
Physicochemical Characterization of the Catalytic Unit of Hammerhead Ribozyme and Its Relationship with the Catalytic Activity

 , , and
, , and 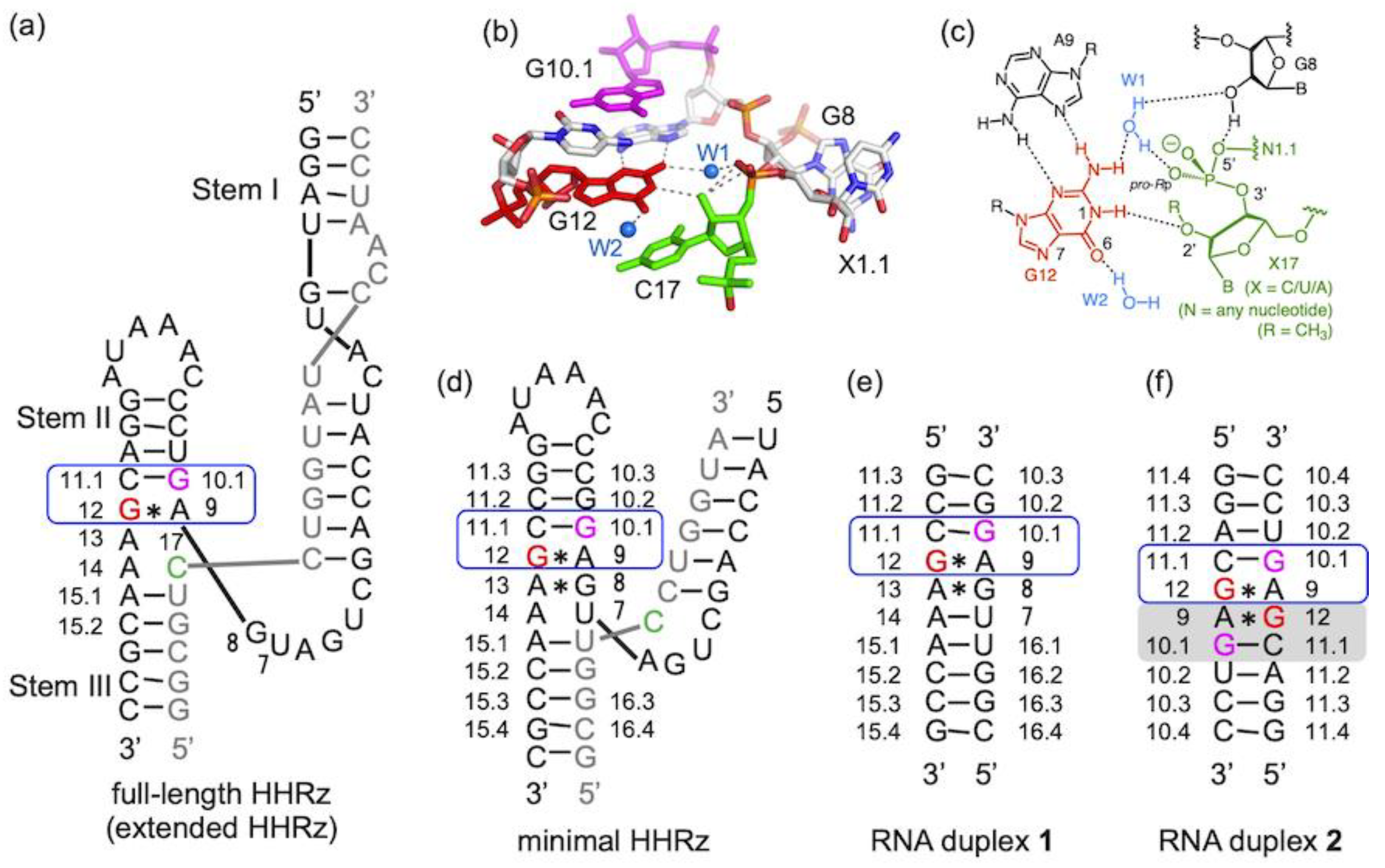{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
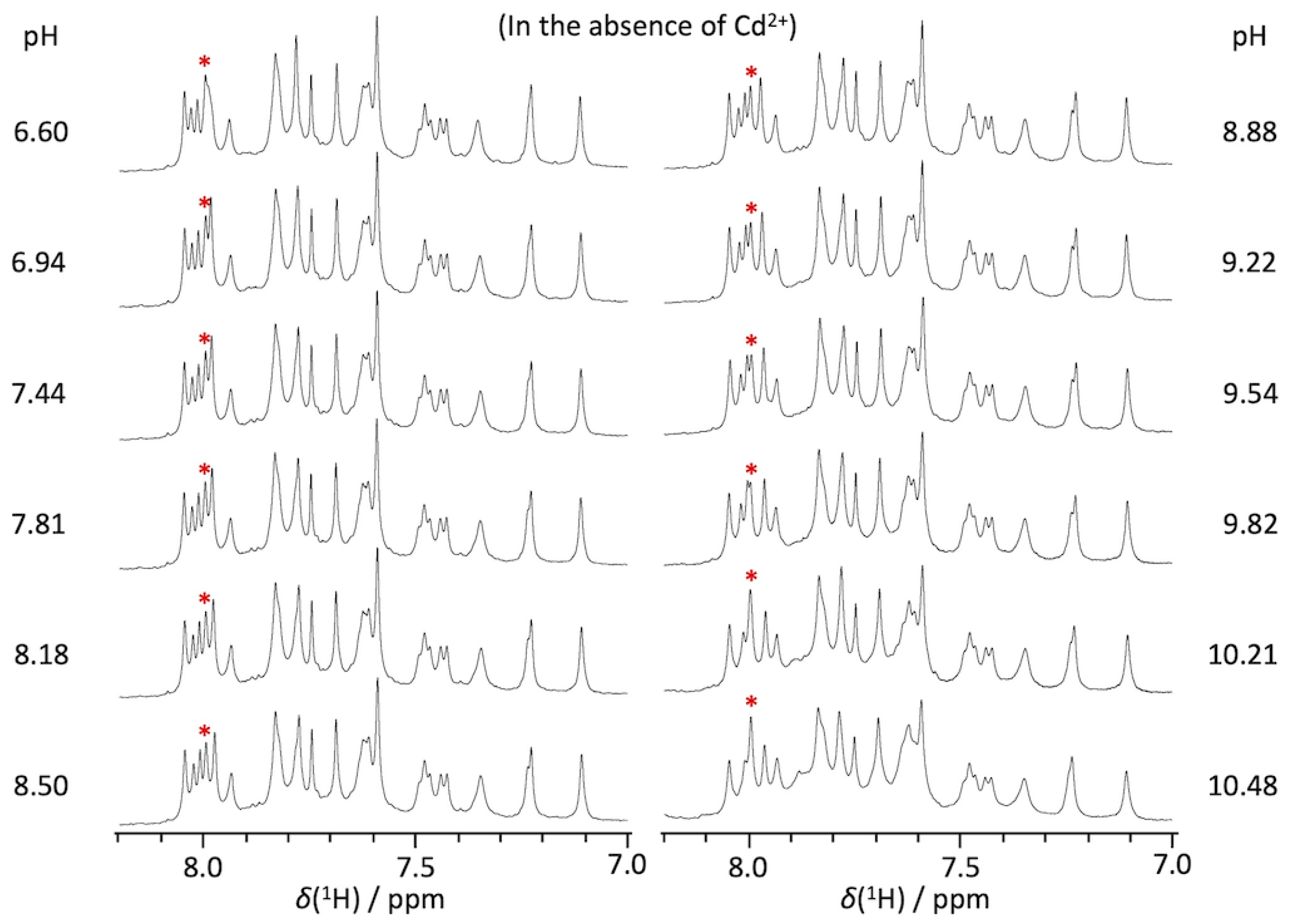
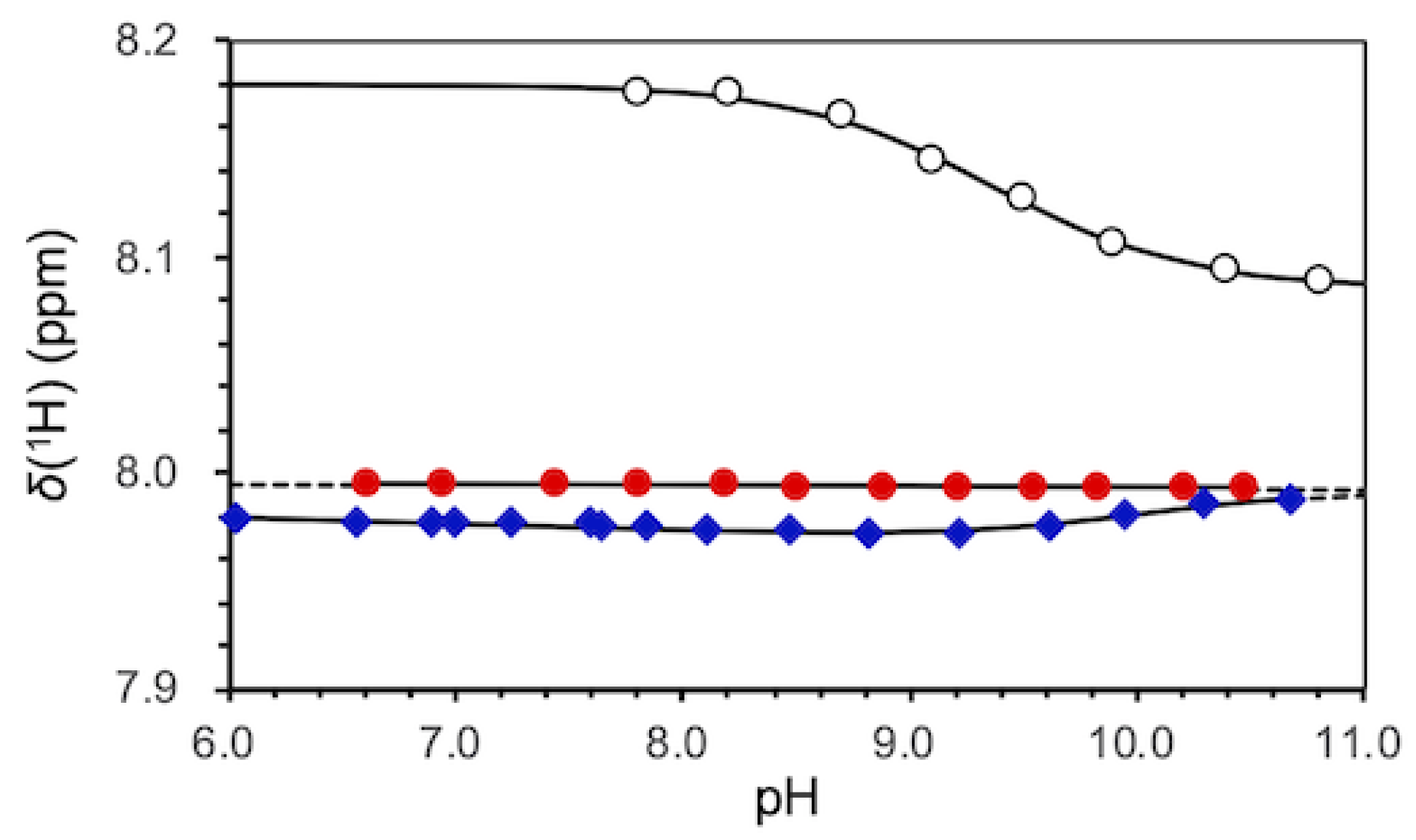
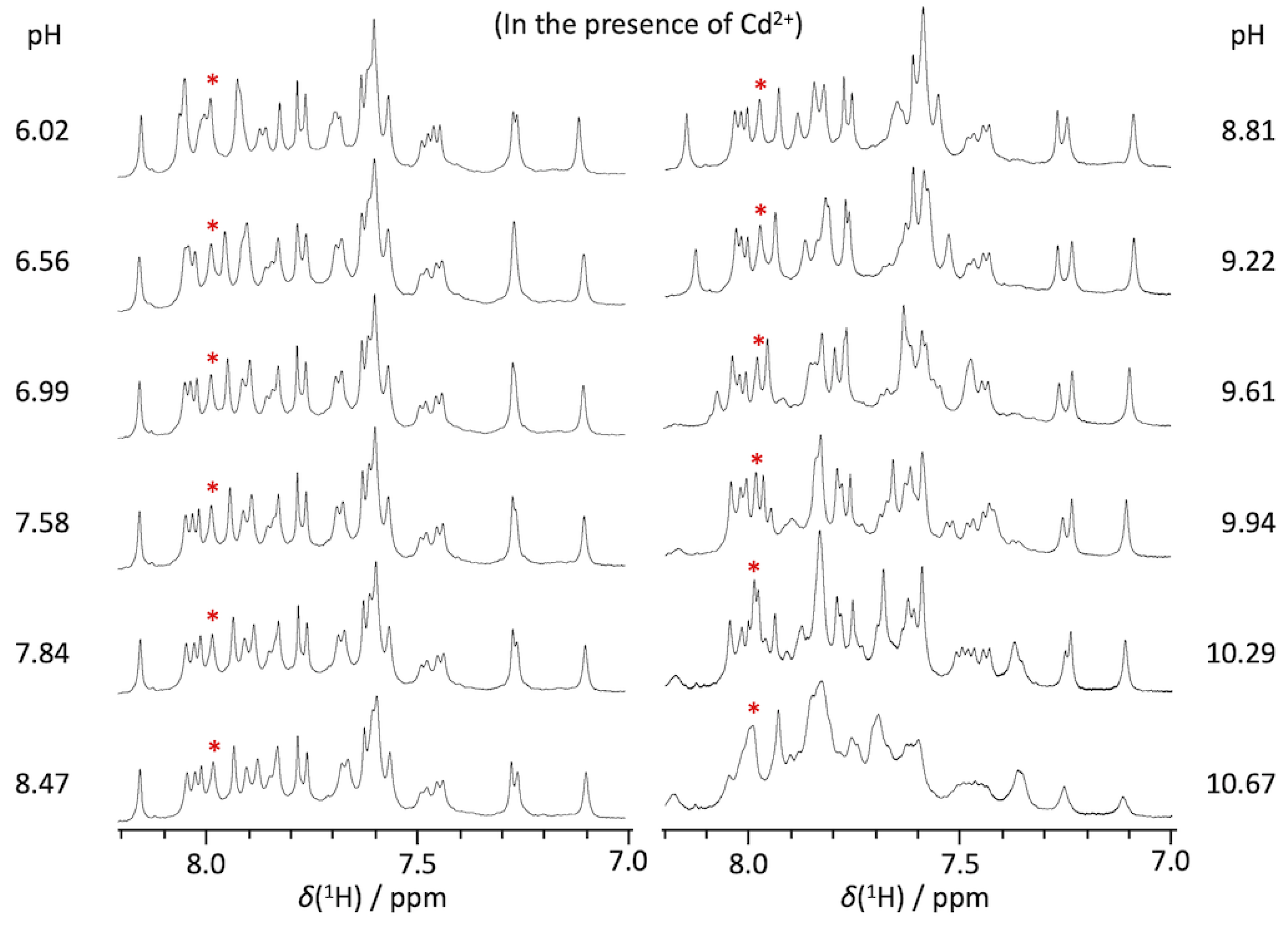
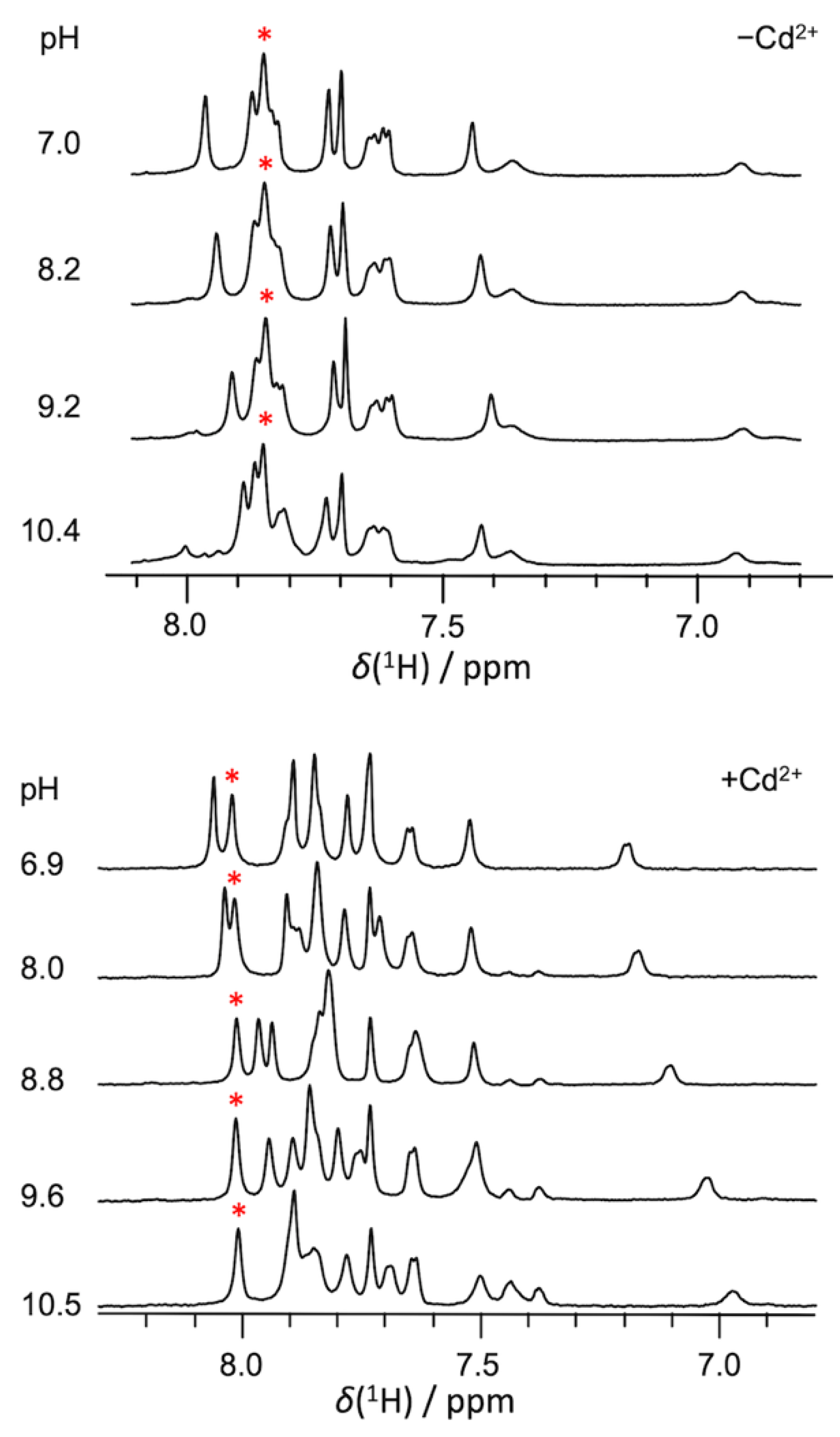
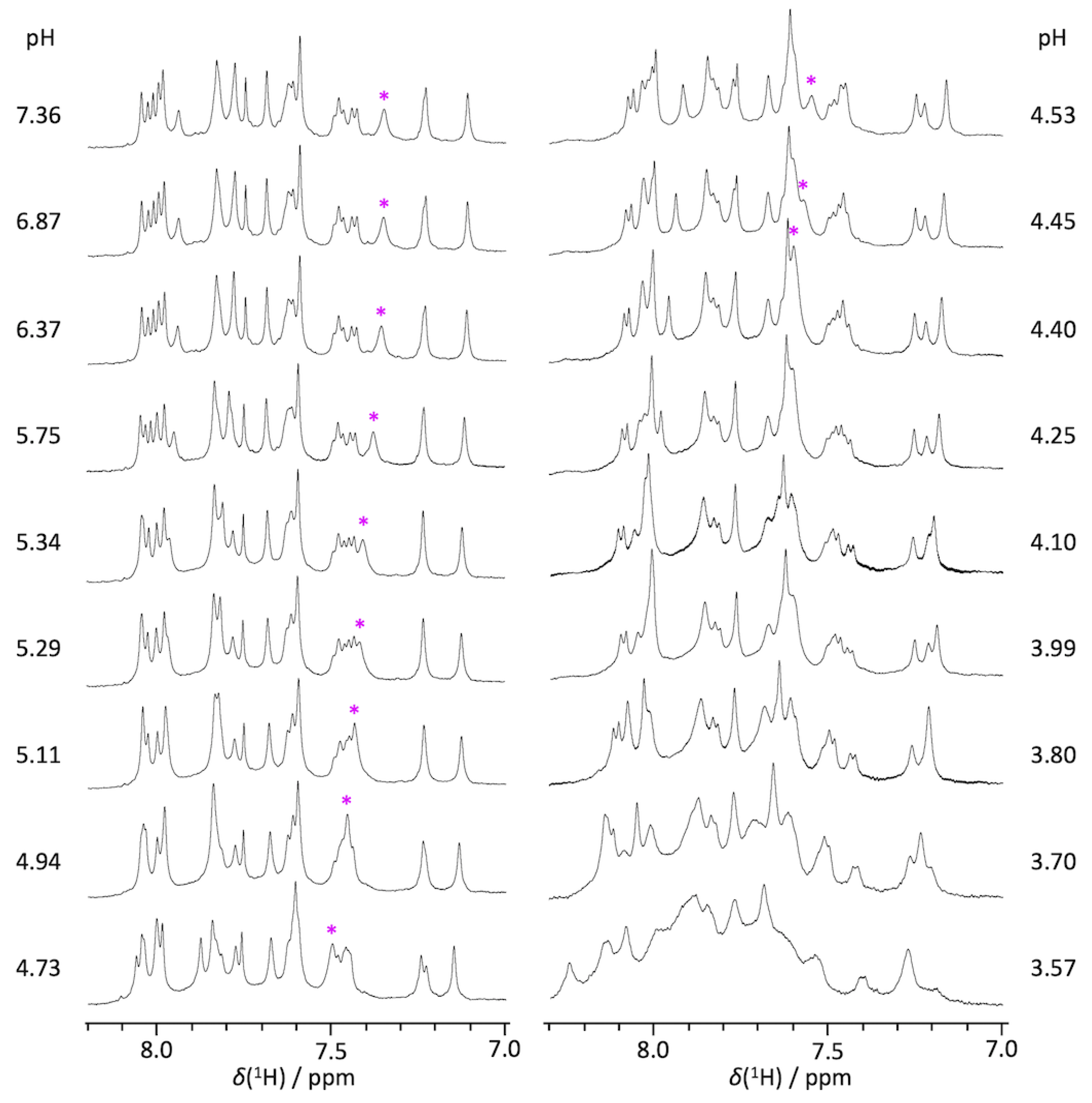
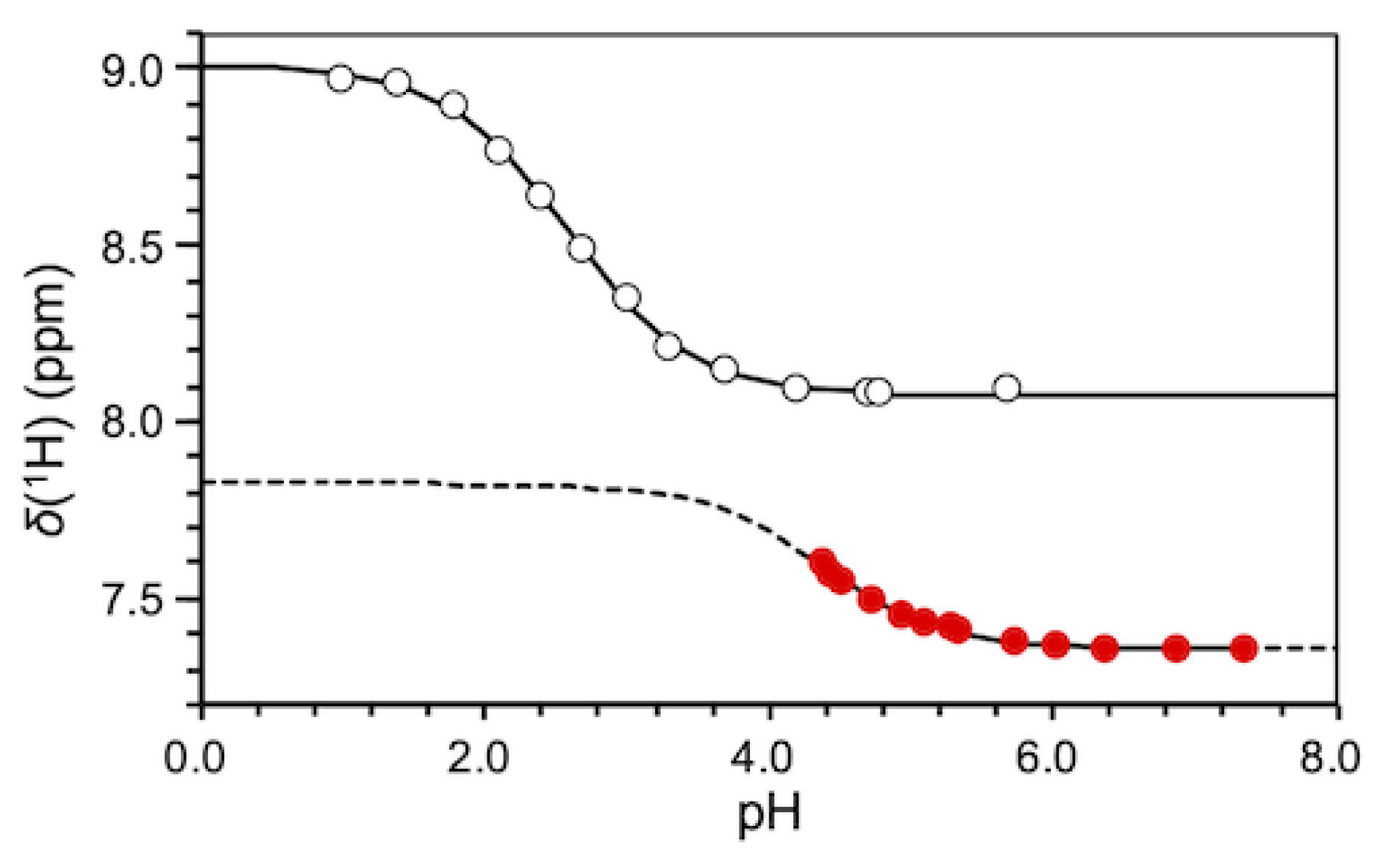
2.1. pKa Determination of G12-Corresponding Residue
2.2. pKa Determination of G10.1-Corresponding Residue
3. Discussion
3.1. Experimental pKa Values of Functionally Important Residues
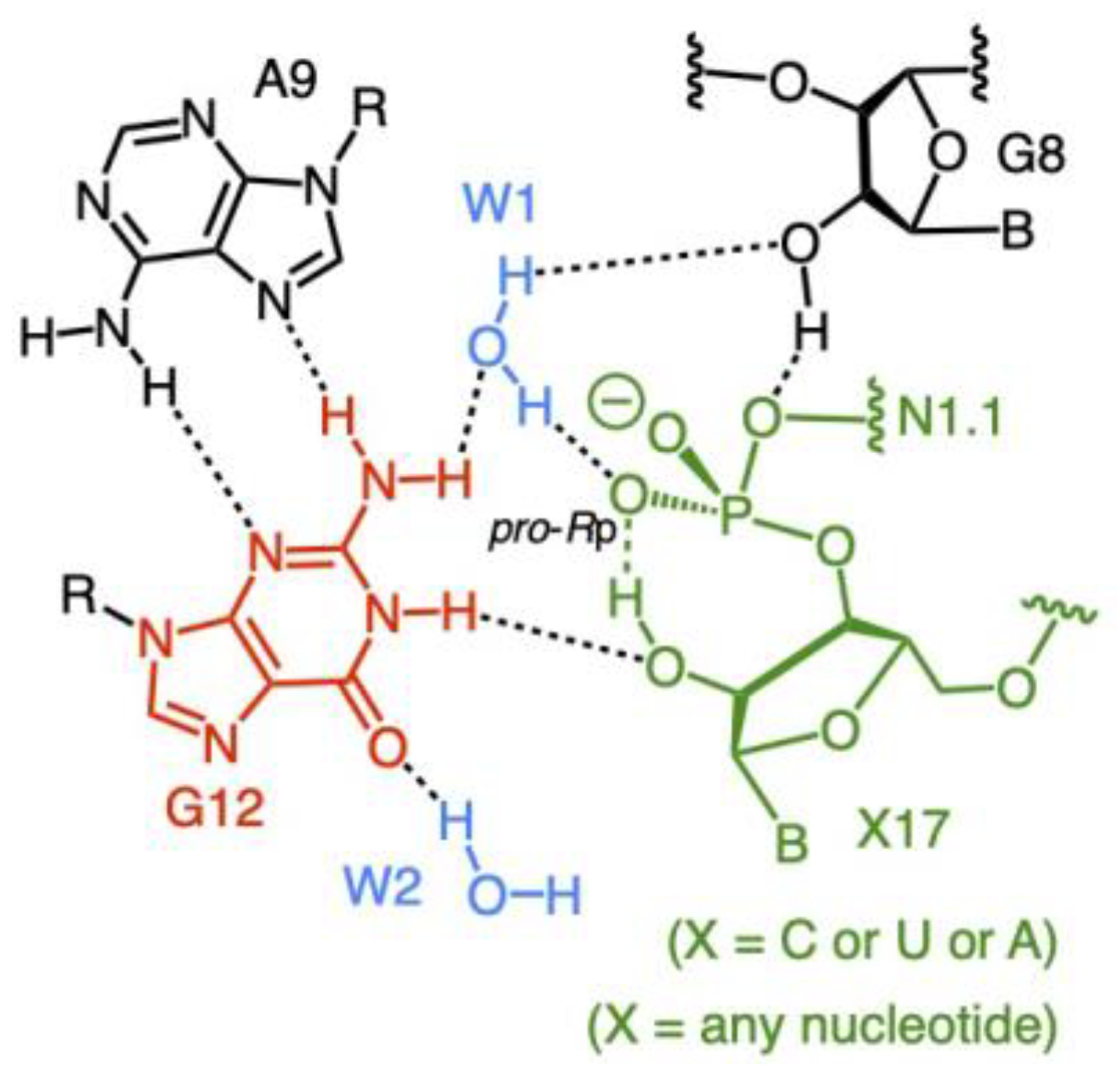
3.2. Ground State Structure Inferred from the Crystal Structure
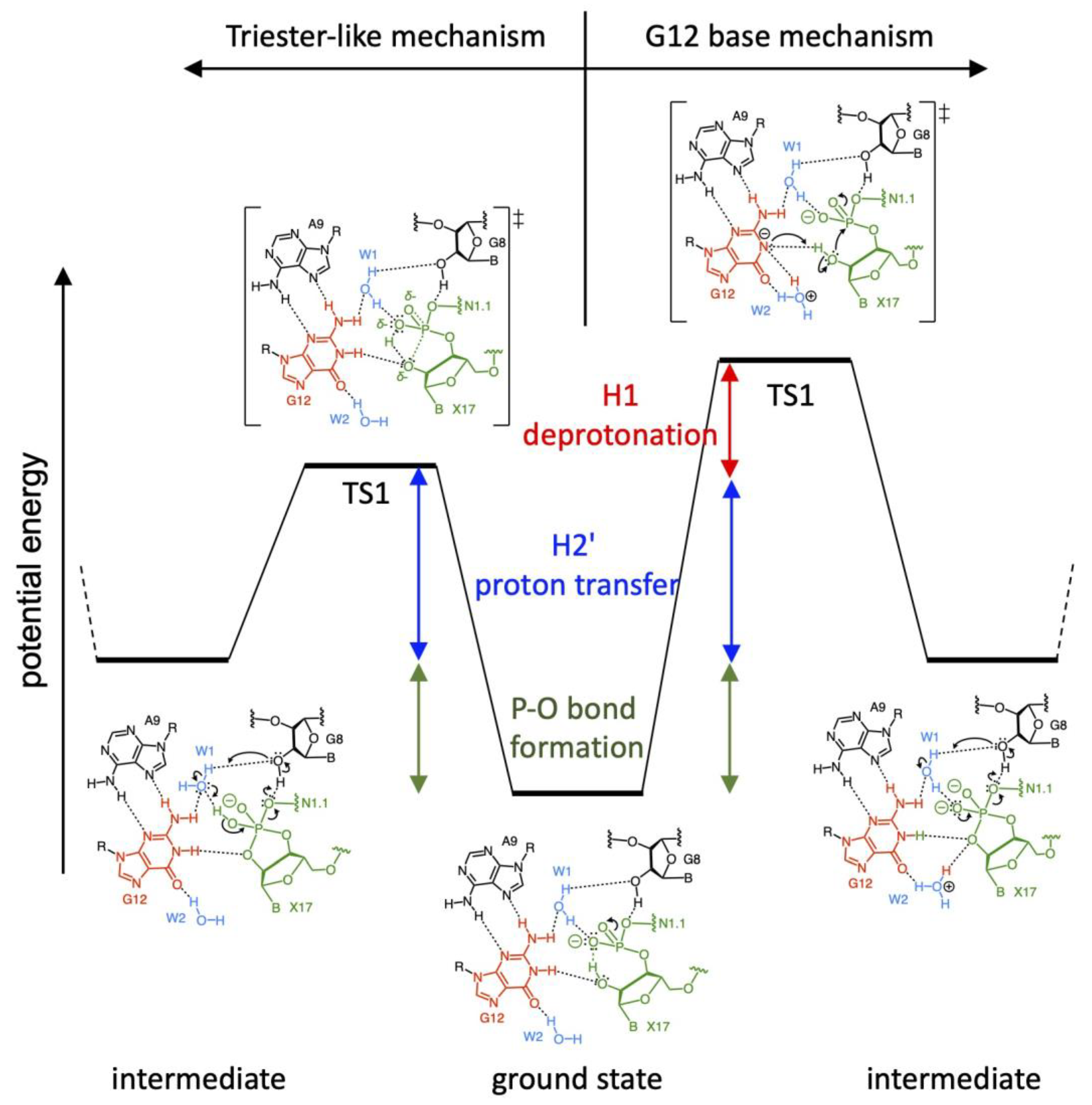
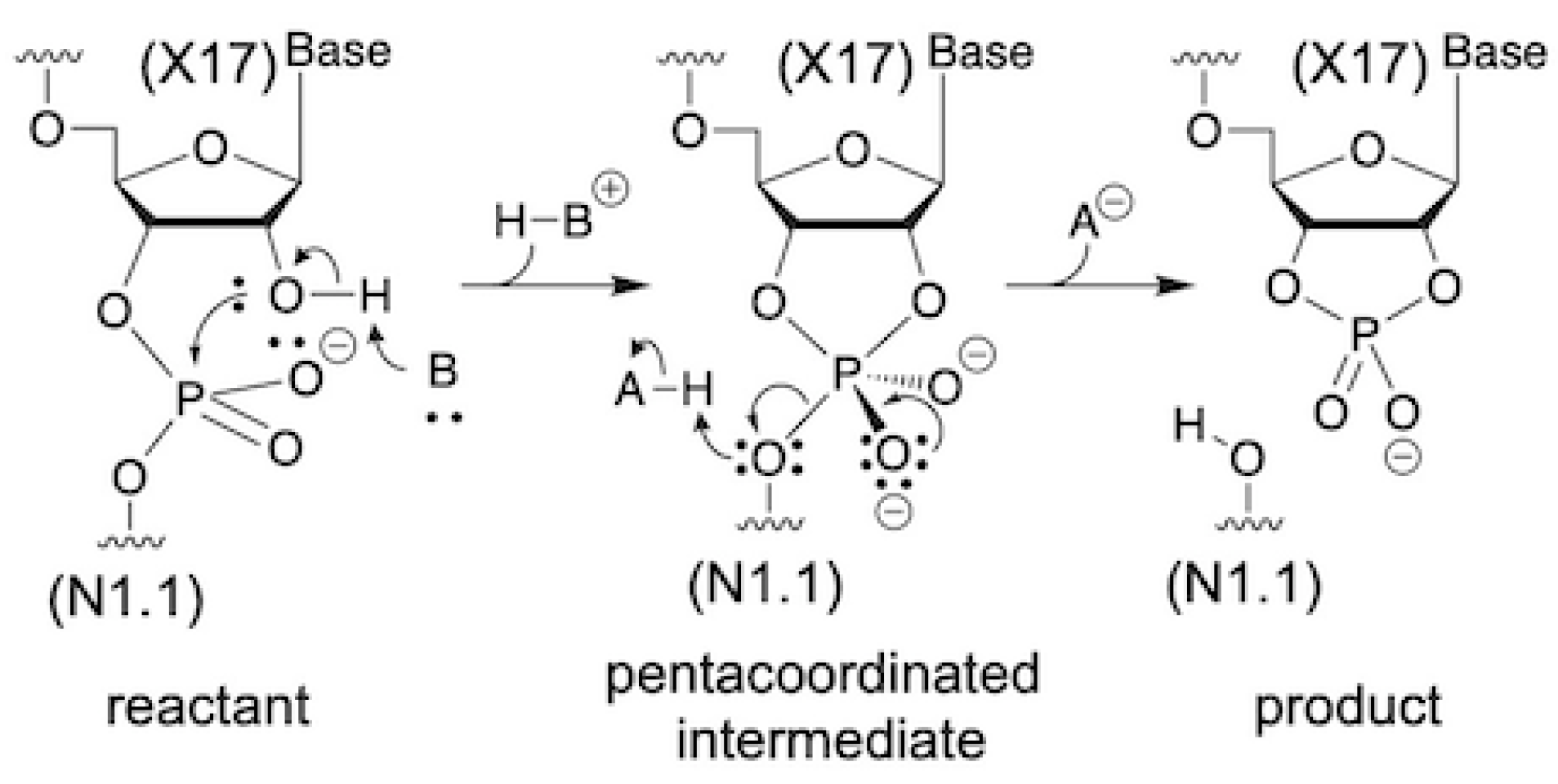
3.3. Two Major Possible Mechanisms
3.4. Correlation of Possible Reaction Mechanisms with Kinetic Data
3.5. Correlation of Possible Reaction Mechanisms with Thio-Effect
3.6. Correlation of Possible Reaction Mechanisms with Their Intermediate and Product States
3.7. Possible Reaction Mechanisms
3.8. Conclusions
4. Materials and Methods
4.1. Preparations of RNA Oligomers and NMR Measurements
4.2. Equilibrium Analyses for Titration Data
4.3. Theoretical Calculations of NMR Chemical Perturbations
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cech, T.R.; Zhang, A.J.; Grabowski, P.J. In vitro splicing of the ribosomal RNA precursor of Tetrahymena: Involvement of a guanosine nucleotide in the excision of the intervening sequence. Cell 1981, 27, 487–496. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Prody, G.A.; Bakos, J.T.; Buzayan, J.M.; Schneider, I.R.; Bruening, G. Autolytic processing of dimeric plant virus satellite RNA. Science 1986, 231, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, C.J.; Rathjen, P.D.; Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNA transcripts of Avocado sunblotch viroid. Nucleic Acids Res. 1986, 14, 3627–3640. [Google Scholar] [CrossRef]
- Epstein, L.M.; Gall, J.G. Self-cleaving transcripts of satellite DNA from the newt. Cell 1987, 48, 535–543. [Google Scholar] [CrossRef]
