
Epigenetic Distribution of Recombinant Plant Chromosome Fragments in a Human–Arabidopsis Hybrid Cell Line

 ,
,  and
and
Abstract
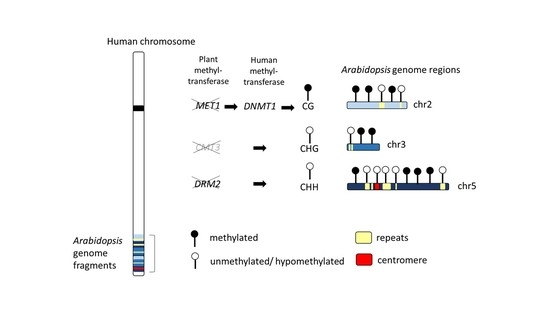
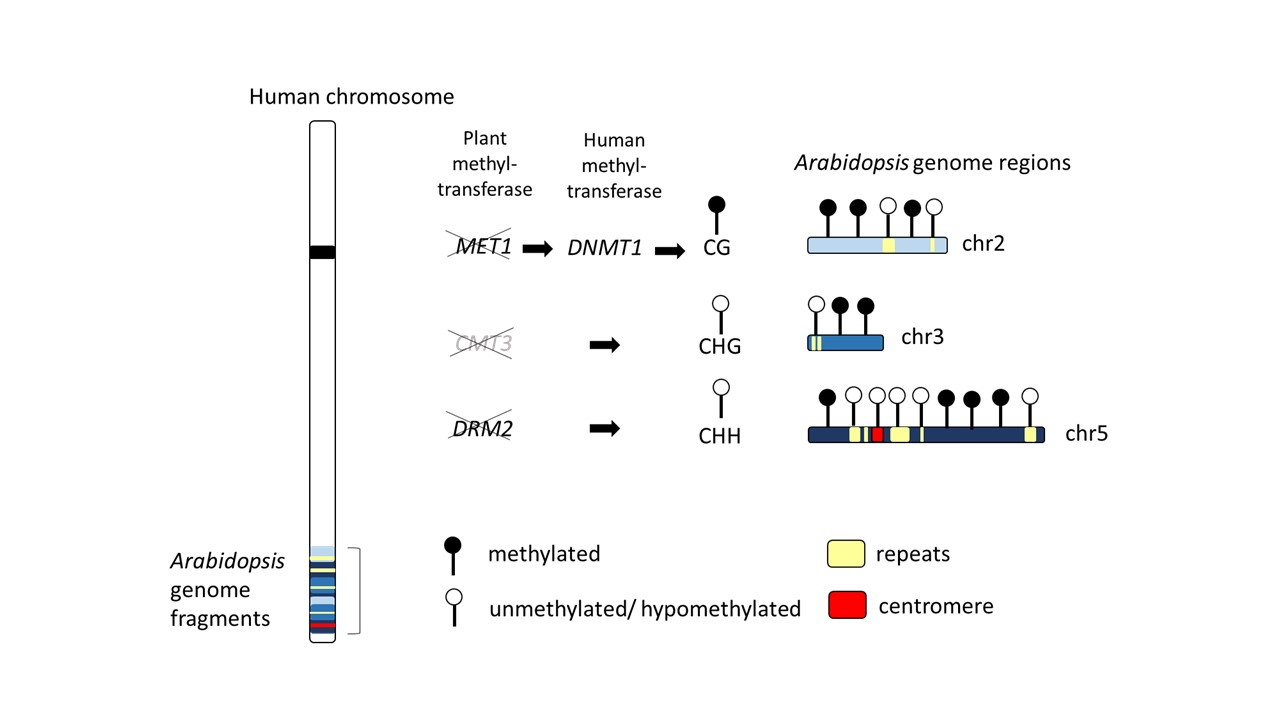
:
1. Introduction
2. Results
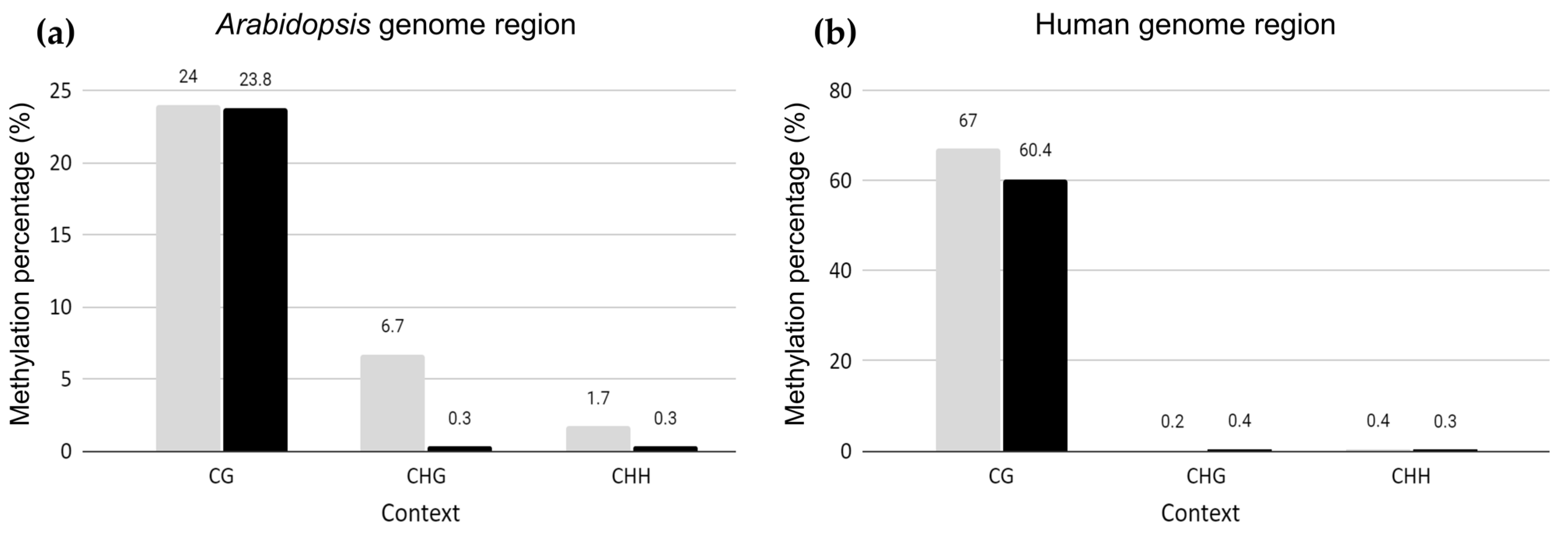
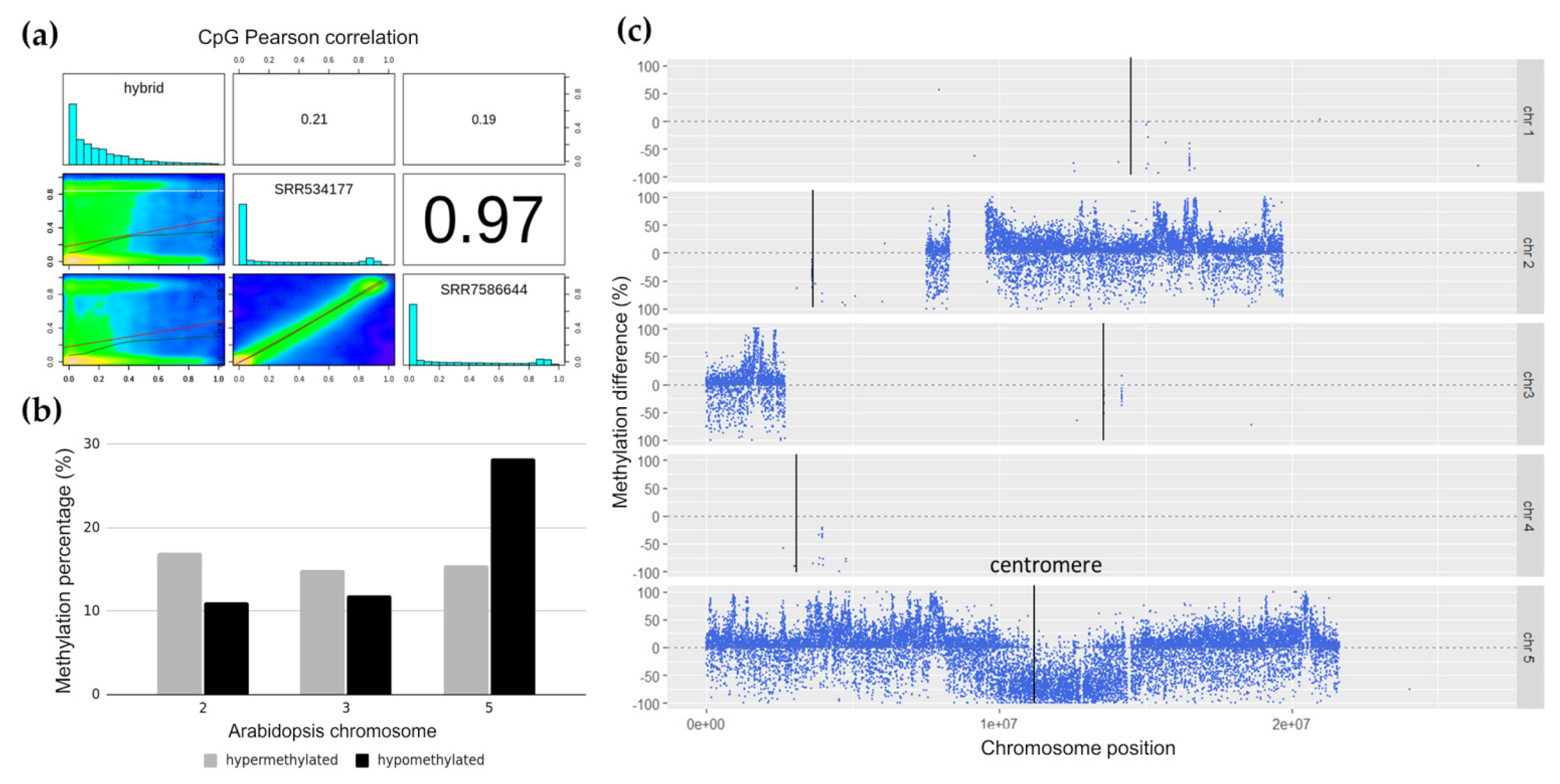
2.1. Genome-Wide Methylation Landscape of Plant Genome Fragments in Human Hybrid Cells
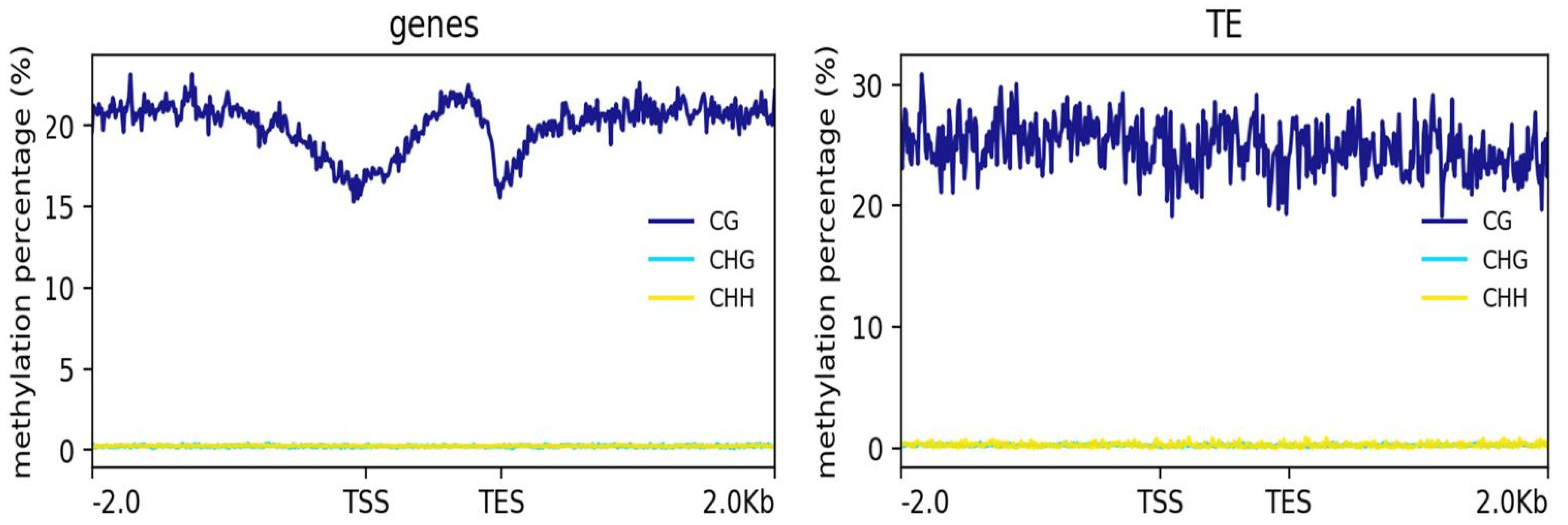
2.2. Patterns of DNA Methylation in Genes and TEs
2.3. Differentially Methylated Arabidopsis Genome Segments in Hybrid Cell Lines
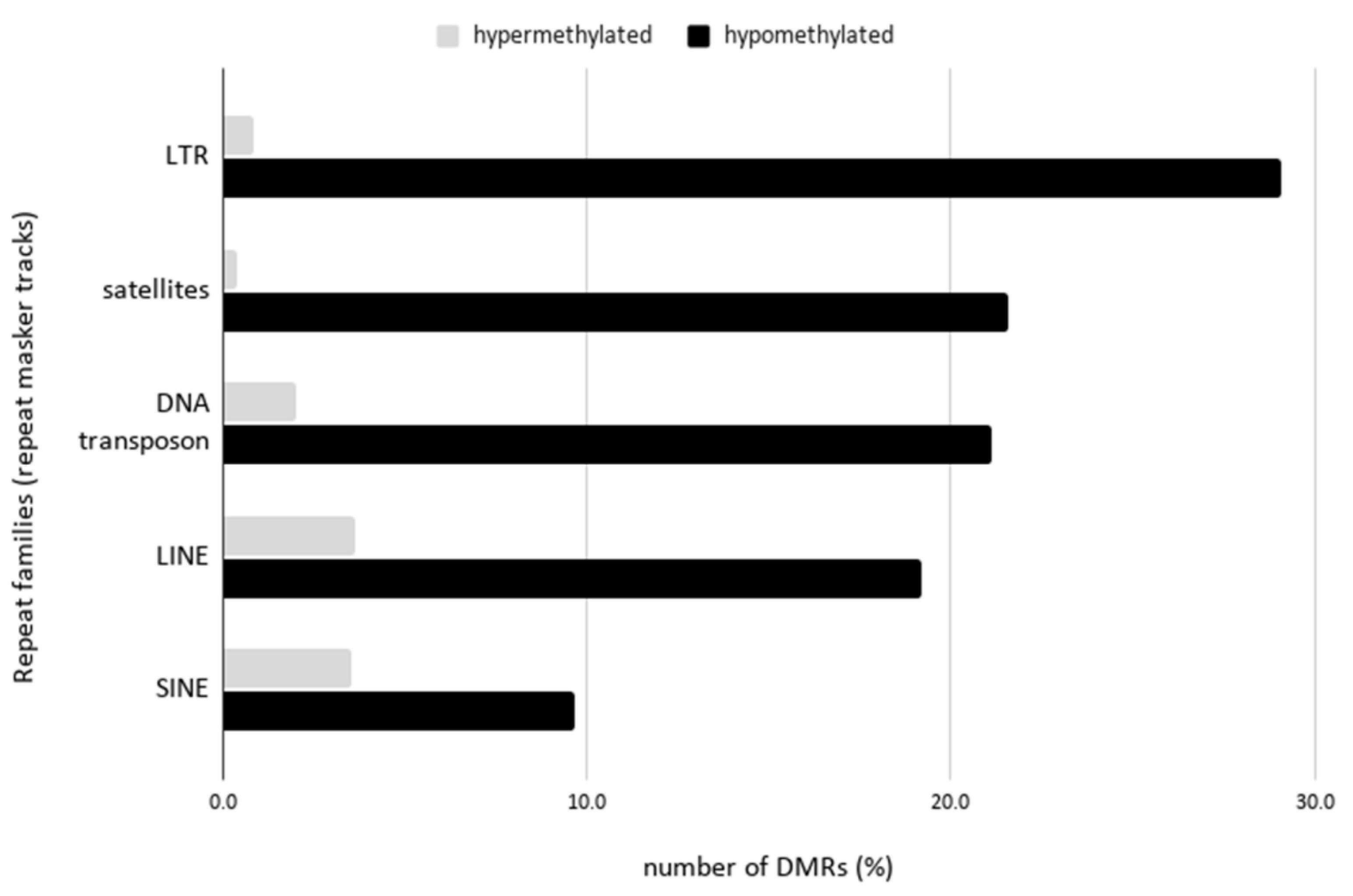
2.4. Hypomethylation of Arabidopsis Repetitive Elements in the Human Genome Background
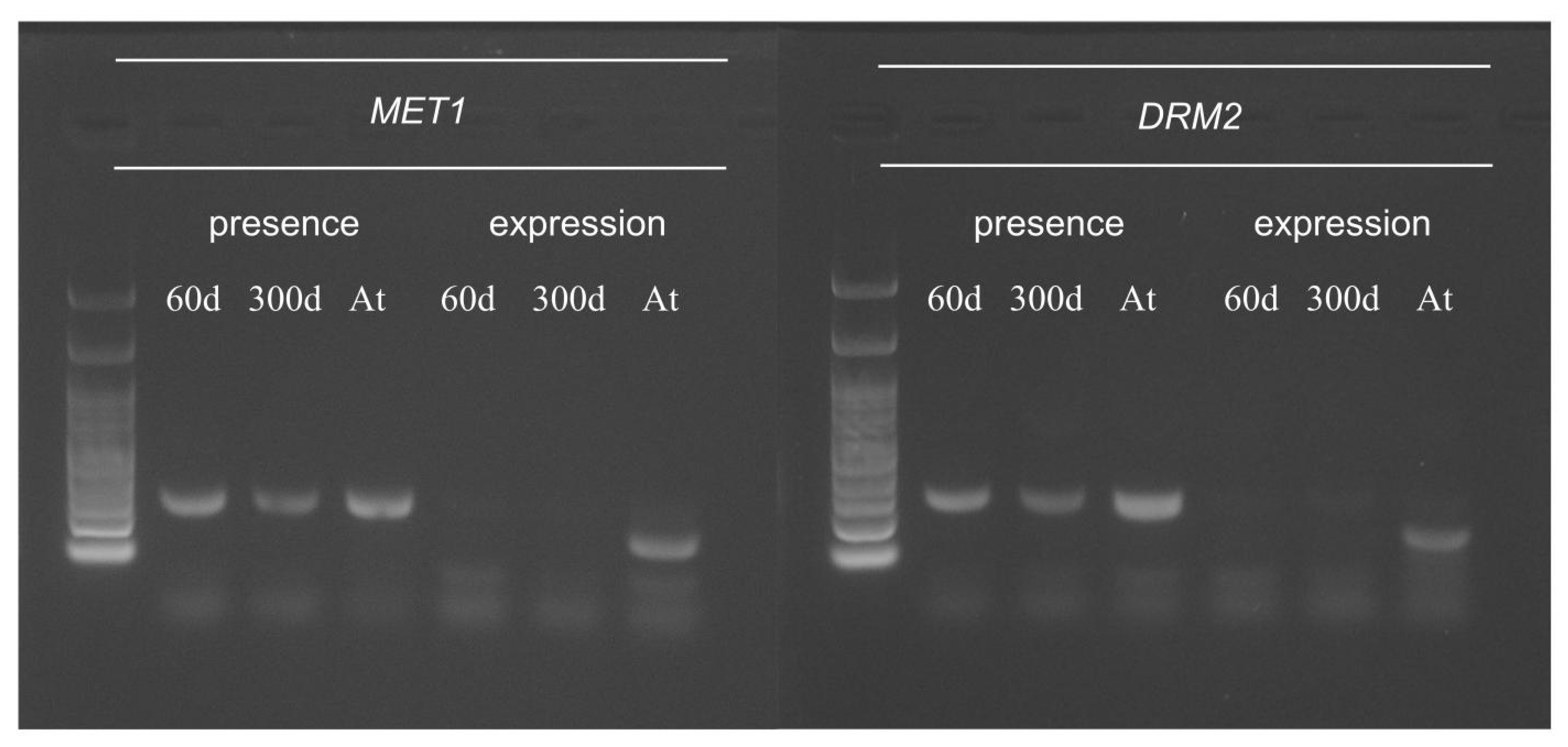
2.5. Function of Arabidopsis Methyltransferase in Hybrid Cells
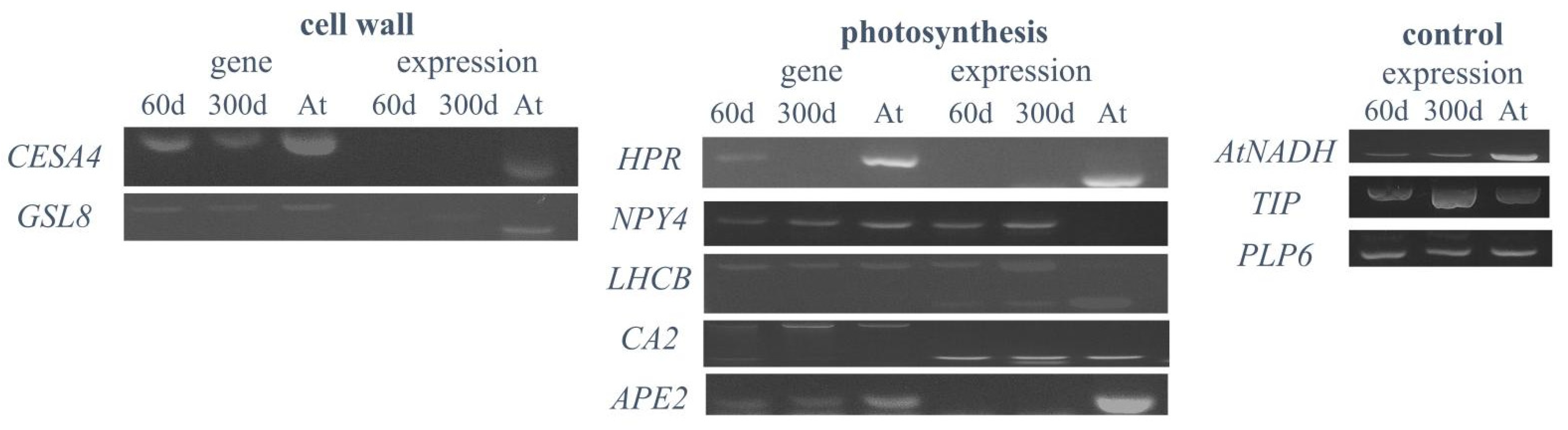
2.6. Expression Analysis of Plant-Specific Genes in Hybrid Cells
3. Discussion
3.1. Conservation of Genome-Wide Arabidopsis CG Methylation Level in Human Genome Background
3.2. Hypomethylation of Arabidopsis Centromere in 300-Day-Old Hybrid Cells
3.3. Hypomethylation of Arabidopsis Repetitive Elements in the Human Genome Background
3.4. No Preferential Expression of Original Arabidopsis Genes in Human Genome Background
4. Materials and Methods
4.1. Cell Culture
4.2. Plant Material and DNA Extraction
4.3. Whole-Genome Bisulfite Sequencing (WBGS)
4.4. WGBS Data Processing
4.5. Differential Methylation Analysis
4.6. Gene Expression Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Blomen, V.A.; Boonstra, J. Stable Transmission of Reversible Modifications: Maintenance of Epigenetic Information through the Cell Cycle. Cell. Mol. Life Sci. 2011, 68, 27–44. [Google Scholar] [CrossRef] [Green Version]
- Feng, J.X.; Riddle, N.C. Epigenetics and Genome Stability. Mamm. Genome 2020, 31, 181–195. [Google Scholar] [CrossRef]
