
Abstract
The emerging use of qPCR and dPCR in regulated bioanalysis and absence of regulatory guidance on assay validations for these platforms has resulted in discussions on lack of harmonization on assay design and appropriate acceptance criteria for these assays. Both qPCR and dPCR are extensively used to answer bioanalytical questions for novel modalities such as cell and gene therapies. Following cross-industry conversations on the lack of information and guidelines for these assays, an American Association of Pharmaceutical Scientists working group was formed to address these gaps by bringing together 37 industry experts from 24 organizations to discuss best practices to gain a better understanding in the industry and facilitate filings to health authorities. Herein, this team provides considerations on assay design, development, and validation testing for PCR assays that are used in cell and gene therapies including (1) biodistribution; (2) transgene expression; (3) viral shedding; (4) and persistence or cellular kinetics of cell therapies.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Gene therapies (GTx) and cell therapies (CTx) are novel therapeutic modalities that have been on the rise in recent years for the treatment of various diseases. Gene therapy is a technique used to modify a gene to treat or cure disease by gene replacement, inactivation of a disease-causing gene, or introduction of a new transgene product into the patient (1). Gene therapy developers harness various delivery tools such as viral vectors, lipid nanoparticles, or various polymers to deliver DNA or RNA therapeutics into target cells (2, 3). Cell therapies introduce modifications to cells isolated from the patient themselves (autologous) or from a donor source (allogeneic). These ex vivo edited cells are then introduced into the patient to elicit a therapeutic response (4,5,6).
GTx and CTx modalities have advanced rapidly, this, together with their inherent complexity can be overwhelming; therefore, a well-considered bioanalytical strategy to measure exposure as determinants of efficacy and safety of these therapeutics is critical (7). Because the basis of the GTx and CTx are nucleic acids (DNA or RNA), molecular biology-based technologies, including quantitative polymerase chain reaction (qPCR), have become an important platform for bioanalysis supporting GTx/CTx drug development.
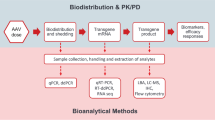
Figure 1 illustrates the molecular assays that are commonly used to support cell and gene therapies. Biodistribution, often as part of the pharmacokinetic analysis for GTx/CTx, characterizes the distribution and persistence of a gene/cell therapy product from the site of administration to target and non-target tissues and biofluids (8, 9) by measuring the vector genome or expression of the transgene, as well as drug-derived gene products. Vector shedding can be quantified by PCR and is described as the release of virus-based gene therapy products from the patient through excreta (feces), secreta (urine, saliva, nasopharyngeal fluids, etc.), and skin (pustules, sores, wounds) to understand the exposure of the virus to the environment and to potentially other humans (10). Persistence or cellular kinetic assays are conducted to measure the pharmacokinetics of the gene or cell therapy after administration and can be determined by PCR or flow cytometry (11, 12).

The molecular assays that are commonly used to support cell and gene therapies
There have been several pivotal publications that have served as landmark recommendations for PCR assays. One of the influential publications was recommendations for including minimum information required for publication of quantitative real-time PCR experiments, known as the MIQE guidelines. These recommendations had the intention of raising the standard for publishing reproducible and repeatable data in scientific journals (13), and later, MIQE guidelines were released for dPCR (14). Following the increase in cell and gene therapy drug development, several manuscripts and white papers from various authors in the industry have been published with recommendations and best practices for developing and validating bioanalytical PCR assays (15,16,17,18,19,20,21,22). International Organization for Standardization (ISO) standards have also been published and available (23). There have also been resourceful publications with recommendations specific for biodistribution and vector shedding (15) and cell therapies (e.g., CAR-T, chimeric antigen receptor) that utilize qPCR and dPCR for persistence and biodistribution (11, 12). These papers have been helpful resources and have provided framework and considerations for the industry to build on for validating PCR assays used in regulated bioanalysis, especially given the absence of regulatory guidance documents for molecular assay validations.
The purpose of this publication is to provide a framework for assay design considerations and best practices for validating regulated quantitative PCR (qPCR) and digital PCR (dPCR) assays that are used to support cell and gene therapy drug development. The assays that are within scope are related to cell and gene therapy drug development and include vector or transgene biodistribution, transgene expression, viral shedding, and cellular kinetics. Assays to support other therapeutic modalities such as vaccines, RNA-based therapeutics, or oligonucleotides are out of scope. In addition, other molecular platforms like branched DNA (bDNA) assays, hybridization assays, and sequencing or next-generation sequencing (NGS) are also out of scope of these recommendations.
To date, there has not been a regulatory guidance document with a descriptive approach on the validation of molecular assays that are used in regulated bioanalysis to support these novel modalities. Current regulatory guidance documents for bioanalytical method validation (24,25,26) are specific to ligand binding and chromatographic assay methods which differ greatly in workflow and mechanism from molecular methods, thus difficult to use as recommendations for PCR assays. To our knowledge, this is the first industry-wide manuscript dedicated to PCR assay validations to support gene therapies written by a consensus of scientists in pharma, biotech, and contract research organizations working in this field. These considerations seek to promote a harmonized and scientifically sound approach by presenting examples that are consistent with best practices but do not preclude alternative approaches. The proposed recommendations acknowledge that experimental design and acceptance criteria depend largely on the context of use (COU) of the assay. Ultimately, assays should be designed and validated to fit the purpose for which they are intended in support of the development of the therapeutic. The authors present best practice examples as a result of collective discussions. These examples are consistent with current standards and would suffice for most qPCR and dPCR assays that are for cell and gene therapy support.
Herein, the authors present their consolidated discussions on topics from assay design, critical reagents, and method validation for PCR assays supporting cell and gene therapy bioanalysis. Considerations specific to qPCR, RT-qPCR, dPCR, and RT-dPCR applications are presented where applicable.
Assay Design and Development Considerations
Primer and Probe Design
Design and selection of primers and probe for a bioanalytical PCR assay are critical for method development and validation. Continued discussion of best practices is warranted (15,16,17, 27), and the recommendations presented here seek to build upon that discussion through industry-driven consensus. Because a multitude of sources exist which cover the nuanced aspects of PCR primer and probe design, we will limit our scope to highlight the most relevant to regulated bioanalysis supporting cell and gene therapies.
With the advent of various design software, the selection of primers and probes for PCR assays requires less time and effort than it has in the past. Current design software (e.g., PrimerQuest™ from Integrated DNA Technologies, Primer Express™, Geneious™, and the public software Primer3) can select primer and probe sets from user-provided nucleic acid sequences through application of either default or customized PCR parameters (cation, dNTP concentrations, etc.). While the default parameters may generate primer and probe sets which prove functional, the user may customize their in silico design through specifying reagent concentrations, amplification conditions, and desired characteristics for primers, probes, and amplicons. Customization of these PCR parameters during in silico design may facilitate efficient empirical screening of candidate primer and probe sets since performance predictions are tailored to expected experimental conditions. It is generally advised to design and empirically test at least 3 primer and probe sets because performance predicted by in silico design may not always occur in actual use.
Online tools such as NCBI’s Primer Blast may also be employed preliminarily to ascertain the specificity of target amplification against the host genome/transcriptome, but specificity must be confirmed empirically in genomic DNA (gDNA) or total RNA extracted from naïve host tissues. It is also prudent and efficient from a therapeutic development program perspective to screen relevant candidate primers and probe sets in anticipated non-clinical and clinical biological matrices when possible. At minimum, it is recommended to screen relevant candidates in target tissues/biofluids from each of the species planned for use in non-clinical studies, as well as human target tissues/biofluids.