- Forster, A.C.; Symons, R.H. Self-cleavage of plus and minus RNAs of a virusoid and a structural model for the activate site. Cell 1987, 49, 211–220. [Google Scholar] [CrossRef]
- Uhlenbeck, O.C. A small catalytic oligonucleotide. Nature 1987, 328, 596–600. [Google Scholar] [CrossRef]
- Hertel, K.J.; Pardi, A.; Uhlenbeck, O.C.; Koizumi, M.; Ohtsuka, E.; Uesugi, S.; Cedergren, R.; Eckstein, F.; Gerlach, W.L.; Hodgson, R.; et al. Numbering system for the hammerhead. Nucleic Acids Res. 1992, 20, 3252. [Google Scholar] [CrossRef]
- PabonPena, L.M.; Yi, Z.; Epstein, L.M. Newt satellite-2 transcripts self-cleavage by using an extended hammerhead structure. Mol. Cell. Biol. 1991, 11, 6109–6115. [Google Scholar]
- Garrett, T.A.; PabonPena, L.M.; Gokaldas, N.; Epstein, L.M. Novel requirements in peripheral structures of the extended satellite 2 hammerhead. RNA 1996, 2, 699–706. [Google Scholar]
- Zhang, Y.; Epstein, L.M. Cloning and characterization of extended hammerheads from a diverse set of caudate amphibians. Gene 1996, 172, 183–190. [Google Scholar] [CrossRef]
- Jimenez, R.M.; Delwart, E.; Lupták, A. Structure-based Search Reveals Hammerhead Ribozymes in the Human Microbiome. J. Biol. Chem. 2011, 286, 7737–7743. [Google Scholar] [CrossRef] [Green Version]
- Perreault, J.; Weinberg, Z.; Roth, A.; Popescu, O.; Chartrand, P.; Ferbeyre, G.; Breaker, R.R. Identification of Hammerhead Ribozymes in All Domains of Life Reveals Novel Structural Variations. PLoS Comput. Biol. 2011, 7, 13. [Google Scholar] [CrossRef]
- Hammann, C.; Lupták, A.; Perreault, J.; de la Pena, M. The ubiquitous hammerhead ribozyme. RNA 2012, 18, 871–885. [Google Scholar] [CrossRef]
- Khvorova, A.; Lescoute, A.; Westhof, E.; Jayasena, S.D. Sequence elements outside the hammerhead ribozyme catalytic core enable intracellular activity. Nat. Struct. Biol. 2003, 10, 708–712. [Google Scholar] [CrossRef]
- Canny, M.D.; Jucker, F.M.; Kellogg, E.; Khvorova, A.; Jayasena, S.D.; Pardi, A. Fast cleavage kinetics of a natural hammerhead ribozyme. J. Am. Chem. Soc. 2004, 126, 10848–10849. [Google Scholar] [CrossRef]
- Osborne, E.M.; Schaak, J.E.; DeRose, V.J. Characterization of a native hammerhead ribozyme derived from schistosomes. RNA 2005, 11, 187–196. [Google Scholar] [CrossRef]