- Ehrlich, M.; Gama-Sosa, M.A.; Huang, L.-H.; Midgett, R.M.; Kuo, K.C.; McCune, R.A.; Gehrke, C. Amount and Distribution of 5-Methylcytosine in Human DNA from Different Types of Tissues or Cells. Nucleic Acids Res. 1982, 10, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, Maintaining and Modifying DNA Methylation Patterns in Plants and Animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Zhang, X.; Yazaki, J.; Sundaresan, A.; Cokus, S.; Chan, S.W.-L.; Chen, H.; Henderson, I.R.; Shinn, P.; Pellegrini, M.; Jacobsen, S.E.; et al. Genome-Wide High-Resolution Mapping and Functional Analysis of DNA Methylation in Arabidopsis. Cell 2006, 126, 1189–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feinberg, A.P.; Vogelstein, B. Hypomethylation Distinguishes Genes of Some Human Cancers from Their Normal Counterparts. Nature 1983, 301, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P.; Gehrke, C.W.; Kuo, K.C.; Ehrlich, M. Reduced Genomic 5-Methylcytosine Content in Human Colonic Neoplasia. Cancer Res. 1988, 48, 1159–1161. [Google Scholar]
- Jin, Z.; Liu, Y. DNA Methylation in Human Diseases. Genes Dis. 2018, 5, 1–8. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA Methylation in Mammals. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W.-L.; Henderson, I.R.; Jacobsen, S.E. Gardening the Genome: DNA Methylation in Arabidopsis Thaliana. Nat. Rev. Genet. 2005, 6, 351–360. [Google Scholar] [CrossRef]
- Pikaard, C.S.; Mittelsten Scheid, O. Epigenetic Regulation in Plants. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Dennis, E.S. Isolation and Identification by Sequence Homology of a Putative Cytosine Methyltransferase from Arabidopsis Thaliana. Nucleic Acids Res. 1993, 21, 2383–2388. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Springer, N.M.; Muszynski, M.G.; Phillips, R.L.; Kaeppler, S.; Jacobsen, S.E. Conserved Plant Genes with Similarity to Mammalian de Novo DNA Methyltransferases. Proc. Natl. Acad. Sci. USA 2000, 97, 4979–4984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goll, M.G.; Bestor, T.H. Eukaryotic Cytosine Methyltransferases. Annu. Rev. Biochem. 