Design of primers and probes does not depend on the PCR platform. The process of amplification is the same between qPCR and dPCR, and the two platforms only differ in how the amplification products are measured and employed to determine starting template concentrations. If a set of primers and probe function well in a qPCR assay, there is no intrinsic reason it would not function in a dPCR assay, or vice versa. It is worth noting, however, that the tolerance of each method for differences in PCR efficiency may impact the selection of candidate primers and probe sets. For example, a set which is determined to have suboptimal PCR efficiency on a qPCR platform may yet be viable on a dPCR platform if positive partitions are still distinguishable from negative partitions. It is also worth noting that dye selection for the PCR probe may be dependent on the PCR reader instrument. Digital PCR platforms often require specific mastermixes with additives that affect reaction conditions. Primers and probes developed for qPCR shall therefore be validated for the dPCR platform using the appropriate dPCR mastermix.
Both qPCR and dPCR rely on fluorescent dyes or probes in the reaction to quantify the amplification of target sequences. The most used detection methods for PCR-based assays are double-stranded DNA (dsDNA) intercalating dyes (e.g., SYBR Green) or hydrolysis probe conjugated to a fluorescent reporter (e.g., TaqMan® probes). Relative to dsDNA binding dyes, probe-based detection methods have the advantage of additional specificity and the ability to monitor the amplification of multiple products in the same reaction (multiplexing). However, fluorescent probes require additional assay design considerations and tend to be more expensive.
At the strategic level of designing primers and probes, one should consider the assay purpose and COU (Fig. 2). For example, a PCR assay for a gene therapy which aims to measure a vector containing a non-codon-optimized transgene in a background of gDNA from the transgene’s species of origin. It may be advantageous in this case to target an assay primer or probe to an exon-exon junction or the junction of the transgene with an untranslated region specific to the vector construct. However, when detecting the expressed transcript, specificity for the vector-derived transcript could be conferred by targeting the junction of the transgene and neighboring vector component (e.g., promoter/5′ or 3′ untranslated region) which would be expressed in the vector-derived transcript but not in the endogenous transcript. This highlights the need to ensure that your developed assay can distinguish between vector-derived transcript and contaminating vector DNA, as well as endogenous transcript. In cases where DNA viral vector-derived transcripts cannot be distinguished from identically sequenced vector DNA, DNA removal steps prior to RT-PCR must be included, and a “No Reverse Transcriptase” (NRT) control should be included in sample testing to allow for the correction of RT-PCR results for copies of contaminating vector DNA. In the case of an exon-skip therapy, primers or probe should instead be targeted to the exon-exon junctions of the non-skipped and skipped forms of the gene. In dealing with gene-insertion therapies, assay development might require additional nuance in primer and probe design to permit the determination and quantification of sense insertions specifically. Another example of strategic design would be designing primers and probe specific to a vector backbone if the same backbone is used in vectors delivering different transgenes, unless transgene-specific quantification is desired. This would reduce method development burden in early program stages through application of a universal PCR assay. In all these application examples, proper targeting of the PCR amplicon and constituent primers and probe is dependent on the COU.

Recommended primer and probe sets targeting area. When the drug target gene is the same as the endogenous gene (not codon-optimized), the suggested PCR assay site is the promoter-target gene junction for detecting the vector only, or the target gene-WPRE (post-translation enhancement element, if any) junction (A), or the target gene-UTR junction (B), to detect the vector and transcript. If the target gene is codon-optimized (C), then the PCR and RT-PCR assay can target the area within the entire target gene with the least similarity to the endogenous sequence. PROM, promotor; WPRE, woodchuck hepatitis virus posttranscriptional regulatory element; PolyA, long chain of adenine nucleotides; UTR, untranslated region; Co-Opti, codon-optimized
In contrast, at the tactical level of PCR primer and probe design, development guidelines are driven by the mechanics of PCR amplification and have been refined naturally over time as collective PCR experience grows. A wealth of literature and resources exists for the technical aspects of PCR primer and probe design (15, 28, 29), so while we will not provide a detailed discussion of design considerations here, we will highlight a few of the most common design best practices:
-
Cas (CRISPR-associated proteins) provide, through complementary annealing, greater target specificity than intercalating dyes (e.g., SYBR) which bind double-stranded DNA independent of sequence. Double-quenched probes permit longer probe lengths to be used and stronger quenching, which can increase assay specificity and signal/noise ratio. Single-quenched probes have historically offered weaker quenching than double-quenched probes due to the distance between the fluorophore located at the 5′ end of the oligo and the quencher located on the 3′ half of the oligo. However, technological improvements such as incorporation of a minor groove binder (MGB)-coupled quencher can stabilize probe-target hybridization and increase Tm (melting temperature), allowing for shorter probes with quenching performance on par with longer double-quenched probes. The carboxyfluorescein (FAM) fluorescence label is typically used for singleplex assays due to the high signal intensity, and it is also typically the label of choice for low abundance targets in multiplexed PCR assays. The selected quencher for the probe should be compatible with the fluorophore. Depending on the modification of the hydrolysis probes, the availability may be limited to certain fluorophore-quencher combinations.
-
Amplicon length of 60–150 bp is typical due to having adequate space for annealing of primers and probes, as well as being a length that is easily amplified within common thermal cycling conditions for the most common Taq polymerases. Longer amplicons are possible but are subject to selected polymerase processivity constraints and thermal cycling optimization (e.g., increasing duration of extension step in a cycle). Designing for shorter amplicons may be advantageous for multiplex assays since shorter amplicons require shorter primers and probes, which are consequently less likely to interact with each other.
-
Target a Tm of 50–65°C for primers. The forward and reverse primer should not differ by more than 5°C when possible, to allow for both primers to anneal to the template at the same time. The annealing temperature during thermal cycling should be no more than 5°C lower than the Tm of your primers but can be optimized as needed.
-
Target a probe Tm which is 5–10°C higher than the primers to ensure target template sequences are saturated with probe at each amplification cycle.
-
Target 18–30 bases for primers and 20–30 bases for conventional single-quenched probes. Single-quenched probes longer than 30 bases may yield high signal background due to the large separation between the fluorescence dye at the 5′ end and the quencher at the 3′ end. Double-quenched probes typically have a second internal quencher relatively near to the 5′ end to decrease assay noise. Probes with modifications like MGB incorporation may permit probe lengths shorter than 20 bases.
-
Target a GC content of 35–65% for primers and probes, with 50% being ideal. In probe selection, avoid placing a G at the 5′ end since it can quench the conjugated fluorophore at the 5′ end, even after hydrolysis. Include more G and C bases at the 3′ end of primers when possible, to promote stronger binding at the 3′ end which supports priming of polymerase. Avoid G or C repeats longer than 3 bases when possible.
-
Primers and probe should be free of secondary structure (e.g., hairpins), and software should be employed to screen for homo- or heterodimerization among primers and probes to avoid intra- or inter-complementarity among all components, particularly in multiplexing.
-
When feasible, BLAST primer and probe sequences to ensure primer pairs are specific to your target and to ensure non-specific probe binding is minimized (which would affect reaction efficiency). When performing a BLAST check of primer pairs targeting exogenous sequences which are specific to drug or drug-effect, one should not observe potential productive amplicons in the genome or transcriptome of the host species. While in silico alignment checks can be beneficial early in screening, selected candidate sets must be tested empirically in the appropriate host nucleic acid background to confirm specificity.