- Kuriyama, M.; Kondo, Y.; Tanaka, Y. Pseudoknot interaction-mediated activation of type I hammerhead ribozyme: A new class of gene-therapeutic agents. Nucleosides Nucleotides Nucleic Acids 2019, 33, 466–480. [Google Scholar] [CrossRef]
- Yamada, M.; Tanaka, Y. Structure-activity relationship of pseudoknot-type hammerhead ribozyme reveals key structural elements for enhanced catalytic activity. Nucleosides Nucleotides Nucleic Acids 2020, 39, 245–257. [Google Scholar] [CrossRef]
- Martick, M.; Scott, W.G. Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 2006, 126, 309–320. [Google Scholar] [CrossRef]
- Martick, M.; Lee, T.S.; York, D.M.; Scott, W.G. Solvent structure and hammerhead ribozyme catalysis. Chem. Biol. 2008, 15, 332–342. [Google Scholar] [CrossRef]
- Chi, Y.I.; Martick, M.; Lares, M.; Kim, R.; Scott, W.G.; Kim, S.H. Capturing hammerhead ribozyme structures in action by modulating general base catalysis. PLoS Biol. 2008, 6, e234. [Google Scholar] [CrossRef]
- Anderson, M.; Schultz, E.P.; Martick, M.; Scott, W.G. Active-site monovalent cations revealed in a 1.55-A-resolution hammerhead ribozyme structure. J. Mol. Biol. 2013, 425, 3790–3798. [Google Scholar] [CrossRef]
- Mir, A.; Chen, J.; Robinson, K.; Lendy, E.; Goodman, J.; Neau, D.; Golden, B.L. Two divalent metal ions and conformational changes play roles in the hammerhead ribozyme cleavage reaction. Biochemistry 2015, 54, 6369–6381. [Google Scholar] [CrossRef] [PubMed]
- Mir, A.; Golden, B.L. Two active site divalent ions in the crystal structure of the hammerhead ribozyme bound to a transition state analogue. Biochemistry 2016, 55, 633–636. [Google Scholar] [CrossRef]
- Kuimelis, R.G.; McLaughlin, L.W. Nucleic Acids and Molecular Biology; Eckstein, F., Lilley, D.M.J., Eds.; Springer: Berlin/Heidelberg, Germany, 1996; Volume 10, pp. 197–215. [Google Scholar]
- Han, J.; Burke, J.M. Model for General Acid-base catalysis by the hammerhead ribozyme: pH-activity relationships of G8 and G12 variants at the putative active site. Biochemistry 2005, 44, 7864–7870. [Google Scholar] [CrossRef]
- Nelson, J.A.; Uhlenbeck, O.C. Hammerhead redux: Does the new structure fit the old biochemical data? RNA 2008, 14, 605–615. [Google Scholar] [CrossRef]