2005, 74, 481–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colot, V.; Rossignol, J.-L. Eukaryotic DNA Methylation as an Evolutionary Device. BioEssays 1999, 21, 402–411. [Google Scholar] [CrossRef]
- Baulcombe, D.C.; Dean, C. Epigenetic Regulation in Plant Responses to the Environment. Cold Spring Harb. Perspect. Biol. 2014, 6, a019471. [Google Scholar] [CrossRef]
- Greaves, I.K.; Gonzalez-Bayon, R.; Wang, L.; Zhu, A.; Liu, P.-C.; Groszmann, M.; Peacock, W.J.; Dennis, E.S. Epigenetic Changes in Hybrids. Plant Physiol. 2015, 168, 1197–1205. [Google Scholar] [CrossRef] [Green Version]
- Laporte, M.; Luyer, J.L.; Rougeux, C.; Dion-Côté, A.-M.; Krick, M.; Bernatchez, L. DNA Methylation Reprogramming, TE Derepression, and Postzygotic Isolation of Nascent Animal Species. Sci. Adv. 2019, 5, eaaw1644. [Google Scholar] [CrossRef] [Green Version]
- Hochstein, N.; Muiznieks, I.; Mangel, L.; Brondke, H.; Doerfler, W. Epigenetic Status of an Adenovirus Type 12 Transgenome upon Long-Term Cultivation in Hamster Cells. J. Virol. 2007, 81, 5349–5361. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, R.J.W.; O’Neill, M.J.; Graves, J.A.M. Undermethylation Associated with Retroelement Activation and Chromosome Remodelling in an Interspecific Mammalian Hybrid. Nature 1998, 393, 68–72. [Google Scholar] [CrossRef]
- Remus, R.; Kämmer, C.; Heller, H.; Schmitz, B.; Schell, G.; Doerfler, W. Insertion of Foreign DNA into an Established Mammalian Genome Can Alter the Methylation of Cellular DNA Sequences. J. Virol. 1999, 73, 1010–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doerfler, W. Epigenetic Consequences of Foreign DNA Insertions: De Novo Methylation and Global Alterations of Methylation Patterns in Recipient Genomes. Rev. Med. Virol. 2011, 21, 336–346. [Google Scholar] [CrossRef]