-
Non-specific amplification or the formation of primer-dimers may also impact the PCR reaction due to the competition for PCR reaction reagents. For example, the formation of primer-dimers may pose a significant issue when quantifying low levels of target template with a probe-based qPCR assay since a reduced reaction efficiency results in a reduced assay sensitivity. It is therefore recommended to employ non-specific DNA binding dyes (e.g., SYBR Green) and perform melting curve analysis as a confirmatory measure early in probe-based qPCR assay development to discern if non-specific products are formed in addition to the specific PCR product (30).
-
Chemical modifications to primer and probe oligos can also offer significant PCR advantages. One of the most notable examples, Locked Nucleic Acids (LNA), are modified single nucleotides in a “locked” steric configuration which affects Tm and consequently allows for greater fine-tuning of complementary annealing. The use of LNA and other chemical modifications can be seen in various applications including guide RNA design for CRISPR, SNP detection, and stem-loop PCR for detection of short oligos.
Single and Multiplex Assays
PCR assays can be designed to quantify a single nucleotide sequence (singleplex reaction) or multiple nucleotide sequences (multiplex reaction). FAM generally constitutes the most common fluorescent dye of choice in most singleplex and multiplex reactions given its high fluorescence intensity (and hence the ability to detect even low abundance targets) along with the availability of appropriate optics on all instruments. While multiplex reactions offer potential advantages of reduced sample requirement, cost and time for assay set-up, the COU of the assay should be considered when designing a single versus multi-target assay. Multiplex assays are particularly attractive when measuring multiple targets in a single sample. Although most PCR systems can multiplex up to 4 different targets plus a passive dye, there is additional complexity to assays with more than one target that can result in assay challenges as well as extended development time. Choice of dye combinations that can be multiplexed is critical to ensure specificity while reducing background and improving signal/noise ratio. Care should be taken to ensure the use of appropriate excitation and emission filters to avoid overlap in signal between the targets of interest and/or the passive dye. The reliability of data from poorly designed multiplexed reactions could be compromised due to potential interactions between various primer probe pairs, targets and amplicons in question, preferential amplification of one target over another, reagent exhaustion, changes in primer concentration due to polymerase saturation, etc. Various free tools to ensure lack of hairpins, homo- and heterodimer formations among primers and probes, selection of appropriate dyes that can be multiplexed are available online and could be a useful resource while designing such assays (e.g., PrimeTime™ multiplex dye selection tool).
The decision to multiplex a PCR assay should be based on the COU of the assay. Assays where the target can be quantified based on input nucleic acid concentration (e.g., AAV-based) may not require any additional targets based on the COU of the assay such as biodistribution. Other assays such as RT-qPCR for gene expression, cellular therapies, or biodistribution, where one might desire a reportable result in terms of copies/cell, may require a reference gene to determine relative gene expression levels in comparison to constitutively expressed targets or to define the number of vector copies per cellular genome. In these cases, a multiplex assay allows for the simultaneous measurement of two targets or more, which creates efficiencies in the assay process and can decrease variability that could occur when comparing single target assays. However, care must be exercised in the application of reference gene normalization when analyzing samples originating from different tissues and biofluids as invariance might not be uniform among matrices and treatment. Because multiplex assays can be challenging, it is important to ensure comparable assay performance of each target in singleplex and multiplex format during the development stage. PCR efficiency in multiplex vs. singleplex should both be 90–110% with similar curve slopes, have similar limits of detection (LODs), and similar precision and accuracy performance. When multiplexing in dPCR, ensure that the droplets/partitions can be distinguished between the two targets and that the fluorophores do not compete. Similar principles apply for reference/housekeeping genes. If multiplexing results in an observed bias, Cq (quantification cycle, Ct) shifts, or imprecision in copy number calculations, it should be investigated during development.
Care should also be taken in the selection of the fluorophores based on available instrument settings and in the fluorophore selection for any potential cross-reactivity within the multiplex signal. This is especially important when there are targets of high abundance (e.g., reference gene/endogenous control) and low abundance (e.g., gene/target of interest) in the reaction. The use of high-quenching or double-quenched probes in such instances may help minimize crosstalk. Targets of low abundance should be reserved for channels with more robust signal (e.g., FAM) with minimal signal overlap, and targets with high abundance should be reserved for channels with lower signal (e.g., HEX, SUN). It might also be necessary to reduce primer concentration (primer-limited) of the more abundant target to prevent one target from likely outcompeting the other for reagents and “hijacking” the reaction or choosing a reference gene that is less abundant. Other troubleshooting steps to consider during optimization could include, but are not limited to, thermocycling conditions, and potential redesign.
Assay Platforms and Instrumentation Classes
Understanding the COU should drive assay design for developing a suitable and appropriate assay characterized for its intended purpose, including the selection of the most appropriate assay platform and detection chemistry. Quantitative PCR has been around for several decades and is still considered one of the most sensitive technology platforms used in drug development. In recent years, the rise of new technologies like digital PCR (dPCR) has also garnered a lot of interest due to its ability for absolute quantitation without requiring a standard curve. A comparison between traditional qPCR and dPCR has been discussed and summarized in previous publications (17) with certain advantages for each platform.
Both qPCR and dPCR can provide accurate and precise quantification of a nucleic acid target of interest. Thus, the selection of platform between qPCR and dPCR is also driven by considerations for COU. Quantitative PCR has a broader dynamic range than dPCR (7 orders of magnitude versus 5, respectively) and detects changes as low as twofold, while dPCR has an advantage for higher precision for quantifying rare targets and for detecting changes as low as 10%. Additionally, dPCR reactions are less susceptible to the presence of inhibitors that may be present in complex biological matrices such as feces (31). Thus, dPCR platforms may be preferable for analyzing challenging matrices. Initially, dPCR instruments were capable of detection of just one or two colors limiting the potential for assay multiplexing. However, newer dPCR platforms detect 4–6 colors, allowing for the multiplexing targets in a single well like qPCR systems available.
The binary quantification in dPCR might be particularly advantageous in certain applications, such as monitoring exon skipping in an antisense oligo (ASO) therapy, where it has been shown that qPCR over-estimates the shorter skipped transcript, but dPCR enables accurate and precise quantification of both non-skipped and skipped transcripts (32). Digital PCR might also offer an advantage over qPCR when quantifying sequences with secondary structures (e.g., inverted terminal repeat sequences) or when there is some unavoidable PCR inhibitor. Certain dPCR platforms can also support multiplex assays which allow one to determine if two amplicons are linked on the same template in a partition, permitting determination of sequence contiguity or the “intactness” of a target. dPCR is also seeing increased use as a characterization method to establish nominal therapeutic concentration in production (e.g., viral genome titers).
By the same token, qPCR might offer advantages over dPCR depending on COU. For example, one limitation of dPCR is that the application of Poisson statistics to the quantification mechanism assumes that analyte templates are independently assorted into partitions. In the case of possible concatemerized vector genomes in a gene therapy, a dPCR assay would be unable to distinguish a single episome from vector genome concatemers without targeted enzymatic digestion or assay designs specific for concatemer forms. Quantitative PCR is also less costly than dPCR in reagent, and instrument cost currently and generally offers higher sample throughput. This may be desirable, for example, to support a large non-clinical biodistribution study where many tissues and biofluids will require analysis. The larger dynamic range of qPCR would also offer the potential for more efficient sample testing since high-concentration samples would fall within the quantitative range of the assay without the need for dilution and reanalysis of samples which initially report as above the limit of quantification (ALQ). A comparison of advantages of qPCR versus dPCR for cell and gene therapies is summarized in Table1.
