




Abstract
We recently showed that Saccharomyces cerevisiae telomeric DNA can fold into an unprecedented pseudocircular G-hairpin (PGH) structure. However, the formation of PGHs in the context of extended sequences, which is a prerequisite for their function in vivo and their applications in biotechnology, has not been elucidated. Here, we show that despite its ‘circular’ nature, PGHs tolerate single-stranded (ss) protrusions. High-resolution NMR structure of a novel member of PGH family reveals the atomistic details on a junction between ssDNA and PGH unit. Identification of new sequences capable of folding into one of the two forms of PGH helped in defining minimal sequence requirements for their formation. Our time-resolved NMR data indicate a possibility that PGHs fold via a complex kinetic partitioning mechanism and suggests the existence of K+ ion-dependent PGH folding intermediates. The data not only provide an explanation of cation-type-dependent formation of PGHs, but also explain the unusually large hysteresis between PGH melting and annealing noted in our previous study. Our findings have important implications for DNA biology and nanotechnology. Overrepresentation of sequences able to form PGHs in the evolutionary-conserved regions of the human genome implies their functionally important biological role(s).
INTRODUCTION
DNA G-hairpins are generally defined as fold-back structures stabilized by G:G base pairs. G-hairpins were originally postulated as intermediates in the folding of G-rich sequences into four-stranded G-quadruplex structures (Figure 1A) (1–6). In 2014, Sugiyama et al. demonstrated that stable G-hairpins can be formed under enforced conditions of DNA origami (Figure 1B) (7). A G-hairpin stabilized in the DNA origami frame converted into a G-quadruplex upon binding of a pyridine-based ligand (8). These observations established G-hairpins as alternative drug targets for processes involving G-quadruplex motifs, including the regulation of gene expression and/or telomeric DNA maintenance (9–13).
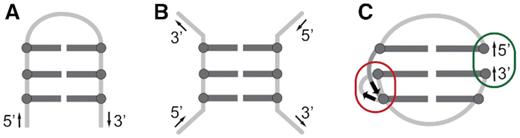
Schematic representations of (A) G-hairpin, a proposed G-quadruplex folding intermediate, (B) a model of a G-hairpin imposed by DNA origami framework, and (C) a pseudocircular G-hairpin (PGH) adopted by SC11 sequence (PDB ID: 5M1W). Characteristic structural features of PGH fold include a chain reversal, responsible for mixed parallel/antiparallel backbone progression (red ellipse) and 5′-to-3′ stacking of terminal residues (dark green ellipse). G:G pairs are represented with dark grey, while loop regions are light grey.
Recently, we discovered a novel mixed parallel/antiparallel fold-back structure formed by a native 11-nt 5′-d(GTGTGGGTGTG)-3′ fragment from the telomeric DNA of Saccharomyces cerevisiae (herein referred to as SC11) that was stabilized by three dynamic G:G base pairs (14). Although the structure of SC11 has been consistent with the general definition of G-hairpins, the high-resolution NMR structure revealed that its folding topology was fundamentally distinct from those postulated for G-quadruplex folding intermediates as well as those formed in DNA origami. The G-hairpin formed by SC11 displayed a number of distinct features, including a pseudocircular topology, which arose from the stacking of the 5′- and 3′-terminal guanine residues, and a chain reversal, which is a structural element responsible for the mixed parallel/antiparallel nature of the structure (Figure 1C).
Observation of the stable pseudocircular G-hairpin (PGH) structure formed by a natively occurring sequence in the telomeric DNA of S. cerevisiae has indicated the importance of this structure in yeast telomere maintenance (14–16). Telomere maintenance in most eukaryotes is associated with the ability of telomeric DNA to form a G-quadruplex structure, which is based on regular tandem G-rich repeats. Formation of the PGH structure, which is topologically distinct from the G-quadruplex structure, might suggest that an unknown mechanism is responsible for the maintenance of S. cerevisiae telomeric DNA based on irregular T(G1–3) repeats. Nevertheless, an active participation of PGHs in the telomere biology is implicitly determined by their ability to fold in the context of extended single-stranded (ss) DNA. As indicated by the structure of SC11 PGH, this ability could be decisively impaired by one of most prominent structural characteristics of PGH, specifically, its ‘circular’ nature resulting from the stacking of terminal guanines (Figure 1C). Due to the potential relevance of PGH structures for the maintenance of genomic stability and integrity in yeast, which is a frequently used model organism in telomere biology, we deemed it essential to explore the structural space of extended SC11-based constructs emulating natively present telomeric segments in S. cerevisiae.
Here, using NMR and CD spectroscopy, we studied impact of both extensions and point mutations of the parent SC11 sequence on formation of the PGH structure (14), referred to as form I. Notably, a high-resolution NMR analysis of one of the extended sequences not only provided unprecedented details on the junction between protruding 3′-ssDNA and the pseudocircular core of PGH but also unexpectedly revealed the existence of a novel folding PGH topology, named form II. The data from the spectral analysis of the point mutants of form I and II allowed us to define minimal sequence requirements for PGH formation and subsequently perform genome-wide bioinformatic screening for putative PGH sites in genomic DNA. The results of the bioinformatic search shows that sequences with PGH formation potential are both abundant and non-randomly distributed in yeast telomeric DNA as well as in evolutionary conserved regulatory regions of metazoan genomes. Our findings have important implications for DNA biology and nanotechnology. They also provide important insight into principles of the folding of G-rich oligonucleotides.
MATERIALS AND METHODS
Sample preparation
The isotopically unlabeled oligonucleotides employed for the 1D 1H NMR and CD measurements were synthesized and purified as previously described (14). The concentration of the desalted samples was determined by UV absorption at 260 nm using a UV/VIS spectrophotometer from Varian (CARY-100 BIO UV–VIS). The extinction coefficients were determined by the nearest neighbour method. The oligonucleotides were dissolved in phosphate buffer (10 mM potassium phosphate, pH 7.0 and 100 mM KCl) to a final oligonucleotide concentration of 100 μM. The samples were then heated to 90°C for 10 min, gradually cooled to room temperature overnight, and then incubated for at least 24 h at 4°C before data acquisition. The samples of T-SC11, SC11-T and GT-SC11-T employed for 2D NMR spectroscopy were diluted in the same buffer to final oligonucleotide concentrations of 0.6 mM, heated to 90°C for 10 min, gradually cooled to room temperature overnight and stored at 4°C for 4 days prior to data acquisition.
Preparation of 15N/13C-labeled oligonucleotides
The residue-specific, isotopically 15N/13C-labeled oligonucleotides of GT-SC11-T were synthesized on a K&A Laborgeraete GbR DNA/RNA Synthesizer H-8 using standard phosphoramidite chemistry in DMT-on mode. The material was purified by reversed-phase HPLC, and the DMT group was removed by treatment with 80% AcOH for 30 min. The oligonucleotides were then extracted with pure ethanol and 8 M LiCl. After rinsing with 70% ethanol, the samples were dissolved in pure water. The pH value of each sample solution was adjusted to ∼7 using LiOH. The samples were subsequently desalted using an ultrafiltration cell with a 1 kDa cellulose membrane. The oligonucleotides were then dissolved in phosphate buffer to final concentrations of 0.5 mM and handled analogously to the T-SC11, SC11-T and GT-SC11-T samples (see above).
Synthesis of 4′-alkoxy site-specifically modified oligonucleotides
4′-Alkoxy phosphoramidite T and G monomers used for synthesis of 4′-alkoxy site-specifically labeled SC11 were prepared according to published protocols (17,18). For general procedure see Supplementary Scheme S1. 4′-Alkoxy modified oligodeoxyribonucleotides were synthesized on a GeneSyn synthesizer (IOCB, Prague) by standard DNA phosphoramidite protocol. Oligonucleotides were synthesized in 3′→5′ direction on a 0.5 μmol scale by standard DMTr-off phosphoramidite method using 3′-O-dimethoxytrityl-5′-hemisuccinyl-nucleoside-modified LCAA-CPG solid support. Removal of the acyl-protecting groups and release of the oligonucleotides from the support were achieved by treatment with gaseous ammonia (0.7 MPa) at room temperature for 24 h.
Oligonucleotides were purified at 55°C on a DNAPac PA100 (9 × 250 mm; Dionex) nucleic acid column at a flow rate of 3 ml/min using a linear gradient of sodium chloride (20–500 mM, 60 min) in 50 mM sodium acetate buffer (pH 7.0) containing 10% (v/v) of acetonitrile. Alternatively, pure oligonucleotides were desalted on a Luna C18 (2) column (10 μm; 10 × 50 mm; Phenomenex) at a flow rate of 3 mL/min using a linear gradient of acetonitrile (0–25%, 30 min) in 0.1 M aq. ammonium acetate (pH 7). Purified oligonucleotides were freeze dried and characterized by MALDI-TOF analysis.
CD spectroscopy
The CD spectra were recorded on a Jasco J-815 spectropolarimeter equipped with a Jasco PTC-423S temperature controller using 1-mm path-length cuvettes. The experiments were performed at an oligonucleotide concentration of 100 μM in phosphate buffer. The CD spectra were recorded at 20°C (unless stated otherwise) in the wavelength range of 220–320 nm. CD spectra correspond to an average of three scans. The scan rate was set at 50 nm/min with a response time of 4 s and a bandwidth of 2 nm. The buffer baseline was subtracted from each spectrum.
Native PAGE
Non-denaturing PAGE was performed in phosphate buffer in a thermostated SE-600 instrument (Hoefer Scientific, San Francisco, CA, USA). Gels (16% PA, 29:1 mono/bis ratio, 14 × 16 × 0.1 cm in size) were run at 40–50 V and 7°C for 16 h unless indicated otherwise. Low-molecular-weight markers (10–100 nt, Affymetrix) were used as a DNA size ladder. A total of 1 nmol of DNA was loaded into each lane. The DNA was visualized by Stains-all (Sigma-Aldrich) staining.