- Weber, S.; Hofmann, A.; Herms, S.; Hoffmann, P.; Doerfler, W. Destabilization of the Human Epigenome: Consequences of Foreign DNA Insertions. Epigenomics 2015, 7, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Fitz-James, M.H.; Tong, P.; Pidoux, A.L.; Ozadam, H.; Yang, L.; White, S.A.; Dekker, J.; Allshire, R.C. Large Domains of Heterochromatin Direct the Formation of Short Mitotic Chromosome Loops. eLife 2020, 9, e57212. [Google Scholar] [CrossRef]
- Wada, N.; Kazuki, Y.; Kazuki, K.; Inoue, T.; Fukui, K.; Oshimura, M. Maintenance and Function of a Plant Chromosome in Human Cells. ACS Synth. Biol. 2017, 6, 301–310. [Google Scholar] [CrossRef]
- Liu, Y.; Liaw, Y.M.; Teo, C.H.; Cápal, P.; Wada, N.; Fukui, K.; Doležel, J.; Ohmido, N. Molecular Organization of Recombinant Human-Arabidopsis Chromosomes in Hybrid Cell Lines. Sci. Rep. 2021. [Google Scholar] [CrossRef]
- Xi, Y.; Li, W. BSMAP: Whole Genome Bisulfite Sequence MAPping Program. BMC Bioinform. 2009, 10, 232. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F.; Andrews, S.R. Bismark: A Flexible Aligner and Methylation Caller for Bisulfite-Seq Applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun Bisulphite Sequencing of the Arabidopsis Genome Reveals DNA Methylation Patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.C.; Pope, B.J.; Candiloro, I.L.; Korbie, D.; Trau, M.; Wong, S.Q.; Mikeska, T.; Zhang, X.; Pitman, M.; Eggers, S.; et al. MethPat: A Tool for the Analysis and Visualisation of Complex Methylation Patterns Obtained by Massively Parallel Sequencing. BMC Bioinform. 2016, 17, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akalin, A.; Kormaksson, M.; Li, S.; Garrett-Bakelman, F.E.; Figueroa, M.E.; Melnick, A.; Mason, C.E. MethylKit: A Comprehensive R Package for the Analysis of Genome-Wide DNA Methylation Profiles. Genome Biol. 2012, 13, R87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chèneby, J.; Ménétrier, Z.; Mestdagh, M.; Rosnet, T.; Douida, A.; Rhalloussi, W.; Bergon, A.; Lopez, F.; Ballester, B. ReMap 2020: A Database of Regulatory Regions from an Integrative Analysis of Human and Arabidopsis DNA-Binding Sequencing Experiments. Nucleic Acids Res. 2020, 48, D180–D188. [Google Scholar] [CrossRef] [PubMed]