- Nelson, J.A.; Uhlenbeck, O.C. Minimal and extended hammerheads utilize a similar dynamic reaction mechanism for catalysis. RNA 2008, 14, 43–54. [Google Scholar] [CrossRef]
- Thomas, J.M.; Perrin, D.M. Probing general base catalysis in the hammerhead ribozyme. J. Am. Chem. Soc. 2008, 130, 15467–15475. [Google Scholar] [CrossRef]
- Frankel, E.A.; Strulson, C.A.; Keating, C.D.; Bevilacqua, P.C. Cooperative interactions in the hammerhead ribozyme drive pKa shifting of G12 and its stacked base C17. Biochemistry 2017, 56, 2537–2548. [Google Scholar] [CrossRef]
- Lilley, D.M.J. Classification of the nucleolytic ribozymes based upon catalytic mechanism. F1000Research 2019, 8, 1462. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Yamanaka, D.; Morioka, S.; Yamaguchi, T.; Tomonari, H.; Kojima, C.; Tanaka, Y. NMR spectroscopic characterization of a model RNA duplex reflecting the core sequence of hammerhead ribozymes. Nucleosides Nucleotides Nucleic Acids 2018, 37, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Pley, H.W.; Flaherty, K.M.; McKay, D.B. Three-dimensional structure of a hammerhead ribozyme. Nature 1994, 372, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.G.; Murray, J.B.; Arnold, J.R.; Stoddard, B.L.; Klug, A. Capturing the structure of a catalytic RNA intermediate: The hammerhead ribozyme. Science 1996, 274, 2065–2069. [Google Scholar] [CrossRef]
- Baeyens, K.J.; De Bondt, H.L.; Pardi, A.; Holbrook, S.R. A curved RNA helix incorporating an internal loop with G·A and A·A non-Watson–Crick base pairing. Proc. Natl. Acad. Sci. USA 1996, 93, 12851–12855. [Google Scholar] [CrossRef]
- Tanaka, Y.; Morita, E.H.; Hayashi, H.; Kasai, Y.; Tanaka, T.; Taira, K. Well-conserved tandem G-A pairs and the flanking C-G pair in hammerhead ribozymes are sufficient for capture of structurally and catalytically important metal ions. J. Am. Chem. Soc. 2000, 122, 11303–11310. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kojima, C.; Morita, E.H.; Kasai, Y.; Yamasaki, K.; Ono, A.; Kainosho, M.; Taira, K. Identification of the metal ion binding site on an RNA motif from hammerhead ribozyme using 15N NMR spectroscopy. J. Am. Chem. Soc. 2002, 124, 4595–4601. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kasai, Y.; Mochizuki, S.; Wakisaka, A.; Morita, E.H.; Kojima, C.; Toyozawa, A.; Kondo, Y.; Taki, M.; Takagi, Y.; et al. Nature of the chemical bond formed with the structural metal ion at the A9/G10.1 motif derived from hammerhead ribozymes. J. Am. Chem. Soc. 2004, 126, 744–752. [Google Scholar] [CrossRef]