NMR spectroscopy
NMR experiments on isotopically unlabeled samples were performed on Bruker Avance III HD spectrometers at 600, 700 and 850 MHz using either a room temperature inverse (1H-BB) probe or triple-resonance (1H–13C–15N) cryogenic probes. 1D 15N-edited HSQC experiments were performed on an Agilent DD2 600 MHz spectrometer on 6% residue-specific 15N/13C-labeled oligonucleotides. 1D 1H NMR spectra of all 11-nt long constructs were recorded at 20°C (unless indicated otherwise). All other NMR spectra were recorded at 10°C. The NOE build-up procedure and the assignments of exchangeable proton resonances were carried out based on 2D NOESY spectra (mixing times of 50, 100, 150 and 250 ms) recorded in 90% H2O/10% 2H2O. 2D NOESY (mixing times of 50, 150 and 250 ms) and JR-HMBC spectra in 100% 2H2O were used to assign the nonexchangeable proton resonances. 2D TOCSY (mixing time of 40 ms) and DQF-COSY experiments were used to predict the sugar conformations. The spectra were processed with TopSpin v3.1 (Bruker, Germany) or VNMRJ software (Agilent Technologies, USA). The resonances were assigned and integrated with a Gaussian fit procedure using SPARKY software (UCSF) (19). The NOE distance restraints for the nonexchangeable protons of SC14 were obtained from the 2D NOESY spectrum (mixing time of 150 ms) recorded in 100% 2H2O. Only nonoverlapping cross-peaks were integrated and used for distance restraint calculations. The average volume of the nonoverlapping intraresidual H8-H1′ NOE correlations of guanines with anti-glycosidic conformations was used as a reference and assigned to a distance of 3.9 Å. Based on the reference, the remaining signals in the NOESY spectrum were classified into three categories: strong (1.8–3.6 Å), moderate (2.6–5.0 Å) and weak (3.5–6.5 Å). The NOE distance restraints for exchangeable protons were obtained from the 2D NOESY spectrum acquired in 90% H2O/10% 2H2O with a mixing time of 150 ms. Cross-peaks were classified as moderate (2.6–5.0 Å) or weak (3.5–6.5 Å) based on their intensities. All peaks in the 2D NOESY spectrum of SC14 that were inconsistent with a single G:G base pair, such as the mutual restraints between H8 and H1 of the residues that form a G:G base pair, were omitted from distance restraints used in the structure calculations (14).
The torsion angle χ along the glycosidic bond of SC14 was restrained to between 25° and 95° for syn and between 200° and 280° for anti guanine residues. The torsion angle χ for all thymine residues was restrained to between 170° and 310°. The endocyclic torsion angle ν2 was used to restrain the sugar puckering to the C2′-endo conformation (for residues G−II, T−I, G1, T2, G3, T4, G7, T8 and G9) and to the C3′-endo conformation (for residues G5 and T10) based on analysis of the NOESY and/or DQF-COSY NMR spectra. The sugar conformations for G6, G11 and T+I were left unrestrained. Six distance restraints were utilized when calculating the hydrogen bond lengths between O6 and H1 atoms of residues from the individual G:G base pairs. Weak planarity restraints were used to keep the residues of the G5:G1 and G7:G−II base pairs aligned. Each base pair was restrained with three planarity restraints. The planarity of the G6:G9 base pair was left unrestrained due to the indication of the buckling of G9 with respect to G6.
Structure calculations
Structure calculations were performed with AMBER 14 software (20) using the parmbsc0 force field (21,22) with parmχOL4 (23) and parmϵζOL1 (24) modifications. The initial extended structure of the GT-SC11-T oligonucleotide was obtained with the LEAP module of the AMBER 14 program. A total of 100 structures were calculated in 120 ps of NMR restraint-based simulated annealing (SA) simulations using the generalized Born implicit model to account for solvent effects. The cut-off for nonbonding interactions was 20 Å, and the SHAKE algorithm for hydrogen atoms was used with a tolerance of 0.0005 Å. SA calculations were initiated with random velocities. After 5 ps at 300 K, the temperature was raised to 1000 K in the next 10 ps and held constant at 1000 K for 35 ps. The temperature was decreased to 100 K in the next 55 ps and then to 0 K in the last 15 ps using Langevin thermostat. The restraints used in the calculation involved NOE-derived (force constant of 20 kcal mol−1 Å−2) and hydrogen bond distance restraints (force constant of 50 kcal mol−1 Å−2), torsion angle χ and ν2 restraints (force restraints of 200 kcal mol−1 Å−2) and planarity restraints between residues involved in the G5:G1 and G7:G−II base pairs (force constant of 20 kcal mol−1 Å−2). A family of 10 structures with the lowest energies was selected. The data were visualized and the figures were prepared with UCSF Chimera software (25). The coordinates of the SC14 pseudocircular G-hairpin (PGH) have been deposited in the Protein Data Bank with the accession code 6R8E. The list of chemical shifts has been deposited in the Biological Magnetic Resonance Data Bank with the accession code 34386.
Unrestrained molecular dynamics simulations
Unrestrained MD simulations of SC14 were performed using the NMR determined structure of SC14 (PDB ID: 6R8E). The modeling was performed in the xleap module of AMBER 18 software (26) using the parmOL15 force field (27). The models were solvated in an octahedral box of water molecules with 100 mM KCl. We used TIP3P (28) SPC/E (29) and OPC (30) water models with the appropriate ion parameters (TIP4PEw ions for OPC) (31). After equilibration (for protocol details, see next section paragraph of Material and Methods), each system was subjected to MD simulations (32). The trajectories were calculated at a constant temperature of 300 K and pressure of 1 atm using the weak-coupling algorithm (33). A time step of 4 fs was used with SHAKE (34), SETTLE (35) and hydrogen mass repartitioning schemes (36). We applied HBfix potential on selected hydrogen bonds in each simulation (Supplementary Table S1) to compensate for the known underestimated stability of the hydrogen bonds in this force field (37). We accumulated three independent 5 μs long trajectories for each water model for every system.
Equilibration protocol (unrestrained MD simulations)
Equilibration of each system was started by 500 steps of steepest descent minimization followed by 500 steps of conjugate gradient minimization with 25 kcal mol−1 Å−2 position restraints on DNA atoms. Then the system was heated from 0 to 300 K during 100 ps with position restraints of 25 kcal mol−1 Å−2 on DNA and with constant volume. Afterwards, the system underwent minimization with 5 kcal mol−1 Å−2 restraints on DNA atoms using 500 steps of steepest descent method followed by 500 steps using conjugate gradient. Continuing in position restraints on DNA atoms of 5 kcal mol−1 Å−2, the system was equilibrated during 50 ps at a constant temperature of 300 K and pressure of 1 atm. Then, an analogous series of alternating minimizations and equilibrations was performed, consecutively using decreasing position restraints of 4, 3, 2 and 1 kcal mol−1 Å−2. Finally, equilibration using position restraints of 0.5 kcal mol−1 Å−2 and starting velocities from the previous equilibration step, followed by a short 50 ps free MD, was applied.
Sequence logo alignment
Sequence logo is a graphical representation of nucleotide alignments showing conservation of residues in sequence. The relative size of letters displays frequency of a letter in the alignment. The total height of the letters shows the information content at given position. The information content is measured in bits and for DNA the range is from 0 to 2. Positions that are highly conserved and have a low tolerance for substitutions correspond to high information content, while positions with a high tolerance for substitutions correspond to low information content. Thus, at position at which just one base type can occur has an information content 2. In position at which we can incorporate all DNA bases with the same probability has information content 0.
Genome-wide bioinformatic search
Genome-wide PGH-forming sequence analysis was conducted for eighteen organisms, including different viruses, bacteria and eukaryotes (Supplementary Table S2). The corresponding genome sequences were retrieved from the NCBI genome database. The bioinformatic query covered eighteen sequences displaying the potential to form PGHs, namely, form I, form I_T2C, form I_G3A, form I_G3C, form I_G3T, form I_T4A, form I_T4C, form I_T8A, form I_T8C, form I_T10A, form I_T10C and form II, form II_T-IC, form II_G3C, form II_G3A, form II_G3T, form II_T4A, form II_T4C.
Additionally, the genome of Homo sapiens was subjected to a more thorough analysis involving elucidation of the distribution of sequences able to form PGHs in biologically important genomic locations: promoter, 5′-UTR, 3′-UTR, exons, introns, and CDS. The DNA sequences of transcription-related genomic features were retrieved from the UCSC database by UCSC Table Browser (38). The assembly GRCm38 (for H. sapiens) and the NCBI RefSeq annotation track have been used. The promoter region was defined as the area 1 kb upstream from a transcription start site (TSS). The genomes were scanned for PGH sites using our own Python scripts with existing BioPython packages (Supplementary Table S2) (39).
RESULTS AND DISCUSSION
Effect of 5′- and 3′-end extensions on the PGH formation propensity
Despite being formed by short 11-nt oligonucleotide 5′-d(GTGTGGGTGTG)-3′, the SC11 PGH has rather complex topology (Figure 1C). The structure consists of a G:G core, comprised of three G:G base pairs (underlined in the sequence), while other residues represent loop residues. The central G:G core features two distinguished structural elements: (i) chain reversal between residues from the GGG tract, which locally changes directionality of the backbone from anti-parallel to parallel and (ii) stacking of 5′- and 3′-terminal residues that is responsible for pseudocircular character of the structure. The ability to adopt PGH fold in the context of extended ssDNA forms a fundamental prerequisite for its potential biological function including that in the ss-3′-overhang of S. cerevisiae telomeric DNA. Potential steric hindrances related to spatial proximity of both terminal residues evoke a question: To what extend are the 5′- and/or 3′-end protrusions of the parent SC11 sequence compatible with the formation of the pseudocircular structure(s)? To experimentally address the possibility of the PGH formation in the context of a long ssDNA, we designed a set of four constructs bearing different one- or two-sided extensions of the SC11 sequence (Table 1). Importantly, all the constructs emulate situations that occur within native ss-3′-overhang of telomeric DNA of S. cerevisiae.