- Klimopoulos, A.; Sellis, D.; Almirantis, Y. Widespread Occurrence of Power-Law Distributions in Inter-Repeat Distances Shaped by Genome Dynamics. Gene 2012, 499, 88–98. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.-Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and Divergence of Methylation Patterning in Plants and Animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef] [Green Version]
- Su, Z.; Han, L.; Zhao, Z. Conservation and Divergence of DNA Methylation in Eukaryotes. Epigenetics 2011, 6, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Long, H.K.; King, H.W.; Patient, R.K.; Odom, D.T.; Klose, R.J. Protection of CpG Islands from DNA Methylation Is DNA-Encoded and Evolutionarily Conserved. Nucleic Acids Res. 2016, 44, 6693–6706. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.V. Insights into Epigenome Evolution from Animal and Plant Methylomes. Genome Biol. Evol. 2017, 9, 3189–3201. [Google Scholar] [CrossRef] [PubMed]
- Lienert, F.; Wirbelauer, C.; Som, I.; Dean, A.; Mohn, F.; Schübeler, D. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011, 43, 1091–1097. [Google Scholar] [CrossRef] [PubMed]
- Krebs, A.R.; Dessus-Babus, S.; Burger, L.; Schübeler, D. High-throughput engineering of a mammalian genome reveals building principles of methylation states at CG rich regions. Elife 2014, 3, e04094. [Google Scholar] [CrossRef] [PubMed]
- Kohli, R.M.; Zhang, Y. TET Enzymes, TDG and the Dynamics of DNA Demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Gallego-Bartolomé, J.; Gardiner, J.; Liu, W.; Papikian, A.; Ghoshal, B.; Kuo, H.Y.; Zhao, J.M.-C.; Segal, D.J.; Jacobsen, S.E. Targeted DNA Demethylation of the Arabidopsis Genome Using the Human TET1 Catalytic Domain. Proc. Natl. Acad. Sci. USA 2018, 115, E2125–E2134. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genom. Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef] [Green Version]