- Wang, G.; Gaffney, B.L.; Jones, R.A. Differential Binding of Mg2+, Zn2+, and Cd2+ at two sites in a hammerhead ribozyme motif, determined by 15N NMR. J. Am. Chem. Soc. 2004, 126, 8908–8909. [Google Scholar] [CrossRef]
- Liu, H.; Yu, X.; Chen, Y.; Zhang, J.; Wu, B.; Zheng, L.; Haruehanroengra, P.; Wang, R.; Li, S.; Lin, J.; et al. Crystal structure of an RNA-cleaving DNAzyme. Nat. Commun. 2017, 8, 2006. [Google Scholar] [CrossRef]
- Cepeda-Plaza, M.; McGhee, C.E.; Lu, Y. Evidence of a general acid-base catalysis mechanism in the 8-17 DNAzyme. Biochemistry 2018, 57, 1517–1522. [Google Scholar] [CrossRef] [PubMed]
- Velikyan, I.; Acharya, S.; Trifonova, A.; Földesi, A.; Chattopadhyaya, J. The pKa’s of 2′-hydroxyl group in nucleosides and nucleotides. J. Am. Chem. Soc. 2001, 123, 2893–2894. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Acharya, P.; Földesi, A.; Chattopadhyaya, J. Cross-Modulation of Physicochemical Character of Aglycones in Dinucleoside (3′→5′) Monophosphates by the Nearest Neighbor Interaction in the Stacked State. J. Am. Chem. Soc. 2002, 124, 13722–13730. [Google Scholar] [CrossRef] [PubMed]
- Lupták, A.; Ferré-D’Amaré, A.R.; Zhou, K.; Zilm, K.W.; Doudna, J.A. Direct pKa measurement of the active-site cytosine in a genomic hepatitis delta virus ribozyme. J. Am. Chem. Soc. 2001, 123, 8447–8452. [Google Scholar] [CrossRef]
- Guo, M.; Spitale, R.C.; Volpini, R.; Krucinska, J.; Cristalli, G.; Carey, P.R.; Wedekind, J.E. Direct Raman measurement of an elevated base pKa in the active site of a small ribozyme in a precatalytic conformation. J. Am. Chem. Soc. 2009, 131, 12908–12909. [Google Scholar] [CrossRef]
- Liu, L.; Cottrell, J.W.; Scott, L.G.; Fedor, M.J. Direct measurement of the ionization state of an essential guanine in the hairpin ribozyme. Nat. Chem. Biol. 2009, 5, 351–357. [Google Scholar] [CrossRef]