Oligonucleotides based on S. cerevisiae telomeric DNA used in this study.
| Construct name | Sequence | ||
|---|---|---|---|
| 5′- | SC11-core | −3′ | |
| −II −I | 1 2 3 4 5 6 7 8 9 10 11 | +I +II | |
| SC11 | G T G T G G G T G T G | ||
| T-SC11 | T | G T G T G G G T G T G | |
| SC11-T | G T G T G G G T G T G | T | |
| SC14 (GT-SC11-T) | G T | G T G T G G G T G T G | T |
| T-SC11-TG | T | G T G T G G G T G T G | T G |
| Construct name | Sequence | ||
|---|---|---|---|
| 5′- | SC11-core | −3′ | |
| −II −I | 1 2 3 4 5 6 7 8 9 10 11 | +I +II | |
| SC11 | G T G T G G G T G T G | ||
| T-SC11 | T | G T G T G G G T G T G | |
| SC11-T | G T G T G G G T G T G | T | |
| SC14 (GT-SC11-T) | G T | G T G T G G G T G T G | T |
| T-SC11-TG | T | G T G T G G G T G T G | T G |
Oligonucleotides based on S. cerevisiae telomeric DNA used in this study.
| Construct name | Sequence | ||
|---|---|---|---|
| 5′- | SC11-core | −3′ | |
| −II −I | 1 2 3 4 5 6 7 8 9 10 11 | +I +II | |
| SC11 | G T G T G G G T G T G | ||
| T-SC11 | T | G T G T G G G T G T G | |
| SC11-T | G T G T G G G T G T G | T | |
| SC14 (GT-SC11-T) | G T | G T G T G G G T G T G | T |
| T-SC11-TG | T | G T G T G G G T G T G | T G |
| Construct name | Sequence | ||
|---|---|---|---|
| 5′- | SC11-core | −3′ | |
| −II −I | 1 2 3 4 5 6 7 8 9 10 11 | +I +II | |
| SC11 | G T G T G G G T G T G | ||
| T-SC11 | T | G T G T G G G T G T G | |
| SC11-T | G T G T G G G T G T G | T | |
| SC14 (GT-SC11-T) | G T | G T G T G G G T G T G | T |
| T-SC11-TG | T | G T G T G G G T G T G | T G |
Imino regions of NMR spectra of extended sequences were compared to SC11 imino region fingerprint, characterized by seven well-resolved peaks between δ 11.2 and 12.4 ppm (Figure 2). Six of them belong to imino protons of guanine residues involved in G:G base pairs, while the signal at δ 12.0 ppm corresponds to imino hydrogen of T2 (14). Evaluation of structural changes upon sequence extensions was complemented by comparison of CD spectrum marked by a maximum and a shoulder at 278 and 299 nm, respectively, and a minimum at around 258 nm (Supplementary Figure S1). The 1D 1H NMR and CD spectral fingerprints of T-SC11 and SC11-T, representatives of one-sided 5′- or 3′-extensions of SC11, indicate the formation of PGH structures (Figure 2 and Supplementary Figure S1). The observed imino-imino NOE connectivities in the 2D NOESY spectra of T-SC11 and SC11-T closely resemble those of SC11 and are consistent with a structure stabilized by three G:G base pairs (for elaborated discussion on the topology determination of T-SC11 and SC11-T with respect to SC11 see Supplementary Figures S2–S4). Detailed analysis of the NOESY spectra of T-SC11 and SC11-T reveals the presence of all the principal structural features of a PGH, i.e., the chain reversal and the stacking of most terminal guanine residues, indicating that neither 5′- nor 3′-sequence extension (by a thymine residue) limits the ability of the extended sequence to form a PGH structure.
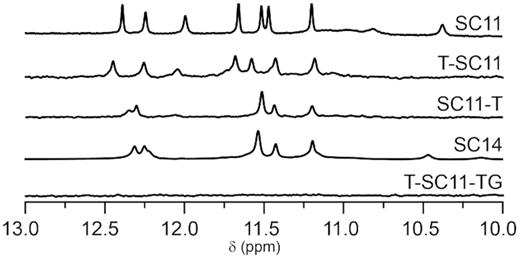
Imino regions of 1D 1H NMR spectra of SC11 and its extended sequences.
Notable differences were observed between spectra of constructs with extensions on both 5′- and 3′-ends of the parent SC11 sequence, namely SC14 and T-SC11-TG (Figure 2 and Supplementary Figure S1). While CD and NMR spectra of T-SC11-TG display signatures typical of unfolded DNA, a spectroscopic analysis of SC14 suggests formation of a folded species: the CD spectrum displays characteristic PGH pattern (Supplementary Figure S1); the imino region of the 1D 1H NMR spectrum shows five well resolved signals between δ 11.2 and 12.4 ppm corresponding to six imino proton resonances consistent with the three G:G base pairs formation (Figure 2). Imino signal of T2, which resonated at δ 12.0 ppm in SC11, shifts downfield upon sequence elongation to around δ 12.3 ppm and is partially overlapped by two other imino signals of SC14 that resonate between δ 12.3 and 12.4 ppm.
We performed the Ferguson analysis (40) of migration data of SC14. The electrophoretic migration of SC14 was compared with those of SC11, 11-nt hairpin, 11-nt TBA G-triplex, 22-nt long heteroduplex, and 19-, 21- and 24-mers forming intramolecular G-quadruplex structures (for details, see Supplementary Figure S5). The slope of the Ferguson plot of SC14 corresponded to those of SC11 and importantly, 11-nt TBA triplex, a validated monomeric structure (Supplementary Figure S5). This observation is consistent with a monomeric nature of SC14. The melting profile of SC14 structure was similar to that of SC11 (Supplementary Figure S6).
NOESY spectrum of SC14 discloses significant topological differences in comparison to SC11
The imino-imino region of the 2D NOESY NMR spectrum of SC14 displays a complex network of NOE connectivities substantiating that the core of the structure consists of G5:G1, G7:G−II and G6:G9 base pairs (Figure 3A; for unambiguous assignment of SC14 imino protons, see Supplementary Figure S7). In addition to the imino-imino NOE connectivities characteristic of N1-carbonyl symmetric base pair arrangements (tWW), aromatic-imino NOEs typical of N1-carbonyl, N7-amino (WC-H and H-WC) G:G base pairs were observed (Figure 3A; for a detailed definition of tWW, WC-H and H-WC base pairs, see Figure 3B). The concurrent observation of NOE connectivities representative of different and mutually exclusive base pair arrangements indicates that the G:G base pairs in the SC14 are dynamic and switch between different base pairings (see below), similar to the G:G base pairs in SC11 (14). Note: The isochronous chemical shifts of H1 protons from G−II and G7 and the dynamic character of the G:G base pairs in SC14 might impact the interpretation of the intensities of the NOE cross-peaks among the protons from the core-forming guanines in terms of proton-proton distances. This was considered during quantification of the related NOE data.
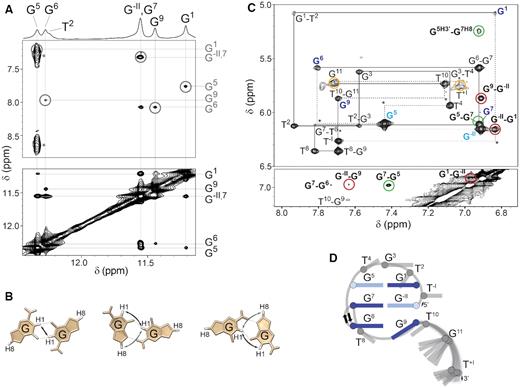
(A) Imino region of the 1D 1H NMR spectrum of SC14 with the assigned imino proton resonances (top). Aromatic-imino (middle) and imino-imino (bottom) regions of the 2D NOESY spectrum (τm = 150 ms). The assignments of the aromatic and imino protons of the G:G core-forming residues are indicated on the right side of the NOESY spectrum in gray and black, respectively. The H8-H1 NOEs characteristic of N1-carbonyl, N7-amino base pair arrangements are marked with gray circles. (B) Schematic of N1-carbonyl symmetric (left) and N1-carbonyl, N7-amino (middle and right) hydrogen bond arrangements in a Hoogsteen–Watson–Crick (H-WC)-type and Watson–Crick–Hoogsteen (WC-H)-type arrangements, respectively. The aromatic-imino and imino-imino NOEs characteristic of the individual base pairing arrangement are indicated by gray and black arrows, respectively. (C) Aromatic-anomeric (top) and aromatic-aromatic (bottom) regions of the NOESY spectrum (τm = 150 ms) of SC14 in 2H2O. The sequential walk is indicated by solid lines. The missing cross-peaks are designated with asterisks, while respective correlations are indicated by dashed lines. Intraresidual H1′–H8 NOE cross-peaks belonging to guanines in anti and syn glycosidic conformations are depicted in dark and light blue, respectively. The cross-peaks indicative of a chain reversal, the formation of a new discontinuous G-tract and a flexible 3′-tail are shown in green, dark red and orange, respectively. (D) Schematic presentation of PGH topology adopted by SC14. The anti and syn G:G base pair-forming guanines are shown in dark and light blue, respectively. The loop residues and residues forming the flexible 3′-tail are colored in gray.