- Erdmann, R.M.; Picard, C.L. RNA-Directed DNA Methylation. PLoS Genet. 2020, 16, e1009034. [Google Scholar] [CrossRef] [PubMed]
- Kenchanmane Raju, S.K.; Ritter, E.J.; Niederhuth, C.E. Establishment, Maintenance, and Biological Roles of Non-CG Methylation in Plants. Essays Biochem. 2019, 63, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matzke, M.A.; Mosher, R.A. RNA-Directed DNA Methylation: An Epigenetic Pathway of Increasing Complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, K.; Tomioka, S.; Suzuki, Y.; Nakamura, R.; Doi, K.; Yoshimura, J.; Kumagai, M.; Inoue, Y.; Uchida, Y.; Irie, N.; et al. Centromere Evolution and CpG Methylation during Vertebrate Speciation. Nat. Commun. 2017, 8, 1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinley, K.L.; Cheeseman, I.M. The Molecular Basis for Centromere Identity and Function. Nat. Rev. Mol. Cell Biol. 2016, 17, 16–29. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The Dark Side of Centromeres: Types, Causes and Consequences of Structural Abnormalities Implicating Centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secco, D.; Wang, C.; Shou, H.; Schultz, M.D.; Chiarenza, S.; Nussaume, L.; Ecker, J.R.; Whelan, J.; Lister, R. Stress Induced Gene Expression Drives Transient DNA Methylation Changes at Adjacent Repetitive Elements. eLife 2015, 4, e09343. [Google Scholar] [CrossRef] [Green Version]
- Wan, Z.Y.; Xia, J.H.; Lin, G.; Wang, L.; Lin, V.C.L.; Yue, G.H. Genome-Wide Methylation Analysis Identified Sexually Dimorphic Methylated Regions in Hybrid Tilapia. Sci. Rep. 2016, 6, 35903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, T.; Schranz, M.E. Network-Based Microsynteny Analysis Identifies Major