- Liberman, J.A.; Guo, M.; Jenkins, J.L.; Krucinska, J.; Chen, Y.; Carey, P.R.; Wedekind, J.E. A transition-state interaction shifts nucleobase ionization toward neutrality to facilitate small ribozyme catalysis. J. Am. Chem. Soc. 2012, 134, 16933–16936. [Google Scholar] [CrossRef]
- Dawson, R.M.C.; Elliott, D.C.; Elliott, W.H.; Jones, K.M. Data for Biochemical Research, 5th ed.; Clarendon Press: Oxford, UK, 1959; Chapter 5; p. 109. [Google Scholar]
- Murray, J.B.; Seyhan, A.A.; Walter, N.G.; Burke, J.M.; Scott, W.G. The hammerhead, hairpin and vs. ribozymes are catalytically proficient in monovalent cations alone. Chem. Biol. 1998, 5, 587–595. [Google Scholar] [CrossRef]
- Roychowdhury-Saha, M.; Burke, D.H. Distinct reaction pathway promoted by non-divalent-metal cations in a tertiary stabilized hammerhead ribozyme. RNA 2007, 13, 841–848. [Google Scholar] [CrossRef]
- Boots, J.L.; Canny, M.D.; Azimi, E.; Pardi, A. Metal ion specificities for folding and cleavage activity in the Schistosoma hammerhead ribozyme. RNA 2008, 14, 2212–2222. [Google Scholar] [CrossRef]
- O’Rear, J.L.; Wang, S.; Feig, A.L.; Beigelman, L.; Uhlenbeck, O.C.; Herschlag, D. Comparison of the hammerhead cleavage reactions stimulated by monovalent and divalent cations. RNA 2001, 7, 537–545. [Google Scholar] [CrossRef]
- Takagi, Y.; Inoue, A.; Taira, K. Analysis on a cooperative pathway involving multiple cations in hammerhead reactions. J. Am. Chem. Soc. 2004, 126, 12856–12864. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Karbstein, K.; Peracchi, A.; Beigelman, L.; Herschlag, D. Identification of the hammerhead ribozyme metal ion binding site responsible for rescue of the deleterious effect of a cleavage site phosphorothioate. Biochemistry. 1999, 38, 14363–14378. [Google Scholar] [CrossRef] [PubMed]
- Yoshinari, K.; Taira, K. A further investigation and reappraisal of the thio effect in the cleavage reaction catalyzed by a hammerhead ribozyme. Nucleic Acids Res. 2000, 28, 1730–1742. [Google Scholar] [CrossRef] [PubMed]
- Katahira, M.; Kanagawa, M.; Sato, H.; Uesugi, S.; Fujii, S.; Kohno, T.; Maeda, T. Formation of sheared G:A base pairs in an RNA duplex modelled after ribozymes, as revealed by NMR. Nucleic Acids Res. 1994, 22, 2752–2759. [Google Scholar] [CrossRef]