Next to three G:G base pairs, several sugar-aromatic and aromatic-aromatic NOE contacts between the core guanines confirm the presence of all main structural elements characteristic of a PGH fold, i.e., a chain reversal arrangement of the backbone and the formation of a discontinuous G1–G−II–G9 G-tract (Figure 3C). The unusual arrangement of the backbone chain responsible for placing residue G7 between residues G5 and G6 was confirmed by the NOE cross-peaks between the sugar protons of G5 and H8 of G7 (dark green cross-peaks in the aromatic-anomeric region of the NOESY spectrum). The stacking of G5 to G7 was corroborated by the strong NOE cross-peak between the H8 protons of G5 and G7 (dark green cross-peak in the aromatic–aromatic region of the NOESY spectrum) and by the absence of sequential H1′–H8 and H8–H8 NOE cross-peaks between G5 and G6. The formation of a discontinuous G-tract involving residues G−II, G1 and G9 was established by the NOE cross-peaks between H1′ of G−II and H8 of G1 and between H8 of G−II and H8 of G1 (dark red cross-peaks). The position of G1 above G−II was further confirmed by the NOE cross-peaks between the sugar H2′/H2″ and H3′ protons of G−II and H8 of G1 as well as between the sugar H4′ and H5′/H5″ protons of G1 and H8 of G−II. The G−II-G9 stacking was indicated by the NOE cross-peaks between H1′ of G9 and H8 of G−II and between H8 of G−II and H8 of G9 (dark red cross-peaks). Cross-peaks involving H1′ and H8 protons of G11 and T+I are broadened, indicating that the residues constituting the 3′-tail are highly dynamic (orange cross-peaks). Intense intraresidual H1′–H8 NOE cross-peaks in the anomeric-aromatic region of the NOESY spectrum suggest syn glycosidic conformations for G−II and G5 (light blue), while for the other Gs of the G:G core, namely, G1, G6, G7 and G9, the intraresidual H1′-H8 NOE intensities are consistent with glycosidic torsion angles for anti orientations.
Despite indications that both SC11 and SC14 form PGH structures with common main structural features, in-depth analysis of the NOESY spectra of SC14 revealed notable topological differences in comparison to SC11. SC11 PGH structure is stabilized by G5:G1, G7:G11 and G6:G9 base pairs, where 5′-G1 participates in the formation of a terminal G:G base pair. In contrast, G7:G−II base pair in SC14, which engages terminal (5′) G, is stacked between G5:G1 and G6:G9 and constitutes the middle base pair (Figure 3D). These topological differences cannot be distinguished by comparison of CD spectra profiles only (cf. Supplementary Figure S1).
Noteworthy, comparative analysis of 1D 1H NMR and CD spectra revealed similarities between NMR and CD profiles of SC11-T and SC14, suggesting that these two constructs might share the same PGH folding topology (Figure 2 and Supplementary Figure S1). Next to sequence-related restraints of SC11-T to occupy the same topology as SC14 (Table 1), the pattern of NOE cross-peaks in 2D NOESY NMR spectra of SC11-T corroborate that the arrangement of G:G base pairs in SC11-T is identical to the G:G core of SC11 (Supplementary Figures S2 and S4). These results show that the spectroscopic observables of SC11-T and SC14 are strongly affected by the structural element shared by both constructs—3′-tail, which influences the appearance of NMR profiles.
High-resolution NMR structure of SC14
To reveal the structural details of SC14, 346 NOE-derived distance restraints, along with 6 hydrogen bonds, 25 torsion angles and 6 planarity restraints, were employed in the structure calculations (for NMR restraints and structure statistics, see Table 2). The resulting structure (Figure 4A) features a compact core consisting of three G:G base pairs, where G5:G1 and G7:G−II preferentially adopt N1-carbonyl symmetric geometries, while G6:G9 forms a nonplanar N1-carbonyl, N7-amino-like base pair in which G6 participates with its Watson–Crick and G9 with its Hoogsteen side (Figure 4B). While the discontinuous G-tract-forming G1 and G−II stack to each other, G9 that forms a buckled base-pair with G6 is tilted for approximately 40° with respect to G−II. With the exception of T−I, which faces away from the core and is completely exposed to solvent, and of both G11 and T+I, which form a flanking disordered 3′-tail, the other loop residues (i.e., T2, G3, T4, T8 and T10) are well-defined and tightly packed around the core. T2 is positioned behind the G5:G1 base pair with its base pointing towards the amino protons of G7 and the sugar edge of T8 (Figure 4A). G3, which stacks on the top of the G5:G1 base pair, is covered by T4. Residues T8 and T10 are located under the G6:G9 base pair and are both well-defined as expected based on the high number of NOE cross-peaks between G6, T8, G9 and T10 (more specifically, 15 NOEs).
NMR statistics of SC14 PGH structure.
| NMR restraints statistics | |
| NOE-derived distance restraints | |
| Total | 346 |
| Intra-residual | 204 |
| Inter-residual | 142 |
| Sequential | 80 |
| Long-range | 62 |
| Hydrogen bond restraints | 6 |
| Torsion angle restraints | 25 |
| Planarity restraints | 6 |
| Structure statistics | |
| Violations | |
| Mean NOE restraint violation (Å) | 0.093 ± 0.031 |
| Max. NOE restraint violation (Å) | 0.145 |
| Max. torsion angle restraint violation (°) | 0 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.0127 ± 0.0001 |
| Angles (°) | 2.27 ± 0.04 |
| Pairwise heavy atom RMSD (Å) | |
| Overall | 1.36 ± 0.50 |
| Overall without G11 and T+I | 0.63 ± 0.18 |
| Overall without T−I, G11 and T+I | 0.49 ± 0.11 |
| G:G base-pairs only | 0.35 ± 0.11 |
| NMR restraints statistics | |
| NOE-derived distance restraints | |
| Total | 346 |
| Intra-residual | 204 |
| Inter-residual | 142 |
| Sequential | 80 |
| Long-range | 62 |
| Hydrogen bond restraints | 6 |
| Torsion angle restraints | 25 |
| Planarity restraints | 6 |
| Structure statistics | |
| Violations | |
| Mean NOE restraint violation (Å) | 0.093 ± 0.031 |
| Max. NOE restraint violation (Å) | 0.145 |
| Max. torsion angle restraint violation (°) | 0 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.0127 ± 0.0001 |
| Angles (°) | 2.27 ± 0.04 |
| Pairwise heavy atom RMSD (Å) | |
| Overall | 1.36 ± 0.50 |
| Overall without G11 and T+I | 0.63 ± 0.18 |
| Overall without T−I, G11 and T+I | 0.49 ± 0.11 |
| G:G base-pairs only | 0.35 ± 0.11 |
NMR statistics of SC14 PGH structure.
| NMR restraints statistics | |
| NOE-derived distance restraints | |
| Total | 346 |
| Intra-residual | 204 |
| Inter-residual | 142 |
| Sequential | 80 |
| Long-range | 62 |
| Hydrogen bond restraints | 6 |
| Torsion angle restraints | 25 |
| Planarity restraints | 6 |
| Structure statistics | |
| Violations | |
| Mean NOE restraint violation (Å) | 0.093 ± 0.031 |
| Max. NOE restraint violation (Å) | 0.145 |
| Max. torsion angle restraint violation (°) | 0 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.0127 ± 0.0001 |
| Angles (°) | 2.27 ± 0.04 |
| Pairwise heavy atom RMSD (Å) | |
| Overall | 1.36 ± 0.50 |
| Overall without G11 and T+I | 0.63 ± 0.18 |
| Overall without T−I, G11 and T+I | 0.49 ± 0.11 |
| G:G base-pairs only | 0.35 ± 0.11 |
| NMR restraints statistics | |
| NOE-derived distance restraints | |
| Total | 346 |
| Intra-residual | 204 |
| Inter-residual | 142 |
| Sequential | 80 |
| Long-range | 62 |
| Hydrogen bond restraints | 6 |
| Torsion angle restraints | 25 |
| Planarity restraints | 6 |
| Structure statistics | |
| Violations | |
| Mean NOE restraint violation (Å) | 0.093 ± 0.031 |
| Max. NOE restraint violation (Å) | 0.145 |
| Max. torsion angle restraint violation (°) | 0 |
| Deviations from idealized geometry | |
| Bonds (Å) | 0.0127 ± 0.0001 |
| Angles (°) | 2.27 ± 0.04 |
| Pairwise heavy atom RMSD (Å) | |
| Overall | 1.36 ± 0.50 |
| Overall without G11 and T+I | 0.63 ± 0.18 |
| Overall without T−I, G11 and T+I | 0.49 ± 0.11 |
| G:G base-pairs only | 0.35 ± 0.11 |
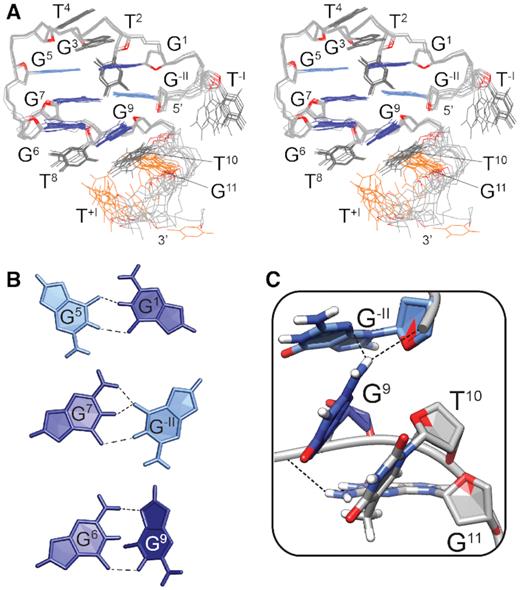
(A) Stereo-view of superposition of the 10 lowest-energy solution structures of SC14 PGH (PDB ID: 6R8E). (B) Arrangements of the G5:G1, G7:G−II and G6:G9 base pairs in the lowest-energy solution-state NMR structure. The hydrogen bonds are marked with dashed lines. (C) A junction between the pseudocircular element and the single stranded 3′-end protrusion in the SC14 PGH structure. The junction is stabilized by stacking of G9 and T10 and by hydrogen bonds between the N3 and O5′ atoms of G−II and the amino proton of G9. The guanines in anti and syn glycosidic conformations are shown in dark and light blue, respectively. The loop residues and residues G11 and T+I, which form the flexible 3′-tail are colored grey. O4′ atoms are colored red.