Sample Collection Considerations
In general, extracted DNA and RNA should be handled, collected, and stored in DNase and RNase-free materials or tubes. For biodistribution assays, tissues are typically collected and stored frozen until processed by homogenization and nucleic acid extraction. General considerations for collecting tissues for non-clinical biodistribution studies are noted in the ICH S12 Guidance (33). For biodistribution and transgene expression assays, it is important to consider the tissue sampling size for collection, especially for RT-qPCR to ensure proper perfusion of RNA preservatives during collection and storage. It is also important to include a collection of samples to be stored for back up if reanalysis is needed.
For shedding assays, the types of matrices that are collected are dependent on various factors including route of product administration, vector tropism, and biodistribution or shedding data from preclinical studies (10). Common matrices can include urine, saliva, tears, and feces. There are various methods, although not an exhaustive list, that can be used for collection of shedding matrices for tears (e.g., Schirmer strips), feces (e.g., OMNIgene™ gut tube, fecal swabs), saliva (e.g., collection cups, sponges), urine (e.g., collection cups), and wound dressing swabs.
For RNA, whole blood or tissues can be collected in the presence of RNA preservatives or stabilizing agents (e.g., PAXgene® tubes, RNAlater®, Qiagen ALLProtect®) (34, 35). Most traditional RNA purification procedures take place in the presence of these RNase inhibitory agents. However, it is typically prior to and after the extraction when RNA integrity is at the highest risk. Stabilizers such as RNAlater® enable conservation of the samples without sacrificing the integrity of the RNA. The use of these RNAlater® solutions for tissue storage is compatible with most RNA extraction procedures.
Regardless of sampling technique, handling, and storage, it is important to know the method of sample collection to ensure the extraction method is optimized for sample analysis appropriate for the way it is received. Different sampling methods can be tested in method development to optimize extraction and understand compatibility and effect of any stabilizing reagent on the extraction or downstream qPCR method.
Extraction Considerations
The sample processing procedure comprised of sample collection, transport, storage, and nucleic acid extraction may contribute more confounding variation than the actual analytical PCR procedure. Sample collection and storage conditions through various mechanisms (e.g., nuclease activity) may impact the integrity of nucleic acids extracted from biological tissues and fluids, but a discussion of these mechanisms is beyond the scope of this publication. As the most proximal step to the analytical PCR procedure, nucleic acid extraction is considered part of the overall method to be included in development and validation of the PCR assay. When developing a DNA or RNA extraction protocol, it is critical to define the characteristics of the tissues/cells from which the material is extracted and then select the tools that will lead to the best results, as effectiveness of the method is directly related to the nature of the sample. The mean efficiency of nucleic acid extraction may differ from sample type to sample type, since the biochemical composition of tissues and fluids differ. In addition, in most cases, it is difficult to measure extraction efficiency of a contrived sample that may not be representative to the biology of the actual study sample that is being analyzed. Variation in the efficiency of nucleic acid extraction is a large contributor to measurement variation during sample analysis of biological tissues and fluids, and it underlies analytical variability delineated in all analytical method parameters evaluated during method development and validation. The abundance and complexity of extraction platforms, methods, and biological sample types prevent the inclusion of extraction efficiency variability when defining analytical variability. Therefore, while we recommend the characterization of extraction efficiency during method development to further understanding of your analytical PCR method, its specific application in method validation is subject to the context of use and should be determined on a per-assay basis. It is considered acceptable if due diligence is performed in the characterization of extraction efficiency to a level that helps understand the reproducibility of the extraction process.
Multiple techniques and tools are available for the homogenization of tissues and extraction of nucleic acids. Homogenization methods are generally grouped into physical methods, enzymatic methods, chemical methods, or a combination of the three. Physical methods typically involve some type of sample grinding or bead-beating to breakdown connective tissues and disrupt the cell membranes or tough tissue, while chemical methods can be used alone with easy-to-lyse materials, such as tissue culture cells or in combination with other methods. Enzymatic methods are often used with more structured starting materials in combination with other methods. The enzymes utilized help to disrupt tissues and tough cell membranes. Depending on the starting material, typical enzymatic treatments include proteinase K, collagenase, and lipase. In many protocols, a combination of chemical and enzymatic disruption is often used since chemical disruption of cells rapidly inactivates proteins, including nucleases. Depending on the sample type from which DNA or RNA must be extracted (i.e., solid tissue, fluid, fatty tissue, stool), a first-step disruption (e.g., homogenization, proteinase digestion), rather than a single processing step, might be required to obtain the best results. The need to process large numbers of samples from various types of tissues, like in the case of biodistribution studies, might also require a trade-off between the level of effective homogenization and throughput. Finally, the weight of tissue or volume of samples as well as their storage should also be considered when determining the best homogenization protocol. This is specifically relevant for methods developed to support clinical studies, in which samples might be limited if they originate from biopsies. Fibrous tissues, tissues rich in proteins, nucleases, and lipids present unique challenges such as complete cell lysis when preparing tissue homogenates. Extensive optimization and modifications of nucleic acid isolation methodologies may be required for the extraction of nucleic acid from the heart, brain, thymus, and spleen. Some examples of recommended modifications are thorough disruption, snap freezing, dilution of lysate, additional extractions, efficient homogenization, and adding tissues to cellular nucleic acid stabilizers. For shedding analysis, it is important to ensure the extraction method can fully open the capsid (viral or non-viral) to release the DNA/RNA therapeutics to be quantified.
The choice of extraction method for nucleic acids has an impact on the overall quality and quantity of the extracted material as well as the target. It is therefore critical during method development to select an optimized method for different sample types. The extraction method used for a fibrous tissue may not be applicable for fatty tissues or sample types with known inhibitors, such as stool. There are generally three basic methods, with some variants, that are used to extract DNA in the context of biodistribution studies: silica, magnetic beads, or organic-based extraction. Silica and magnetic bead extractions both yield highly purified DNA and are amenable to automation, while organic extraction also yields good quality DNA at a low cost but is time consuming and uses hazardous material. Genomic DNA extraction poses several challenges due to the wide range of potential starting material and differences in extraction methodology may ultimately lead to differences in identification of the target sequence by PCR. As such, there is no single “gold standard” by which DNA should be extracted, and optimization of extraction methodologies will be dependent on the required molecular weight of the target DNA, required quantity, and purity. In all cases, one should aim during method development to select a method that will lead to purified target DNA free of contaminants, including proteins, other cellular components, and undesired nucleic acids.
The choice of extraction method is critical for RNA given the susceptibility to RNase manipulation as compared to DNA. As stated above, the extraction protocol must also be optimized to ensure the best quality elution material. Therefore, careful consideration must be given regarding the treatment and handling of samples prior to RNA isolation, storage of the prepared RNA sample, and choice of technologies used to prepare the RNA. Several RNA preparation technologies are widely available that can be classified into four general techniques: organic extraction methods, spin basket formats, magnetic particle methods, and direct lysis methods. While all can be used to prepare high-quality RNA suitable for a wide variety of analysis techniques, there are several factors to consider in selecting the right purification technology such as the following: (1) Difficulty in managing the sample; tissues that are high in nucleases or fatty tissues, and samples with high amounts of inhibitors, can present problems; (2) Quantity of sample for processing; larger sample sizes require kits that contain scalable chemistries. Generally, the larger the sample, the lower the throughput. Moreover, the incorporation of a DNase digestion step to further reduce DNA contamination should be considered if not already integrated in the protocol used. For RT-qPCR, when primers lie within a single exon or when the amplification target sequence appears in both the vector and vector-derived transcripts, DNA removal should be included as a part of the extraction to limit the potential for gDNA carryover as well as ensure there is minimal interference in RNA quantification by contaminating DNA. For shedding assays, enzyme-based or alternate methods can be used to provide greater confidence in the measurement of intact or infectious vector. DNA is typically removed from RNA extractions through the use of gDNA elimination columns or enzymatic digestion (e.g., DNase). If DNA removal columns are used in place of enzymatic digestion, the impact or potential loss of target and assay consistency after column purification must be characterized. The carryover of gDNA or vector DNA and consequent non-transcript amplification can be assessed through the application of a no reverse transcriptase (NRT) control. Significant amplification in the NRT controls will determine whether the current DNA removal steps in the method are sufficient. Pros and Cons of each RNA isolation methods as well as guidance regarding the best method to use depending on the sample type to be processed and downstream analysis are easily accessible on multiple vendor’s site and should be consulted at the time of development.