- Zhou, D.M.; Taira, K. The hydrolysis of RNA: From theoretical calculations to the hammerhead ribozyme-mediated cleavage of RNA. Chem. Rev. 1998, 98, 991–1026. [Google Scholar] [CrossRef]
- Zhou, D.M.; He, Q.C.; Zhou, J.M.; Taira, K. Explanation by a putative triester-like mechanism for the thio effects and Mn2+ rescues in reactions catalyzed by a hammerhead ribozyme. FEBS Lett. 1998, 431, 154–160. [Google Scholar] [CrossRef]
- Loverix, S.; Winqvist, A.; Strömberg, R.; Steyaert, J. Mechanism of RNase T1: Concerted triester-like phosphoryl transfer via a catalytic three-centered hydrogen bond. Chem. Biol. 2000, 7, 651–658. [Google Scholar] [CrossRef]
- Kuzmin, Y.I.; Da Costa, C.P.; Fedor, M.J. Role of an active site guanine in hairpin ribozyme catalysis probed by exogenous nucleobase rescue. J. Mol. Biol. 2004, 340, 233–251. [Google Scholar] [CrossRef]
- Ward, W.L.; DeRose, V.J. Ground-state coordination of a catalytic metal to the scissile phosphate of a tertiary-stabilized hammerhead ribozyme. RNA 2012, 18, 16–23. [Google Scholar] [CrossRef]
- Lopez, X.; Schaefer, M.; Dejaegere, A.; Karplus, M. Theoretical evaluation of pKa in phosphoranes: Implications for phosphate ester hydrolysis. J. Am. Chem. Soc. 2002, 124, 5010–5018. [Google Scholar] [CrossRef] [PubMed]
- Mlýnský, V.; Banáš, P.; Walter, N.G.; Šponer, J.; Otyepka, M. QM/MM studies of hairpin ribozyme self-cleavage suggest the feasibility of multiple competing reaction mechanisms. J. Phys. Chem. B 2011, 115, 13911–13924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, Y.; Tanaka, Y.; Morita, E.H.; Tanaka, Y.; Taira, K. Physicochemical analysis of the interaction between a metal ion and the metal-ion-binding motif in hammerhead ribozymes. Nucleic Acids Res. 2001, (Suppl. S1), 81–82. [Google Scholar] [CrossRef]
- Santa Lucia, J., Jr.; Turner, D.H. Structure of (rGGCGAGCC)2 in solution from NMR and restrained molecular dynamics. Biochemistry 1993, 32, 12612–12623. [Google Scholar] [CrossRef] [PubMed]
- Devlin, F.J.; Finley, J.W.; Stephens, P.J.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields: A Comparison of Local, Nonlocal, and Hybrid Density Functionals. J. Phys. Chem. 1995, 99, 16883–16902. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Harihara, P.C.; Pople, J.A. The influence of polarization functions on molecular-orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Banks, J.L.; Beard, H.S.; Cao, Y.X.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated Modeling Program, Applied Chemical Theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef]