Flexibility of the 3′-tail appears the reason for generally higher overall pairwise heavy atom RMSD value calculated for 10 lowest-energy structures (1.36 Å; Table 2): RMSD value calculated for residues G−II to T10, i.e., upon exclusion of single stranded tail comprising G11 and T+I, gets reduced to 0.63 Å. The G:G base pairs are expectedly the best defined part of the structure with RMSD value of 0.35 Å. Noteworthy, inclusion of loop residues T2, G3, T4, T8 and T10 that embrace the core of the structure does not increase the RMSD value significantly (0.49 Å).
The NMR structure provides unprecedented insights into the junction between the pseudocircular element and protruding 3′-ssDNA: To accommodate the 3′-end protrusion, the G9 base moiety involved in the N1-carbonyl, N7-amino-like geometry with G6 is tilted from the coplanar arrangement adopted by all the other guanines forming the G:G core. This leads to stacking of G9 and T10, which results in partial ‘opening’ of the pseudo circle in the ventral direction. This arrangement at the junction is further stabilized by the set of hydrogen bonds connecting the N3 and O5′ atoms of G−II at the 5′-end and the amino proton of G9 at the 3′-end of the discontinuous G-tract (Figure 4C). Although G11 is part of the flexible 3′-tail, its movement is partially restricted by a hydrogen bond between the amino proton of G11 and the phosphate group of G9. This interaction pulls G11 behind the stacked residues G9 and T10, further preventing steric clashes between the residues forming the junction.
PGH folding requires conformational flexibility at the particular positions
Detailed comparison of previously determined SC11 structure and SC14 structure determined in this study reveals that these two oligonucleotides occupy different topological forms of PGHs – form I and form II, respectively, which are characterized by two major structural differences. The first key difference between two PGH topologies includes altered base pairing register in the G:G cores (Figure 5A). While in form I the 5′-G is involved in the formation of a terminal G:G base pair of the PGH core, in form II the 5′-G participates in the middle G:G base pair. Second, considerable differences between form I and form II topologies exist in the relative orientations of the phosphodiester backbone of the loop-forming T10 with respect to the backbone of residues constituting the G:G core. While in form I PGH the backbone of T10 aligns close to parallel with respect to the backbone of the G:G core (cf. PDB ID: 5M1W), in form II the corresponding backbone is oriented perpendicularly (Figure 4C). Both altered base pairing register and T10 backbone orientation appear important for minimization of steric hindrances between the 5′-end of the pseudocircular core and protruding flexible 3′-tail of form II PGH (Figures 3D, 4A and C). Notably, despite the differences, both form I and form II PGHs share the same distribution of syn/anti glycosidic conformations of the guanines along the G-strands of the G:G core: syn–anti–anti along the G-strand with the reversed backbone chain element and anti–syn–anti along the G-strand containing discontinuous G-tract (Figure 5A). This observation suggests that the PGH structure can only be formed by sequences in which the core guanines can access the required combination of syn/anti glycosidic conformations at the particular positions.
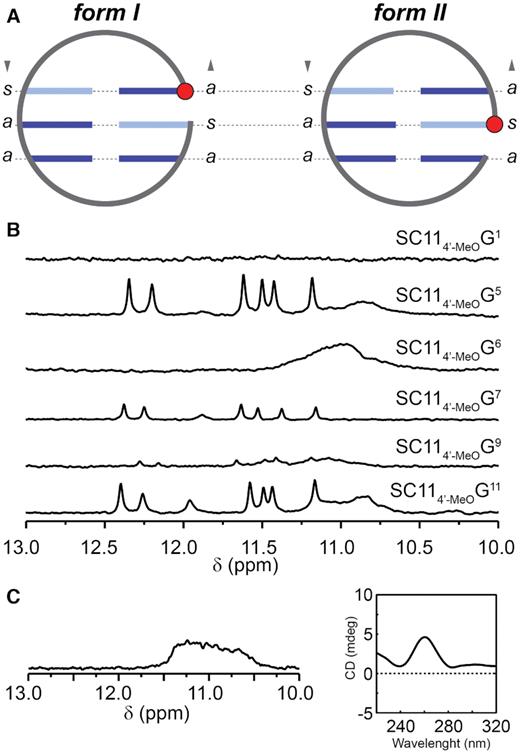
(A) Schematic representations of G:G cores in form I and form II PGHs. Note that both forms share the same distribution of syn (s) and anti (a) glycosidic conformations of guanines of the G:G core. The 5′-G involved in a core G:G base pair is marked with a red filled circle. Note: For the sake of simplicity, the chain-reversal element is not displayed. (B) Imino regions of 1D 1H NMR spectra of SC11-based constructs bearing 4′-alkoxy modification at indicated positions. (C) Imino region of 1D 1H NMR (left) and CD spectrum (right) of RNA counterpart of SC11, 5′-r(GUGUGGGUGUG)-3′.
4′-Alkoxy modifications imply restricted conformational freedom of some of the G:G core-forming residues of SC11
To address stereochemical requirements connected with a sugar unit, we assessed PGH formation for set of SC11 constructs site-specifically modified at the residues forming G:G base pairs (G1, G5, G6, G7, G9 and G11) with 4′-alkoxy group. 4′-alkoxy modified guanines (4′-MeOG) at selected sites were introduced to modulate the local conformational flexibility, namely increase preference for C3′-endo sugar conformation at modified position (17). Imino regions of 1D 1H NMR spectra of the constructs with modified G5 and G11 (Figure 5B) show signal patterns typical for PGH, suggesting that these constructs readily form PGHs. Although signal pattern typical of PGH was observed also for SC114′-MeOG7 construct, the signal intensities are notably reduced implying that the introduction of 4′-alkoxy modification at this position to some extent hinders the PGH formation (Figure 5B). In contrast, no imino signals characteristic of PGH formation were observed upon individual modifications of G1, G6 and G9. These results show that restrictions of conformational freedom at sugar moiety at positions 1, 6 and 9 of SC11 impair the formation of PGH structure (Figure 5B).
Collectively, these data implicate importance of the local conformational flexibility of the G:G core-forming residues for PGH formation and are coherent with the dynamic character of G:G base pairs in SC11 revealed previously using unrestrained MD simulation (14). The dynamic nature of the G:G base pairs in PGH structures that is inherently connected with requirements on local conformational flexibility implies that structurally rigid nucleotides such as those in RNA are to interfere with PGH formation. To address this implication, we also probed RNA analog of SC11, r(GUGUGGGUGUG), using NMR and CD spectroscopy. As evidenced from patterns of NMR and CD spectra (Figure 5C), the RNA analog does not adopt a PGH fold: its NMR and CD spectra have typical signatures of a (parallel) G-quadruplex (41).
Unrestrained MD simulations of SC14 reveal dynamic nature of G:G base pairs
Moving back to SC14, observation of an intricate network of NOE connectivities typical of different and mutually exclusive base pair arrangements indicated that the G:G base pairs in SC14 are also dynamic and switch between different base pairing arrangement (as we already showed previously for SC11). To assess potential interconversions between different base pairing arrangements in the G:G core of SC14, the derived (lowest-energy) NMR structure of SC14 was subjected to unrestrained MD simulations using three distinct explicit solvent models (cf. Material and Methods). Three independent 5 μs long simulations employing the OL15 AMBER force field augmented with HBfix term for G:G base pairing (27,42) were performed in each solvent model (Supplementary Table S1). Regardless of the solvent model, the SC14 structure was stable over the whole simulation period in at least two (out of three) simulations (Supplementary Table S3). All major structural features displayed by the NMR structure were preserved during the MD simulations, including the chain reversal and three G:G base pairs. Most importantly, inspection of the MD trajectories revealed the dynamic nature of the G5:G1 and G7:G−II base pairs, both switching between (major) N1-carbonyl symmetric (tWW) and (minor) N1-carbonyl, N7-amino arrangements (WC-H for G5:G1 and H-WC for G7:G−II) (Table 3). At the time-scale of the simulation (5 μs), almost no transitions were observed for the G6:G9 base pair, which remained in the N1-carbonyl, N7-amino WC-H-like state (Table 3). Lack of base pair transitions for the G6:G9 and H-WC arrangement for the G7:G−II base pair (presence of which was implicated by the NMR data) in the course of the MD simulations is likely to be a consequence of limited ability of MD to sample conformational space, which makes MD simulation outcome strongly dependent on geometry of starting (NMR) structure. The G:G core-forming guanines of SC14 retained their initial syn/anti glycosidic conformations during the unrestrained MD simulations. Most important, G−II did not experience almost any syn/anti reorganization during the MD simulations (Table 4). Interestingly, the situation was different in the case of SC11: guanine at equivalent position to G−II more often reorganized between syn/anti glycosidic conformations; additionally, unrestrained MD simulations of SC11 showed that the base pair at equivalent position (middle base pair) switched between N1-carbonyl symmetric and WC-H (14). These results indicate that the involvement of the 5′-guanine (G−II) in the middle base pair of the PGH energetically disfavors the formation of a base pair in the WC-H arrangement and pushes the equilibrium towards the N1-carbonyl symmetric and H-WC states. Our results of 4′-alkoxy modified SC11 constructs and NOE and MD data for SC14 together with the recent NMR and MD study of SC11 support a notion of inherently dynamic nature of PGH structures. Accordingly, the derived NMR structure of SC14, similarly to SC11 structure (14), should be considered as a representation of ensemble averaged experimental data.
