] [PubMed]
- Vaure, C.; Liu, Y. A Comparative Review of Toll-Like Receptor 4 Expression and Functionality in Different Animal Species. Front. Immunol. 2014, 5, 316. [Google Scholar] [CrossRef] [Green Version]
- Bouřa, E.; Liebl, D.; Špíšek, R.; Frič, J.; Marek, M.; Štokrová, J.; Holáň, V.; Forstova, J. Polyomavirus EGFP-pseudocapsids: Analysis of model particles for introduction of proteins and peptides into mammalian cells. FEBS Lett. 2005, 579, 6549–6558. [Google Scholar] [CrossRef] [Green Version]
- Husebye, H.; Aune, M.H.; Stenvik, J.; Samstad, E.; Skjeldal, F.; Halaas, Ø.; Nilsen, N.J.; Stenmark, H.; Latz, E.; Lien, E.; et al. The Rab11a GTPase Controls Toll-like Receptor 4-Induced Activation of Interferon Regulatory Factor-3 on Phagosomes. Immunity 2010, 33, 583–596. [Google Scholar] [CrossRef] [Green Version]
- Szatmári, Z.; Kis, V.; Lippai, M.; Hegedűs, K.; Faragó, T.; Lőrincz, P.; Tanaka, T.; Juhász, G.; Sass, M. Rab11 facilitates cross-talk between autophagy and endosomal pathway through regulation of Hook localization. Mol. Biol Cell 2014, 25, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Yavuz, B.G.; Gunaydin, G.; Gedik, M.E.; Kosemehmetoglu, K.; Karakoc, D.; Ozgur, F.; Guc, D. Cancer associated fibroblasts sculpt tumour microenvironment by recruiting monocytes and inducing immunosuppressive PD-1 + TAMs. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef]
- Aldinucci, D.; Colombatti, A. The inflammatory chemokine CCL5 and cancer progression. Mediat. Inflamm 2014, 2014, 292376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, C.; Cheng, X.; Lu, B.; Yang, G. Activation of interleukin-6/signal transducer and activator of transcription 3 by human papillomavirus early proteins 6 induces fibroblast senescence to promote cervical tumourigenesis through autocrine and paracrine pathways in tumour microenvironment. Eur. J. Cancer 2013, 49, 3889–3899. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.L.; Macdonald, A. Autocrine STAT3 activation in HPV positive cervical cancer through a virus-driven Rac1—NFκB—IL-6 signalling axis. PLoS Pathog. 2019, 15, e1007835. [Google Scholar] [CrossRef] [Green Version]
- Taraboletti, G.; Belotti, D.; Dejana, E.; Mantovani, A.; Giavazzi, R. Endothelial cell migration and invasiveness are induced by a soluble factor produced by murine endothelioma cells transformed by polyoma virus middle T oncogene. Cancer Res. 1993, 53, 3812–3816. [Google Scholar]
- Yang, H.; Wang, B.; Wang, T.; Xu, L.; He, C.; Wen, H.; Yan, J.; Su, H.; Zhu, X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PLoS ONE 2014, 9, e109980. [Google Scholar] [CrossRef]
- Ou, T.; Lilly, M.; Jiang, W. The Pathologic Role of Toll-Like Receptor 4 in Prostate Cancer. Front. Immunol. 2018, 9, 1188. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Janovec, V.; Ryabchenko, B.; Škarková, A.; Pokorná, K.; Rösel, D.; Brábek, J.; Weber, J.; Forstová, J.; Hirsch, I.; Huérfano, S. TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness. Cancers 2021, 13, 2076. https://doi.org/10.3390/cancers13092076
Janovec V, Ryabchenko B, Škarková A, Pokorná K, Rösel D, Brábek J, Weber J, Forstová J, Hirsch I, Huérfano S. TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness. Cancers. 2021; 13(9):2076. https://doi.org/10.3390/cancers13092076
Chicago/Turabian StyleJanovec, Vaclav, Boris Ryabchenko, Aneta Škarková, Karolína Pokorná, Daniel Rösel, Jan Brábek, Jan Weber, Jitka Forstová, Ivan Hirsch, and Sandra Huérfano. 2021. "TLR4-Mediated Recognition of Mouse Polyomavirus Promotes Cancer-Associated Fibroblast-Like Phenotype and Cell Invasiveness" Cancers 13, no. 9: 2076. https://doi.org/10.3390/cancers13092076