- Bochevarov, A.D.; Harder, E.; Hughes, T.F.; Greenwood, J.R.; Braden, D.A.; Philipp, D.M.; Rinaldo, D.; Halls, M.D.; Zhang, J.; Friesner, R.A. Jaguar: A high-performance quantum chemistry software program with strengths in life and materials sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. [Google Scholar] [CrossRef]
- Friesner, R.A.; Guallar, V. Ab initio quantum chemical and mixed quantum mechanics/molecular mechanics (QM/MM) methods for studying enzymatic catalysis. Annu. Rev. Phys. Chem. 2005, 56, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.P.; Uvdal, P. New scale factors for harmonic vibrational frequencies using the B3LYP density functional method with the triple-zeta basis set 6-311+G(d,p). J. Phys. Chem. A 2005, 109, 2937–2941. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. A comparison of models for calculating nuclear magnetic resonance shielding tensors. J. Chem. Phys. 1996, 104, 5497–5509. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tanaka, Y.; Yamanaka, D.; Morioka, S.; Yamaguchi, T.; Morikawa, M.; Kodama, T.S.; Sychrovský, V.; Kojima, C.; Hattori, Y. Physicochemical Characterization of the Catalytic Unit of Hammerhead Ribozyme and Its Relationship with the Catalytic Activity. Biophysica 2022, 2, 221-239. https://doi.org/10.3390/biophysica2030022
Tanaka Y, Yamanaka D, Morioka S, Yamaguchi T, Morikawa M, Kodama TS, Sychrovský V, Kojima C, Hattori Y. Physicochemical Characterization of the Catalytic Unit of Hammerhead Ribozyme and Its Relationship with the Catalytic Activity. Biophysica. 2022; 2(3):221-239. https://doi.org/10.3390/biophysica2030022
Chicago/Turabian StyleTanaka, Yoshiyuki, Daichi Yamanaka, Saori Morioka, Taishi Yamaguchi, Masayuki Morikawa, Takashi S. Kodama, Vladimír Sychrovský, Chojiro Kojima, and Yoshikazu Hattori. 2022. "Physicochemical Characterization of the Catalytic Unit of Hammerhead Ribozyme and Its Relationship with the Catalytic Activity" Biophysica 2, no. 3: 221-239. https://doi.org/10.3390/biophysica2030022