Populations of G:G base-pair arrangementsa of SC14 and their transitions in the unrestrained MD simulations.b
| Population (%) | Average residence time (ns)c | |||||
|---|---|---|---|---|---|---|
| Base-pair of SC14 | WC-H | tWW | H-WC | WC-H | tWW | H-WC |
| G5:G1 | 3.1 | 94.6 | 0.0 | 23 | 594 | 1 |
| G7:G−II | 0.0 | 86.9 | 9.5 | 6 | 150 | 17 |
| G6:G9 | 98.6 | 0.0 | 0.0 | 4032 | – | – |
| Population (%) | Average residence time (ns)c | |||||
|---|---|---|---|---|---|---|
| Base-pair of SC14 | WC-H | tWW | H-WC | WC-H | tWW | H-WC |
| G5:G1 | 3.1 | 94.6 | 0.0 | 23 | 594 | 1 |
| G7:G−II | 0.0 | 86.9 | 9.5 | 6 | 150 | 17 |
| G6:G9 | 98.6 | 0.0 | 0.0 | 4032 | – | – |
aFor detailed description of base pair arrangements see Figure 3B.
bThe data are calculated over all simulations, taking the portions where PGH was present (see Supplementary Table S3).
cAverage time spent in a given base pair arrangement before transition into another one (or before reaching simulation end). Note that residence times on the order of thousands of nanoseconds are comparable to the length of the simulation; therefore they could be affected by the simulation-end artifact and would probably be longer if the simulations were longer.
Populations of G:G base-pair arrangementsa of SC14 and their transitions in the unrestrained MD simulations.b
| Population (%) | Average residence time (ns)c | |||||
|---|---|---|---|---|---|---|
| Base-pair of SC14 | WC-H | tWW | H-WC | WC-H | tWW | H-WC |
| G5:G1 | 3.1 | 94.6 | 0.0 | 23 | 594 | 1 |
| G7:G−II | 0.0 | 86.9 | 9.5 | 6 | 150 | 17 |
| G6:G9 | 98.6 | 0.0 | 0.0 | 4032 | – | – |
| Population (%) | Average residence time (ns)c | |||||
|---|---|---|---|---|---|---|
| Base-pair of SC14 | WC-H | tWW | H-WC | WC-H | tWW | H-WC |
| G5:G1 | 3.1 | 94.6 | 0.0 | 23 | 594 | 1 |
| G7:G−II | 0.0 | 86.9 | 9.5 | 6 | 150 | 17 |
| G6:G9 | 98.6 | 0.0 | 0.0 | 4032 | – | – |
aFor detailed description of base pair arrangements see Figure 3B.
bThe data are calculated over all simulations, taking the portions where PGH was present (see Supplementary Table S3).
cAverage time spent in a given base pair arrangement before transition into another one (or before reaching simulation end). Note that residence times on the order of thousands of nanoseconds are comparable to the length of the simulation; therefore they could be affected by the simulation-end artifact and would probably be longer if the simulations were longer.
Population of the syn glycosidic conformation of individual bases in the PGH core during the unrestrained MD simulationsa of SC14.
| Baseb | syn population (%) | Base | syn population (%) |
|---|---|---|---|
| G5 | 99.6 | G1 | 0.0 |
| G7 | 0.0 | G−II | 97.4 |
| G6 | 0 | G9 | 0 |
| Baseb | syn population (%) | Base | syn population (%) |
|---|---|---|---|
| G5 | 99.6 | G1 | 0.0 |
| G7 | 0.0 | G−II | 97.4 |
| G6 | 0 | G9 | 0 |
aThe data were calculated over all the simulations, taking the portions where PGH was present (see Supplementary Table S3).
bGuanines in syn glycosidic conformation as indicated by NMR data are in bold.
Population of the syn glycosidic conformation of individual bases in the PGH core during the unrestrained MD simulationsa of SC14.
| Baseb | syn population (%) | Base | syn population (%) |
|---|---|---|---|
| G5 | 99.6 | G1 | 0.0 |
| G7 | 0.0 | G−II | 97.4 |
| G6 | 0 | G9 | 0 |
| Baseb | syn population (%) | Base | syn population (%) |
|---|---|---|---|
| G5 | 99.6 | G1 | 0.0 |
| G7 | 0.0 | G−II | 97.4 |
| G6 | 0 | G9 | 0 |
aThe data were calculated over all the simulations, taking the portions where PGH was present (see Supplementary Table S3).
bGuanines in syn glycosidic conformation as indicated by NMR data are in bold.
Analysis of MD data for SC11 (14) and SC14 also shows that there are similarities in distribution of counter ions around form I and form II. Both forms have an increased cation density around phosphates (as is typical for nucleic acids) and along the PGH side facing N7 and O6 of guanines and O4 atoms of thymines. Nevertheless, the cation binding appears rather weak and the ions exchange with solvent frequently; their residency times are on the order of dozens of nanoseconds.
Comprehensive analysis of sequential requirements leading to form I and form II PGHs
Identification of the minimal motif able to fold into a form II PGH
Since a compact core of SC14 structure suggests that residues at 3′-end are not crucial for the structure integrity, we sequentially removed up to three 3′-residues succeeding G:G base pair-forming G9 to identify the shortest sequence still able to fold into form II PGH. Matching patterns of imino resonances on 1D 1H NMR spectra between SC14 and its truncated variants reveal that deletions are well tolerated and do not influence the formation of form II PGH (Figure 6). Equivalently to SC11 sequence, which represents the shortest sequential motif that leads to the formation of form I PGH, SC14ΔT10G11T+I construct corresponding to 11-nt long sequence 5′-d(GTGTGTGGGTG)-3′ thus characterizes the minimal motif, which enables the formation of form II PGH (Figure 6).
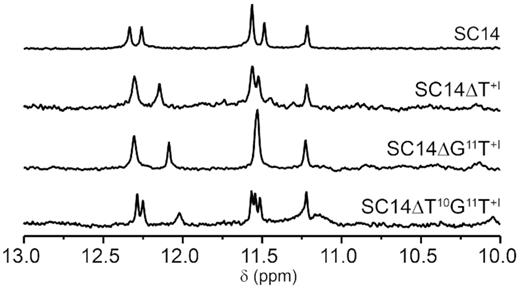
Imino regions of 1D 1H NMR spectra of SC14 and its truncated variants, namely ΔT+I, ΔG11T+I and ΔT10G11T+I.
Both topological forms of PGHs (form I and II) are characterized by compact G:G cores, which implies that the base pairing of six guanine residues play the most important role in PGH formation, while residues involved in the loop regions appear less crucial. To directly assess the influence of individual residues on PGH formation propensity, we analyzed two sets of 33 sequence variants corresponding to the single point mutants of the shortest (11-nt long) sequences forming form I and form II PGHs, respectively (see Supplementary Tables S4 and S5). The sequence requirements for the form I and form II PGH formation in relation to their position in the PGH structure is depicted in Figure 7A and can be schematically summarized in a form of sequence logos (Figure 7B). For both topological forms, constructs bearing mutations in one of the six G:G base pairs-forming residues result in 1D 1H NMR spectra devoid of signals in the imino regions (indicating the absence of a folded structure) (Supplementary Figures S8 and S9), which supports critical roles of these residues in the integrity and formation of form I and form II PGHs, respectively. In form I, mutations of residues in loops (T2, G3, T4, T8 and T10) promote the formation of PGH-like structures, as indicated by characteristic NMR and CD spectral fingerprints (Supplementary Figures S8 and S10). The only exceptions are constructs bearing T-to-G mutations (i.e., T2G, T4G, T8G and T10G constructs), which fold, as indicated by CD/NMR spectra and native PAGE into intermolecular G-quadruplex structures with a parallel folding topology (Supplementary Figures S8, S10 and S12). Interestingly, the formation of form I PGH is prevented upon T2A mutation as well, while cytosine or thymine at this position did not perturb the fold. The selectivity of pyrimidine over purine base at position 2 is in accordance with the high-resolution structure of SC11 (cf. PDB ID: 5M1W), where T2 fills the pocket behind the G5:G1 base-pair, leaving no place for purines imidazole moiety.
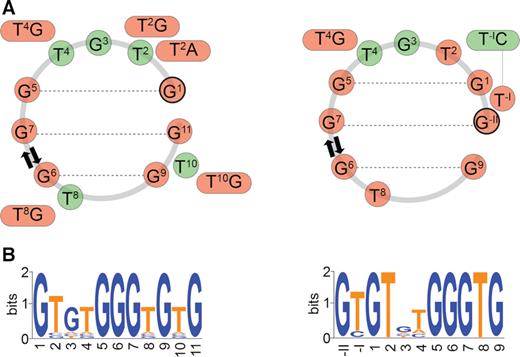
Schematic representation of the sequence requirements for form I (left) and form II (right) PGH formation in relation to their position in the PGH structure (A). Residues labeled with red and green circles correspond to the nucleotides critical and non-critical to PGH formation, respectively. (B) Sequence logos of minimal sequences capable to fold into form I (left) and form II (right) PGHs. For details on sequential logos determination, see Materials and Methods.