Beside analytical factors, the extraction of DNA and RNA can be altered by endogenous factors that might be difficult to control, especially in the context of clinical studies. Indeed, factors related to patient health and sample collection can impact the overall quantity and quality of the extraction. Therefore, during the development of a method, one should consider using samples that are as close as possible to the samples that will be used in a study to better predict the outcome and select the appropriate protocols consequently.
When processing shedding samples, there is a low expectation for DNA or RNA yield. For liquid shedding matrices such as tears, saliva, and urine, a measured volume is input into the extraction method so that the assay’s resulting copy number can be back calculated to be reported as copies per milliliter. For solid shedding matrices such as feces, a measured mass is input into the extraction method so that the assay’s resulting copy number can be back calculated to be reported as copies per milligram. There is no need to measure nucleic acid concentration for these samples. Loading equal undiluted volumes to the PCR assay so as to add the highest concentration of material has the best chance of capturing any rare shedding instances.
Nucleic Acid Quantification and Automation
Accurate quantitation and assessment of sample purity of nucleic acid is critical in molecular workflows. Optical technologies such as photometry (UV) and fluorescence are commonly used. Quantitation methods are varied and have different sensitivities and accuracies. The theoretical dynamic range for a spectrophotometer with a 1 cm path length is 5–75 ng/µL, whereas nanodrops can have path lengths of 0.005 cm equating to theoretical dynamic range of 2–15,000 ng/µL. This range allows for quantitation of high dsDNA concentrations but is not sensitive at the lower end of the range and could be problematic for matrices with low concentrations of nucleic acids such as plasma, cerebrospinal fluid, urine, single cells, and FFPE tissues. In addition to lower sensitivity, lower accuracy is also a disadvantage. Nucleic acids absorb maximally at 260 nm, while other common contaminants absorb strongly at 230 nm and 280 nm. Peaks generated by contaminants may artificially inflate the nucleic acid reading at 260 nm. Other advantages of photometry are simple, minimal to no sample prep, direct measurement of purity ratios (A260/280 and 260/230), and provide information on contaminants. The disadvantage of photometry is that it is not selective. Alternate methods for nucleic acid quantitation include fluorescence-based methods that utilize dyes that bind specifically to target DNA or RNA molecules. A major advantage of fluorometry is the selectivity and accuracy of the method for distinguishing between DNA and RNA.
It is important to ensure the integrity of extracted nucleic acids before downstream processing (qPCR, RT-qPCR, dPCR), especially for RNA which can be rapidly digested in the presence of ubiquitous RNase enzymes. Current technologies to evaluate DNA and RNA integrity use microfluidics systems which enable electrophoresis of the nucleic acids with high sensitivity, minimal sample consumption, and fast time of results. Some commonly used systems, although not an exhaustive list, include Experion (BioRad), Bioanalyzer and Tapestation (Agilent), QIAxcel (Qiagen), and LabChip GX Touch (Perkin Elmer). The advantage of microfluidics systems is the ability to measure the RNA integrity and DNA integrity which are displayed as RNA integrity number (RIN) and DNA integrity number (DIN). The RIN and DIN scale ranges from 0 to 10, with 10 indicating maximum RNA or DNA integrity, respectively (36,37,38). Assessing the RIN and DIN scores during method development is a tool to measure the quality and integrity of the eluted nucleic acid to compare extraction methods. However, it may not be a direct prediction of the qPCR assay’s ability to detect transgene or transgene-derived transcript in the sample, and therefore should not be used for exclusion criteria of samples. For example, in RT-PCR an RNA sample with a low RIN may yield results consistent with a sample with a high RIN when sequence-specific and highly proximal reverse transcription priming is employed but may yield lower copies when primed off a potentially distal poly(A) tail through oligo(dT) priming.
There are multiple opportunities to incorporate automation in the qPCR or dPCR workflow in order to increase efficiency and limit error-prone manual steps. The design of the overall throughput and workflow should be considered during assay development. For large biodistribution or pharmacokinetic studies, there may be a considerable number of tissue samples (> 1000) that require homogenization, nucleic acid extraction, nucleic acid quantitation, and qPCR or dPCR analysis. Steps of the assays that are most conducive to automation include nucleic acid extraction, nucleic acid quantitation, PCR set-up and preparation, as well as qPCR or dPCR analysis, although any step can be automated. The level to which an assay can be automated will be dependent on the systems and liquid handlers available to the developer; however, when designing the assay, it is important to consider throughput as well as the ability to integrate systems when deciding between multiple extraction platforms or quantitation processes.
Critical Reagents
Critical reagents are essential components of analytical methods whose unique characteristics are crucial to assay performance (24). The contents of this section were compiled to give general considerations about the selection and use of critical reagents for qPCR/dPCR assays, not to make suggestions for specific products. While not an exhaustive list for every scenario, this section highlights reagents that would be applicable for most qPCR/dPCR assay development/validation and encompasses extraction through final validation and sample analysis. Specific acceptance criteria as they relate to critical reagents are addressed in the assay development/validation section of this paper. For the purposes of this paper, critical reagents include but are not limited to nucleic acid extraction reagents, kits, surrogate matrices, primers/probes, master mix, calibrators, and reference material. Additional reagents may be required on a case-by-case basis, one example of which could be the inclusion of restriction enzymes for dPCR reactions.
Nucleic Acid Extraction
The nucleic acid extraction method selected must be able to accommodate the matrix/matrices that are to be tested, but also be able to achieve the defined acceptance criteria of the assay. If a workflow for similar drug products is not already established, multiple extraction methods should be evaluated during assay development to identify the kit and platform that yields acceptable quantities of nucleic acid necessary for downstream analysis. If multiple kits are to be evaluated, reagents that most consistently meet the acceptance criteria with the best accuracy and precision or which offer the best extraction recovery and performance for the assay’s purpose based on COU should be selected for further development and/or validation. Ideally, a single kit and platform would be used for all matrices to avoid errors and confusion during testing; however, this is not always feasible, where possible, high throughput, automated extraction methods are preferred as they reduce the risk of human variability.
The performance of the PCR assay should be verified in the event the extraction method is changed, and a partial validation of the PCR method is required if there is a single reagent change within the extraction (e.g., change in kit initiated by manufacturer). If extraction chemistry is changed, the need for a partial or full validation should be determined and is a case-by-case scenario based on the assay COU (Table 2). For example, if the standard curve is processed through the extraction, then a drastic change in the extraction method could impact assay sensitivity and could warrant a full validation with the new extraction method.
Surrogate Matrices
The same qPCR assay may be required for use in multiple sample matrices, making matrix-specific standard curves impractical. The number of matrices, timepoints, comparability, and total samples should be considered when choosing surrogates and for standard curve development. The availability of matrices (i.e., disease state vs. naïve matrix, target vs. off-target tissues) may impact the ability to perform surrogate matrix assessment and furthermore, impact the rest of development and validation. For example, due to the proprietary nature and individual genetic makeup of AAV-based gene therapies, qPCR assays are often validated before patient dosing. This requires the use of surrogate matrices spiked with the AAV vector for validation.