Differences and Genomic Outliers in Mammalian and Angiosperm Genomes. Proc. Natl. Acad. Sci. USA 2019, 116, 2165–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Dai, X.; Cheng, Y.; Zhao, Y. NPY Genes Play an Essential Role in Root Gravitropic Responses in Arabidopsis. Mol. Plant 2011, 4, 171–179. [Google Scholar] [CrossRef] [Green Version]
- Tripp, B.C.; Smith, K.; Ferry, J.G. Carbonic Anhydrase: New Insights for an Ancient Enzyme. J. Biol. Chem. 2001, 276, 48615–48618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.A.; Dykes, D.D.; Polesky, H.F. A Simple Salting out Procedure for Extracting DNA from Human Nucleated Cells. Nucleic Acids Res. 1988, 16, 1215. [Google Scholar] [CrossRef] [Green Version]
- Krueger, F. Trim Galore. A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ Files; Babraham Institute: Cambridge, UK, 2015; pp. 516–517. [Google Scholar]
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, F.; Dündar, F.; Diehl, S.; Grüning, B.A.; Manke, T. DeepTools: A Flexible Platform for Exploring Deep-Sequencing Data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef] [Green Version]
- Simon, A.; Pierre, L.; Brian, H.; Phil, E. Babraham Bioinformatics-FastQC A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 15 January 2021).
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [Green Version]
- Han, S.; Kim, D. AtRTPrimer: Database for Arabidopsis Genome-Wide Homogeneous and Specific RT-PCR Primer-Pairs. BMC Bioinform. 2006, 7, 179. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Arabidopsis Methyltransferase | Methylation Context | Presence | Expression |
|---|---|---|---|
| DDM2/MET1 | CG | Yes | No |
| CMT3 | CHG | No | No |
| DRM2 | CHH | Yes | No |
| Gene | Locus | Encoding Protein | Gene Ontology | Log2 Ratio * | Expression in 60-Day-Old Cells | Expression in 300-Day-Old Cells |
|---|---|---|---|---|---|---|