In comparison to mutations in the sequence of form I, minimal sequence able to fold into a form II PGH is, next to mutations of residues from G:G core, particularly sensitive to mutations of T2 and T8, which completely preclude the folding or lead to G-quadruplex formation (Supplementary Figures S9 and S11). With the exception of T4G construct of form II, which induces the formation of G-quadruplex structure(s), mutations of residues G3 and T4 allow formation of PGH-like folding topologies (Supplementary Figures S9, S11 and S13). Similarly, PGH fold is maintained upon T−IC mutation, while mutations of T−I to adenine or guanine are not tolerated and result in the formation of intermolecular G-quadruplex structures. This effect indicates that bulky base moieties (purines) at T−I position hinder the PGH formation through steric clashes. Together, these results demonstrate that form II is more sensitive to sequential variations in comparison to form I. However, the ability of 16 sequence variations to fold into either form I or form II PGHs demonstrates that the formation of these structures is not restricted to the selected SC11 or SC14 sequences and that other (although closely related) sequences can also adopt PGH structures.
Sequences able to form PGHs are frequent and not randomly distributed in metazoan genomes
The knowledge of minimal sequence requirements for PGH formation allowed us to screen putative PGH sites in genomic DNA across major phyla. We performed genome-wide bioinformatics screening of selected viruses, bacteria, and eukaryotes (including human) for sequences with potential to form PGH structures. The screen unveiled that putative PGH sites, while essentially absent in viruses and bacteria, are frequent in metazoan genomes (Supplementary Table S2). Intriguingly, in the human genome, putative PGH sites were found to be not randomly distributed and to preferentially cluster in gene associated regions, in particular in the introns of human genes (Supplementary Table S6 and Table 5): From 13 383 and 10 053 occurrences of putative PGH sites in the human genome (Supplementary Table S2), 6067 (∼45%) for form I and 4115 (∼40%) for form II are located in introns, respectively (Supplementary Table S6 and Table 5). Taking into account that some of these PGHs are overlapping and that human genome contains ∼22 000 genes, it can be estimated that between 10 and 20% of human genes contain at least one putative PGH site. Yet, some of gene introns might contain much higher number of these sites. For example, there are 210 non-overlapping PGH sites in the first intron of PTPRN2 gene associated with insulin-dependent diabetes mellitus (43); 182 of them have SC11 sequence surrounded by adenine residues (A_form I_A).
Occurrences of minimal sequences for form I and form II PGH formation in the important regulatory regions of human genome, respectively.
| Genomic feature | Occurrences of form I (unique genes) | Occurrences of form II (unique genes) |
|---|---|---|
| Introns | 1638 | 1554 |
| Promoters | 63 | 51 |
| 5′UTR | 23 | 17 |
| 3′UTR | 73 | 61 |
| CDSa | 15 | 16 |
| Exons | 103 | 85 |
| Genomic feature | Occurrences of form I (unique genes) | Occurrences of form II (unique genes) |
|---|---|---|
| Introns | 1638 | 1554 |
| Promoters | 63 | 51 |
| 5′UTR | 23 | 17 |
| 3′UTR | 73 | 61 |
| CDSa | 15 | 16 |
| Exons | 103 | 85 |
aProtein coding sequences.
Occurrences of minimal sequences for form I and form II PGH formation in the important regulatory regions of human genome, respectively.
| Genomic feature | Occurrences of form I (unique genes) | Occurrences of form II (unique genes) |
|---|---|---|
| Introns | 1638 | 1554 |
| Promoters | 63 | 51 |
| 5′UTR | 23 | 17 |
| 3′UTR | 73 | 61 |
| CDSa | 15 | 16 |
| Exons | 103 | 85 |
| Genomic feature | Occurrences of form I (unique genes) | Occurrences of form II (unique genes) |
|---|---|---|
| Introns | 1638 | 1554 |
| Promoters | 63 | 51 |
| 5′UTR | 23 | 17 |
| 3′UTR | 73 | 61 |
| CDSa | 15 | 16 |
| Exons | 103 | 85 |
aProtein coding sequences.
To assess PGH formation at internal sites of genome, we acquired NMR spectra for a representative set of five most frequently occurring PGH sites including their immediate surroundings, i.e., simultaneously extended at both ends by single flanking residue (Figure 8). Absence of signals in imino region of NMR spectra of T_form I_T, A_form I_A, T_form II_T, and A_form II_T suggests inability of these constructs to form PGH (or any stable defined structure). For remaining constructs, the NMR spectra indicate formation of folded species stabilized by Hoogsteen base pairs. However, in these cases, the CD spectra display characteristic shapes for anti-parallel/hybrid or parallel G-quadruplex structure(s) or their mixtures (Supplementary Figure S14) (41). Notably, none of these extended constructs displayed capacity to form PGH structure: While the constructs extended on one or both ends with G(s) generally display tendency to form G-quadruplex-like structures, non-G modifications on both 5′- and 3′-ends mostly prevented formation of any folded species.
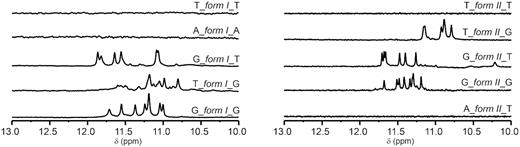
Imino regions of 1D 1H NMR spectra of a minimal PGH forming motifs (form I left, form II right) with their surroundings in human genome sorted by occurrence frequency.
PGH formation is under kinetic partitioning control
As highlighted in our previous study, PGHs can readily form in the presence of KCl, though their formation is diminished in a Na+-based solution (14). Considering that the native (intracellular) environment contains K+ (∼110 mM) and notable amounts of Na+ (∼40 mM), as well as other ions such as magnesium and calcium, gives rise to a logical question: Can PGHs form in complex ion-composed solutions such as the intracellular space? To address this question, we probed the formation of PGHs in buffers containing both KCl and NaCl at various ratios as well as in a complex buffer emulating an intracellular ion composition, referred to here as intracellular (IC) buffer (44). As substantiated by the characteristic PGH pattern of signals in the imino regions of 1D 1H NMR spectra of SC11 and SC14, PGHs readily form in mixed NaCl/KCl-buffers (even at excess concentrations of Na+ ions over K+) as well as in IC buffer containing physiologically relevant K+, Na+, Mg2+ and Ca2+ ion concentrations (Figure 9). Notably, the SC11, but not SC14 PGH, also formed in K+-based buffer supplemented with either Ficoll 70, PEG200 or glycerol, which are synthetic organic compounds commonly used to simulate intracellular molecular crowding (Supplementary Figure S15).
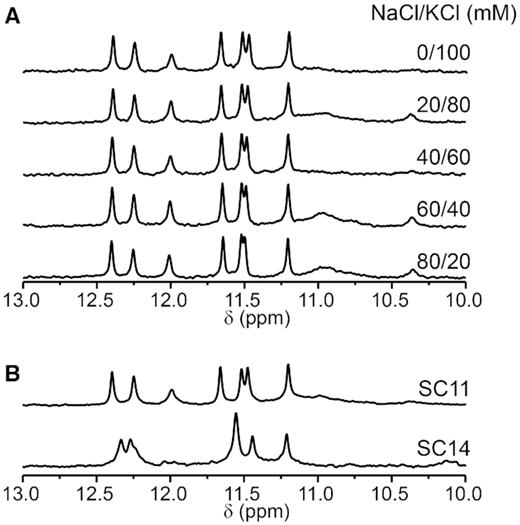
(A) Imino regions of 1D 1H NMR spectra of SC11 acquired at 10°C. Samples were folded in the presence of different ratios (indicated) of NaCl and KCl. (B) Imino regions of 1D 1H NMR spectra of SC11 (top) and SC14 (bottom) acquired at 10°C in a buffer emulating the ion composition of the intracellular space (25 mM KPOi, pH 7, 110 mM KCl, 40 mM NaCl, 1 mM MgCl2 and 130 nM CaCl2).
While these observations are in line with possible PGH formation under physiologically relevant conditions, they do not explain why PGH formation is K+ ion-dependent. We hypothesized that the ion type plays an important role in the stabilization of the folding intermediates rather than in the stabilization of the PGH structure. This hypothesis has been based on the following considerations: (i) the observed differences in the response of SC11 to different types of monovalent ions (K+ versus Na+) cannot be explained solely based on the PGH 3D structure, and performed MD simulations do not reveal any stably associated K+ ions with the structure (14), and (ii) reported previously (14) CD melting and annealing profiles of PGHs acquired in the presence of KCl displayed notable hysteresis, a common hallmark of systems with a complex folding pathway (45–48). To address the existence of potential (ion-type-dependent) folding intermediates, we monitored PGH folding in a time-resolved NMR/CD experiment under K+-/Na+-based conditions, i.e., in the presence of K+-phosphate buffer and KCl or Na+-phosphate buffer and NaCl, respectively (Figure 10 and Supplementary Figure S16). The time-resolved NMR spectra of SC11 acquired at 20°C under K+-based conditions indicated a direct, although remarkably slow, transition between the unfolded and folded (PGH) state (Figure 10A). In a time window spanning ∼ 27 min, the imino region of the 1D 1H NMR spectrum was essentially devoid of any signals, indicating the absence of a hydrogen-bond stabilized structure. Weak imino signals characteristic of PGH formation were detected after 53 min and steadily increased with time. In contrast, performance of the corresponding measurements at 1°C to slow down the folding kinetics and increase populations of the potential intermediate(s) provided a clear indication of a complex folding process. The imino region of the 1D 1H NMR spectrum acquired at 15 min after sample annealing displayed at least two sets of resonances (Figure 10B). The first set included a broad and unresolved hump centered at approximately 11.1 ppm (Figure 10B; gray rectangle) that might be related to the formation of a heterogeneous mixture of G-quadruplex-like species (note: the shapes of the corresponding CD spectra indicated that the G-quadruplex species might have a parallel topology—Supplementary Figure S16). The other set consisted of at least three signals at ∼ 11.3, 11.8 and 11.9 ppm (Figure 10B; solid lines). These signals were indicative of the formation of a species stabilized by either G–G, G–T or T–T base pairs. Most importantly, the intensity of these signals decreased with the simultaneous appearance and gradual increase of imino signals characteristic of PGH formation (Figure 10B; dashed lines). Observation of a coupled transition between the two states suggests the existence of at least one intermediate in the direct folding pathway (Ion) leading to the PGH structure.