When developing and validating the qPCR assay, if the test samples are to come from a single matrix, the standards and QCs should all be spiked into naïve matrix. If multiple matrices are used, standard curve equivalency can be demonstrated by spiking the standards or QCs into a matrix during method development and comparing the assay performance to a standard curve spiked into a surrogate matrix. When performance meets the pre-defined acceptance criteria with regard to slope, precision, and accuracy, a surrogate matrix can be used for the standard curve and QCs during sample analysis. When spiking standard reference material into the matrix, consideration should be given to the COU to determine when appropriate and what reference material to spike based on extraction and method development results. For example, vector shedding assays are designed to primarily quantify intact vector, so the drug product would be preferred. For large biodistribution studies quantifying DNA within cells/tissues, plasmid DNA may be more appropriate. If using plasmid DNA, consideration should also be given to the presence of nucleases in the matrix to define when spike-in should occur (before/after extraction).
qPCR Assay Reagents
High-quality primers and probes are key to the successful development and validation of robust qPCR assays. Depending on the COU, off-the-shelf assays may be available, or a custom primer/probe set may be required. For instances of gene expression and/or reference gene testing, off-the-shelf assays can be used, but performance should still be verified and included in the validation assessments. Considerations for designing custom primers and probes are mentioned earlier in this manuscript, ultimately, the final primer/probe selection should be based on specificity, sensitivity, and optimal performance of the assays tested.
When testing RNA, performance verification of the reverse transcription reaction should be a component of the overall method development and validation (Table 3) (39). When performing the reverse transcription, off-the-shelf kits are typically used and primer selection is contingent upon downstream criteria and quality of RNA in samples. Priming strategy for the RT step should take COU into consideration (i.e., one-step vs. two-step, dT primer, random hexamers, or target-specific primers) and sample quality (RNA integrity score assessment) (40). If multiplexing, consider the most efficient priming strategy. Depending on the target, strand-specific priming could be required. If a two-step RT-PCR is performed, negative RT controls should be included in runs or assessed during development. If issues are determined, alternate extraction methods or further DNA removal steps may be required. Following validation, the need for no RT controls should be evaluated once clinical samples are available to ensure there is no DNA carryover. In the absence of carryover after the analysis of 20 samples, the per sample RT controls can be omitted but should be confirmed periodically. If carryover occurs, include sample-specific RT controls.
Mastermixes contain all the key PCR components aside from primers, probes, and templates. They contain buffers, salts, polymerase, dNTPs, and passive reference dyes if applicable. For most qPCR assays, off-the-shelf mastermixes can be selected for known insensitivity to matrix interference and used per manufacturer’s instructions. Mastermix selection can be incorporated in primer/probe optimization. However, certain applications, such as dPCR, may limit the mastermixes that can be used to proprietary platform-specific mastermixes.
Calibrators, Reference Material, and Quality Controls
Materials selected for calibrators and quality controls are critical to establishing the accuracy and precision of an assay. For dPCR that are absolutely quantitative, standard curves are not required, but QCs must be included during sample analysis. Standard/calibrator curves should consist of at least six data points for each qPCR run. When possible, using the drug product and a full process standard curve is preferable, but the choice of reference material should be guided by assay COU. For example, in shedding studies, encapsulated material is preferred, but if not feasible, extraction efficiency studies should be executed to understand quantification differences between encapsulated material and free-plasmid DNA. For biodistribution studies, plasmid DNA is acceptable given that vector DNA is often no longer encapsidated and may appear in episomal or other forms following in vivo processing. In early assay development, positive control/reference material may consist of synthesized DNA or RNA fragments (e.g., gBlocks by IDT), plasmid DNA (commercially or internally manufactured), or linearized and purified plasmid digests. For RT-PCR assays, employing an RNA template (synthesized or purified from dosed animals in early studies) is advantageous in that it more closely mimics the target nucleic acid molecule and allows an assessment of the entire PCR process from reverse transcription to amplification. However, when suitable RNA material is not available, it is acceptable to use synthetic RNA, or DNA reference materials in an RT-PCR assay with the caveat that the performance of the reverse transcription part of the process will not be monitored. When assay development progresses to assay validation, it is recommended to use drug product or plasmids manufactured for drug product (in the case of cell and gene therapies) when possible. In addition to the standard curve, at least two QCs should be tested (high and low) with a preference for three (high, medium, and low) on each sample analysis run. For dPCR, in the absence of a standard curve, a QC at the LLOQ of the assay could be included to verify the performance of each run at that limit. For studies and samples where confirming negative sample results is critical (i.e., vector shedding), an internal, non-competing pre- or post-extraction spike-in control could be considered. This provides greater confidence that the result is truly negative, and not an extraction failure. Finally, extraction and assay NTCs (no template controls) should be run for all applications (qPCR/dPCR) to confirm that no contamination has occurred.
Reagent Bridging
Reagent lifecycle management is critical to the successful implementation of assays in studies. Bridging should be implemented at any time post-validation when a change is necessary to the reagents (new lot, reagent availability, etc.). Bridging studies are performed to demonstrate comparability between different lots of the critical reagents within the same run. Examples of key reagents would include, but are not limited to, calibrators and QCs, TaqMan Low-Density Array (TLDA) card lot changes, and changes to extraction kit component reagents or procedures. Additional reagents can be bridged if deemed necessary on a case-by-case basis. For traceability, if select reagents are not bridged, lots should be tracked. If possible, order enough off-the-shelf reagents to complete validation and testing at the beginning of the study. For extraction components, switching extraction platforms or entire extraction kits would require a re-validation, while switching single reagents within the extraction kit would constitute a partial validation. Switching individual reagents within a kit/process would constitute a bridging study (Table 2). Bridging should be performed with a minimum of 20 samples and should meet precision and accuracy requirements as outlined in the validation/qualification plan. Lifecycle management of key reagents and material should be based on the manufacturer’s instructions. For internal reagents, QC trending can be implemented, and long-term assay monitoring should be performed.
Assay Validation
Bioanalytical method validation guidance documents are available from health authorities (24,25,26). However, these guidelines are specific to ligand binding or chromatographic assays, and as such, direct application of acceptance criteria that are tailored to those specific technologies may often prove too stringent to capture molecular biology assay performance. In addition, molecular assays can be used to support a variety of endpoints with data that can be used for different applications, thus, the thought of applying the COU to drive assay design, validation, and assigning appropriate acceptance criteria that is fit-for-purpose emerged from the European Bioanalysis Forum (EBF) (19). The COU and purpose of the assay should always guide ascertaining the need for assay qualification or validation, determining the assay parameters to assess, and setting appropriate acceptance criteria. For example, using COU to drive validation of an assay for a proof-of-concept study to demonstrate the presence of a gene therapy product in a target tissue may not require extensive validation of accuracy and precision or other non-relevant assay parameters. For biodistribution or transgene expression assays that are expected to have low copy numbers in tissues may not need to demonstrate dilutional linearity in validation. Another example is assays for transgene expression that use a reference gene for normalization and the level of characterization of that reference gene that would be required to support how it will be used in the assay.