| HPR | AT1G68010 | Hydroxyperuvase reductase | Cellular response to light stimulus, photorespiration, chloroplast | 3.22 | N | Gene absent |
| NPY4 | AT2G23050 | Naked Pins in YUC Mutants 4 | Positive gravitropism | 7.96 | Y | Y |
| LHCB4.3 | AT2G40100 | Light Harvesting Complex Photosystem II | Photosynthesis, light harvesting, response to light stimulus, chloroplast | 4.22 | Y | Y |
| CA2 | AT5G14740 | Carbonic anhydrase 2 | Carbon utilization, chloroplast | 9.06 | Y | Y |
| APE2 | AT5G46110 | Acclimation of Photosynthesis to Environment 2 | Photosynthetic acclimation, chloroplast | 7.01 | N | N |
| CESA4 | AT5G44030 | Cellulose Synthase A4 | Cell wall biogenesis | - | N | N |
| GSL8 | AT2G36850 | Glucan Synthase-Like 8 | Pollen development | - | N | N |
| AtNADH | AT5G11770 | NADH-ubiquinone oxidoreductase | Aerobic respiration | 11.23 | Y | Y |
| GAMMA-TIP | AT2G36830 | Gamma-tonoplast intrinsic protein 1 | Transmembrane transport, response to salt stress | 10.44 | Y | Y |
| PLP6 | AT2G39220 | Patatin-like protein 6 | Hydrolase activity | 10.28 | Y | Y |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liaw, Y.; Liu, Y.; Teo, C.; Cápal, P.; Wada, N.; Fukui, K.; Doležel, J.; Ohmido, N. Epigenetic Distribution of Recombinant Plant Chromosome Fragments in a Human–Arabidopsis Hybrid Cell Line. Int. J. Mol. Sci. 2021, 22, 5426. https://doi.org/10.3390/ijms22115426
Liaw Y, Liu Y, Teo C, Cápal P, Wada N, Fukui K, Doležel J, Ohmido N. Epigenetic Distribution of Recombinant Plant Chromosome Fragments in a Human–Arabidopsis Hybrid Cell Line. International Journal of Molecular Sciences. 2021; 22(11):5426. https://doi.org/10.3390/ijms22115426
Chicago/Turabian StyleLiaw, YengMun, Yikun Liu, CheeHow Teo, Petr Cápal, Naoki Wada, Kiichi Fukui, Jaroslav Doležel, and Nobuko Ohmido. 2021. "Epigenetic Distribution of Recombinant Plant Chromosome Fragments in a Human–Arabidopsis Hybrid Cell Line" International Journal of Molecular Sciences 22, no. 11: 5426. https://doi.org/10.3390/ijms22115426