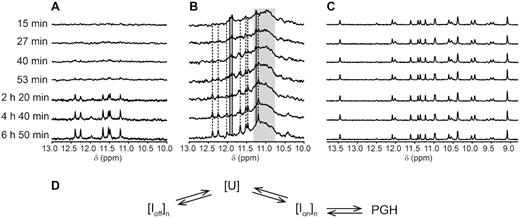
(A and B) Imino regions of 1D 1H NMR spectra of SC11 acquired at 20°C and 1°C as a function of the time after annealing and quenching on ice in potassium phosphate based buffer (10 mM KPOi, pH 7, 100 mM KCl), respectively. Solid and dashed lines are indicative of signal positions of putative K+ ion-dependent intermediate (Ion) and those corresponding to PGH, respectively. Gray rectangle marks the spectral region with overlapped non-resolved signals presumed to correspond to off-folding pathway G-quadruplex-like intermediate(s) (Ioff). (C) Imino regions of 1D 1H NMR spectra of SC11 acquired at 1°C as a function of the time after annealing and quenching on ice in the sodium phosphate based buffer (10 mM NaPOi, pH 7, 100 mM NaCl). (D) Schematic/Simplistic representation of the proposed mechanism of PGH folding. U, Ion and Ioff stand for unfolded ensemble, K+ ion-dependent on-folding pathway intermediate(s), and off-folding pathway G-quadruplex like intermediate(s)/product(s), respectively. n ≥ 1. Note: The structure of species formed under Na+-based conditions has not been investigated in detail as it goes beyond the scope of the present study.
Notably, the NMR spectra patterns acquired at 1°C in the presence of K+ ions were dramatically distinct from those observed in the presence of Na+. The complex patterns of signals in the imino regions of the 1D 1H NMR spectra of SC11 changed over time under K+-based conditions and indicated the coexistence of multiple species. In contrast, the corresponding NMR spectra recorded in the presence of Na+-phosphate buffer and NaCl were time-independent and featured only one set of well-resolved signals with a fundamentally distinct pattern from that observed in the spectra acquired under K+-based conditions (Figure 10C). Altogether, the observed time-resolved spectral patterns for SC11 recorded under K+-based conditions and at 1°C are consistent with the kinetic partitioning mechanism (49–53) of PGH formation, which may involve K+ ion-dependent on-pathway folding intermediate(s) and surely involves off-folding pathway intermediate(s)/product(s), most likely including a heterogeneous mixture of G-quadruplex-like species acting as kinetic traps (Figure 10D). The fact that these species escape NMR detection at 20°C can be explained by fast chemical exchange and/or by their low populations at that temperature. Note that the scheme (Figure 10D) is highly simplified. Both [U] and [Ioff]n could correspond to an extremely complex mixed ensembles of diverse conformations, including dimeric species, with diverse lifetimes. Further, their kinetic accessibilities and mainly lifetimes are assumed to be highly sensitive to ions, temperature and other conditions. Upon given experimental conditions, some components of [Ioff]n may form faster than the PGH; slowing the apparent PGH folding as detected in the experiments. Unambiguous characterization of these ensembles is probably beyond any presently available experimental method. The complicated topology of PGH in addition indicates that its folding time from completely unfolded (unstructured) single-stranded DNA should be longer compared to, e.g., simple stem–loop hairpins requiring straightforward zipping of a Watson–Crick stem. This would increase the likelihood that formation of PGH is kinetically outcompeted by some long-living components of [Ioff]n. Potentially, it may require to pass through a specific on-pathway intermediate (cf. Figure 10D).
The complexity of the SC11 conformational space suggesting the existence of multiple deep free-energy minima (basins on the folding landscape) provides a plausible explanation for the previously observed hysteresis between the SC11 PGH melting and annealing profiles (14). The thermally induced unfolding of a PGH most likely corresponds to a direct and fast transition between the folded and unfolded state; PGH folding corresponds to a slower, intermediate(s)-involving complex process, which is defined by kinetic competition between/among the on- and off-pathway and potentially on-pathway free-energy minima. Consequently, the system is unable to reach a state of thermodynamic equilibrium during the melting/annealing experiment, which leads to the observed hysteresis. The complexity of the folding process needs to be considered when interpreting experimental PGH melting data. At the same time, the indicated formation of K+ ion-dependent intermediate(s) in the folding pathway leading to PGH may provide a plausible explanation for the lack of SC11 PGH formation in the presence of Na+-based buffer and NaCl alone. However, the lack of detection of PGH under the Na+ conditions does not mean that the PGH hairpin is unstable per se (with respect to an unstructured state) but that it is outcompeted by some other structures to such extent that its signal gets below detection limit.
Implications of the stereochemical data for PGH biological relevancy
Our data have shown that PGH formation is not limited to a single (previously described—cf. (14)) sequence from S. cerevisiae telomeric DNA, but that there are a number of the parent sequence variants that readily form PGHs, many of which are overrepresented in evolutionarily conserved internal sites of the human genome. The presence of PGH-forming sequences in evolutionarily conserved and functionally important genomic locations, along with the demonstration of PGH formation in complex physiologically relevant buffers, is suggestive of active, yet unknown biological role(s) of PGHs. However, at the same time, there are several aspects revealed by the present study that argue against the biological relevancy of PGH structures in a genome. First, our data show that PGH folding is compromised by the presence of flanking residues concurrently protruding from both ends of the PGH structure. Second, folding of 3′-terminally extended PGH appears to be hindered in a crowded environment, which is an inherent property of intracellular space. Third, and most importantly, spontaneous folding of a PGH structure takes place on time scale of tens of minutes. This appears to be generally incompatible with the time scale of the vast majority of biological processes that usually operate on a time scale shorter than a minute. Considering these arguments, one plausible explanation for the evolutionary conservation of PGH-forming sequences might be that the PGH-specific sequence rather than the structure is required for a biological function. In this respect, the slow kinetics of PGH folding would maintain PGH formation-prone sequences in an unfolded state at time scales relevant to biological processes and could be a factor subjected to positive evolutionary selection (16). Nevertheless, the fact that the slow folding of PGH has been explained by kinetic partitioning mechanism means that under appropriate conditions (in absence of faster-forming long living competitors), PGH could likely form quickly. This opens a possibility that the PGH structure can act as a condition-dependent molecular switch.
However, it needs to be stressed that while these arguments have negative implications on the putative biological function of PGH structures, they cannot be considered conclusive. Similar to other structural studies, our data relates to the oligonucleotides, whose behavior might differ from that of sequences embedded in a genomic context, where their folding might be modulated by mechanical stress arising from induced DNA superhelicity/unwinding (54,55). It also needs to be pointed out that assessing the impact of the flanking residues on the PGH folding was performed only for a subset of possible nucleotide combinations and that the conditions used to simulate the impact of a crowded environment, i.e., solutions supplemented with PEG200, Ficoll 70, or glycerol, might not necessarily correspond to the natively crowded environment in the cell (56–60). An active biological role of PGHs would also be allowed presuming the existence of (non-)specific intracellular factors (e.g., PGH specific chaperones) that could assist and actively promote PGH formation in cells. In other words, the biological (ir-)relevance of both PGH structures and/or sequences at internal genomic sites remains to be uncovered.
CONCLUSIONS
Altogether, presented data suggest that PGHs, thus far considered a peculiarity of conformational space specific to a single DNA fragment from yeast telomeric DNA, might constitute novel structural class of DNA motifs that can, next to G-triplex and a number of left- and right-handed G-quadruplexes, be formed by G-rich DNA oligonucleotides (61–65). Similar to the G-triplex and left-handed G-quadruplex, the biological relevance of PGH remains to be revealed. However, its unique conformational properties could have relevance for the development of pharmaceuticals, genetic engineering, or DNA-based nanotechnologies. The later was recently demonstrated by exploiting the unique folding kinetics of PGH to calibrate electroporation devices (66). Novel insights into the sequence requirements and structural characteristics of PGHs contribute to our understanding of the structural diversity of G-rich oligonucleotides that could be applied to the prediction of natural and/or the design of artificial recognition DNA elements.
DATA AVAILABILITY
Atomic coordinates and list of chemical shifts have been deposited in the Protein Data Bank with the accession code 6R8E and in the Biological Magnetic Resonance Data Bank with the accession code 34386, respectively.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Czech Science Foundation [19-26041X to L.T., 17-12703S to I.R.]; ‘MSCAfellow2@MUNI’ [CZ.02.2.69/0.0/0.0/18.070/0009846 to M.L.Ž.]; SYMBIT [CZ.02.1.01/0.0/0.0/15_003/0000477 to J.S.] financed by the European Regional Development Fund with financial contribution from the Ministry of Education, Youth, and Sports of the Czech Republic (MEYS CR); Slovenian Research Agency [P1-242, J1-1704 to J.P.]; CERIC-ERIC Consortium and projects CEITEC 2020 [LQ1601]; National Programme for Sustainability II; CIISB [LM2018127] (co-)funded by MEYS CR for access to experimental facilities. Funding for open access charge: Czech Science Foundation [19-26041X].
Conflict of interest statement. None declared.
REFERENCES
Author notes
The authors wish it to be known that, in their opinion, the first three authors should be regarded as Joint First Authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comments