Although there is no regulatory guidance specific to bioanalytical method validation requirements for PCR assays, there are several regulatory documents that include guidance for industry for gene therapy products in drug development (41, 42). The FDA guidance for industry on long-term follow-up after administration of human gene therapy products (41) includes a few paragraphs on assays used to support persistence and biodistribution studies including mention of analyzing samples for vector sequences with quantitative and sensitive assays such as quantitative PCR. The guidance document also includes recommendations for assay sensitivity indicating a recommendation for a target sensitivity limit of < 50 copies/µg genomic DNA. More recently, the FDA has adopted the ICH S12 publication on considerations for conducting non-clinical biodistribution for gene therapy products (33) that lists qPCR and dPCR as standard technologies for quantification of vector genome or transgene. In addition, it states the importance of spike and recovery experiments in method development to demonstrate the ability to detect the target nucleic acid sequence in different tissues/biofluids, as well as “providing a comprehensive description of the methodology and the justification for the technique used, including the performance parameters (e.g., sensitivity and reproducibility) of the method,” without including specific requirements for assay sensitivity.
For viral shedding, the FDA guidance on the design and analysis of shedding studies for virus or bacteria-based gene therapy and oncolytic products (10) mentions the use of at least one quantitative assay such as qPCR to detect the number of genome copies or infectious units to provide a quantitative assessment of shedding. The same guidance document also suggests shedding assays “should be demonstrated to be specific, sensitive, reproducible and accurate. We recommend testing clinical samples in a shedding assay in replicates to determine reproducibility. The specificity of the assay should be well understood to avoid false-positive or false-negative results, particularly since retesting is not always feasible with clinical samples that are limited in quantity, such as nasal or skin swabs. The sensitivity of the assay should be determined in terms of LOD and the limit of quantitation (LOQ), if using a quantitative assay. While the agency does not expect shedding assays to be validated, the assays should be qualified to meet minimal performance capabilities and be suitable for the intended purpose” (10).
Table 4 summarizes a compilation of relevant publications and regulatory guidance documents with recommendations on PCR method validations. The following section will include a summary of relevant performance parameters for assay validation and the author’s consensus on validation experimental design and suggested acceptance criteria for PCR assays supporting cell and gene therapies. A summary of parameters evaluated in method development and validation is included in Table 3.
Precision and Accuracy
Precision and accuracy are assay performance parameters which delineate the contribution of random and systematic error to the measurement of an analyte in a sample. Precision and accuracy are assessed through repeated measurements of relevant biological matrix samples spiked with nominal concentrations of a positive control nucleic acid template (24). The intra and inter-assay accuracy and precision should be determined in method development and confirmed in method validation. Accuracy and precision criteria established in method validation are applied to subsequent routine testing with the method.
Validation Experimental Design
Precision and Accuracy (P&A) should be determined using spikes of positive control template in a background matrix of nucleic acid extracted from target tissues or biofluids from the species of interest. Drug product may be the ideal positive control reference material depending on assay COU, but surrogate templates are acceptable if equivalency is demonstrated. Refer to section “Calibrators, Reference Material and Quality Controls” for the discussion on selection of a reference material for use as assay calibrators and controls and refer to the discussion on co-linearity assessments in the PCR Efficiency section below for further details on evaluating template equivalency.
When multiple tissues and/or biofluids are to be analyzed in a study, each tissue and biofluid should be evaluated for matrix interference in method development and guide the selection of a representative matrix for validation. Since employing calibrator curves and controls spiked in matrix specific to each study tissue and biofluid is impractical for large studies, it is accepted to use a single relevant biological matrix for validation. Ideally, the matrix will consist of nucleic acids extracted from the target tissue or biofluid, but nucleic acids pooled from several tissues/biofluids may serve as the matrix for the assay validation if equivalency of assay performance is demonstrated in P&A during development. The performance of two matrices is considered equivalent if the measured copy number of candidate matrix spiked controls fall within ± 20%RE of the measured copy number of comparator matrix spiked at equal concentrations. The matrix concentration should be greater or equal to the maximum sample concentration anticipated in preparing study samples for analysis (e.g., 1–100 ng extracted DNA per reaction), and this should be determined in method development. In the case of acellular biofluids with low nucleic acid content, such as urine and saliva, it is acceptable to use the neat nucleic acid extracts since quantification is often not feasible. In that case, care must be taken to report results as per volume of extraction input, since the PCR reactions are not loaded with a specific amount of nucleic acid.
Method accuracy and precision are assessed in validation in the same designated runs, on the same spiked system suitability quality controls (QCs) and limits of quantification (LOQs), and they should be assessed over ≥ 6 runs, ≥ 2 analysts, ≥ 2 days. ULOQ (upper limit of quantification), HQC (high-quality control), MQC (mid-quality control), LQC (low-quality control), and LLOQ (lower limit of quantitation) concentrations should be established in method development and their performance confirmed in method validation. Three occasions/determinations should be made per QC/LOQ level per run, with each QC/LOQ run in at least duplicate per determination. QC samples must be prepared independently from the calibrators and should not share intermediate dilutions from the stock positive control reference material. When determining where to set QCs for monitoring of assay performance, consider the platform detection range, assay limits of quantification, and where study samples are expected to fall within the assay’s quantitative range. In dPCR, where a calibration curve is not required, it could be useful to run the LLOQ during routine sample testing runs along with routine QCs to monitor assay performance at the LLOQ. It is important to note that real-time qPCR measurement is performed in semi-log scale, whereas dPCR is on a linear scale, hence the greater impact on accuracy and precision of low-analyte concentration measurements for qPCR.
Validation Acceptance Criteria
The ideal assay will demonstrate:
-
Intra and inter-assay precision ≤ 30% coefficient of variation (%CV) for QCs and ≤ 50%CV for LOQs in either interpolated qPCR or absolute dPCR copy results.
-
Intra and inter-assay accuracy of − 50 to 100%RE on interpolated copies for qPCR, due to the doubling nature of qPCR reactions, where a difference of 1 Cq can land the interpolated result within 1/2 or 2 × of a given nominal concentration. For dPCR, the inter-assay accuracy for absolute copies measured should be |%RE|≤ 30 for QCs, and ≤ 50 for LOQs.
While most qPCR and dPCR assays can achieve the above recommended precision and accuracy criteria, the assay performance demonstrated in development should guide the establishment of fit-for-purpose assay acceptance criteria to apply in validation. If the acceptance criteria appropriately capture assay performance in validation, then they are maintained for sample testing. This is especially true of multi-step reaction assays such as RT-PCR, where the addition of a reverse transcription step can add an additional layer of variability. Therefore, the COU and assay purpose should always guide the establishment of acceptance criteria for assay performance.
PCR Efficiency and Linearity
PCR efficiency encompasses several parameters which all fundamentally assess the linearity of polymerase chain reaction amplification of a target nucleic acid sequence existing in a sample (43). PCR efficiency is evaluated to assess the amplification performance of the assay, matrix interference, and linearity of test sample dilutions, and whether a surrogate positive control may be used in place of an ideal positive control template (e.g., drug product). PCR efficiency should be determined in development and confirmed in validation. To enable a more rigorous characterization and evaluation of PCR efficiency, it can be presented with a 95% confidence interval (44). PCR efficiency evaluation can serve different purposes in qPCR vs. dPCR due to differences in the measurement principle, which we explore further below.
Quantification in qPCR relies on interpolation of unknown sample concentrations from a calibration curve of known nominal concentrations. The linearity of the interpolation curve is evaluated through linear regression analysis of raw assay signals (Cq) versus the log nominal copy number of the positive control template. The positive control template employed in the calibration curve should be the same as that used for QCs, and it can be the drug product (e.g., encapsidated ssDNA) or a surrogate positive control (e.g., dsDNA plasmid or fragment) if equivalency has been demonstrated (see co-linearity section below). Without PCR interference/inhibition from matrix nucleic acid or extraction reagent carryover in a sample, the theoretical amplification efficiency (calculated from the slope of the linear regression) should be 100% which would result in a doubling of template with every amplification cycle. If the template is diluted in a tenfold serial dilution in the absence of any reaction inhibition, then the slope of the calibrator curve should be − 3.32 when using the Eq. (10(−1/slope) − 1) × 100, and assuming 100% efficiency. While efficiencies of 100% may not always be attainable, efficiencies should be at least 90% (44). Calibrator curve slopes falling within − 3.6 and − 3.1 (90–110% efficiency) have historically been accepted for a typical qPCR assay and deemed acceptable for interpolating unknown samples. A PCR efficiency significantly above 100% (i.e., lower limit of the confidence interval is above 100%) shall be considered suspicious indicating there may be issues with contamination, baseline correction, aberrant product formation, polymerase inhibition, pipetting errors, etc. Efficiencies below 90% may be acceptable depending on assay performance and reproducibility and accuracy of interpolation. While dPCR does not require a calibrator curve and interpolation, it is recommended that linearity still be assessed. The demonstrated linear dynamic range of the assay determines the range of quantification (ROQ). While statistical software is available for the calculation of confidence intervals, Microsoft Excel also allows for the calculation of the confidence interval of the slope, intercept, and efficiency. Linearity can also be validated using the Clinical and Laboratory Standards Institute (CLSI) EP06 test (45).
Validation Experimental Design
Different approaches may be taken to evaluate linearity in a qPCR assay. It is acceptable to spike the positive control into an extracted matrix-specific nucleic acid followed by dilution of that high-concentration calibrator in the same matrix-specific nucleic acid to create a calibrator curve. It is acceptable to spike calibrators at nominal concentrations into biological samples which are then extracted and processed as samples would be. The latter, “full process,” approach factors in extraction efficiency variability into the evaluation of linearity, precision, and accuracy of the calibrator curve. When evaluating a calibrator curve for linearity in a qPCR assay, the curve must include LOQs and is recommended to be set up with a tenfold/logarithmic serial dilution series (e.g., 10−2, 10−3, 10−4, 10−5, 10−6, 10−7, 10−8, and 10−9). The curves are assessed in ≥ 6 runs, ≥ 1 curve per run, ≥ 2 analysts, ≥ 2 days, ≥ 2 PCR instruments, when possible. dPCR design follows the same framework as for qPCR above but is applied to LOQs and QCs instead of a calibrator curve. ROQ is determined empirically by evaluation of precision and accuracy at concentrations near absolute quantification limits of the dPCR instrument. Due to the narrower linear range of multiple dPCR platforms, it is recommended that the dilution series be prepared by setting up a fivefold or tenfold dilution series.
Validation Acceptance Criteria
To be considered linear, a calibrator curve or dilution series (positive control spiked into extracted nucleic acid) must demonstrate:
-
All concentrations within the ROQ should generate a signal above the limit of detection.
-
Demonstrate through linear regression analysis an R2 ≥ 0.98 and slope between − 3.1 and − 3.6 (corresponding to 90–110% efficiency) within the ROQ.
-
To assess linearity, the 95% confidence interval of the estimated reaction efficiency can also be reported (44).
-
Efficiency not meeting these criteria may be acceptable depending on the assay purpose and should be justified.
Dilution Linearity
An assessment of dilutional linearity should be performed in validation if high-concentration samples are expected to require dilution to yield signals within the assay’s ROQ. This assessment would determine if the dilution of high-concentration samples may impact reaction efficiencies, thereby affecting the accuracy of interpolation. Due to the sample partitioning which occurs in digital platforms, dPCR may be less affected by matrix inhibitors in contrast to analog qPCR. Digital PCR technologies are also less sensitive to varying PCR efficiency than qPCR assays since the platform quantifies amplification at endpoint in binary fashion (positive or negative results). If positive amplification partitions are sufficiently distinct from negative amplification partitions, dPCR platforms can quantitate target sequences in a sample. However, while dPCR does not rely on interpolation for quantification, linearity of PCR product to reaction input is still important when considering the dilutional linearity of samples which may have target sequence concentrations above the quantitative range of a dPCR assay and require dilution into the ROQ. Furthermore, while qPCR platforms generally demonstrate an upper limit of quantification (ULOQ) of 1E8 copies per reaction, existing dPCR platforms are limited to lower ULOQs (e.g., 1E5 copies per reaction) due to sample partitioning limitations. Thus, acceptable dilutional linearity of samples should still be demonstrated for dPCR platform assays. There are also considerations for duplex assays where diluting the sample could bring one of the targets below the range of the assay, rendering it below the limit of quantification (BLQ).
Validation Experimental Design
If demonstrating dilutional linearity, a dilution series (e.g., spiking positive control into extracted matrix-specific nucleic acid) should be created to include concentrations above and below the quantitative range. A typical design would include at least one concentration 10 × above the ULOQ (ideally a concentration approximating the anticipated maximum concentration one might find in a study sample), at least three concentrations within the ROQ, and at least one concentration below the LLOQ.
Validation Acceptance Criteria
-
Dilution linearity should be performed should be performed if valuable given the assay’s COU.
-
Precision and accuracy of concentrations within the ROQ conforming to that established in overall assay P&A indicate acceptable dilutional linearity.
Co-linearity
The importance of PCR efficiency also manifests when evaluating co-linearity to support bioanalytical assays. The ideal positive control template will differ with an assay’s intended use. For example, using dsDNA plasmids or fragments may be appropriate for gene therapy biodistribution studies, where delivered encapsidated ssDNA vector constructs are likely to be taken up into cells over time, released from their capsids, and assembled into dsDNA episomes. Therefore, the target analyte template is unlikely to remain encapsidated in tissues and biofluids for the duration of the study, so drug product may not be the reference material which is most representative of study samples. In contrast, in vector shedding studies, the aim is to detect potentially infectious material, so drug product would be the ideal assay positive control material. When it may not be desirable or possible to employ actual drug product for use as a calibrator or QC material in an assay, it becomes necessary to demonstrate that a surrogate reference material (e.g., plasmid DNA) behaves equivalently in the assay. An assessment of linearity between the two reference materials in the same nucleic acid matrix would support the substitution of the test article with the surrogate reference material.
Validation Experimental Design
While co-linearity is not a required assessment, demonstration in validation could offer value depending on the assay’s COU. Co-linearity is typically performed in development to allow for the use of any surrogate template in further development and validation. If an assessment of co-linearity is desirable for validation, perform at least a single run in which identical calibrator curves or dilution series are created with each reference material and perform linear regression analysis on both. While regression parameters may be compared as part of evaluating co-linearity in qPCR, co-linearity assessments in both qPCR and dPCR should ascertain that quantification does not significantly differ between the two reference materials.
Validation Acceptance Criteria
-
Co-linearity should be performed if valuable given the assay’s COU.
-
PCR efficiency is within acceptable criteria for both calibrator curves (if qPCR).
-
To assess linearity, the 95% confidence interval of the estimated reaction efficiencies can also be evaluated and reported (44). Non-overlapping reaction efficiency intervals indicate different reaction efficiencies, the impact of which can be determined by a precision and accuracy assessment.
Sensitivity
Validation of analytical sensitivity is critical for bioanalytical assays supporting therapeutic development. Sensitivity is most broadly defined as the smallest concentration of a target analyte that can be reliably measured with an analytical procedure. Regulatory guidance on assay sensitivity is sparse, and there are only a few regulatory guidance documents which provide specific recommendations. The most noteworthy recommendation states that assays supporting preclinical biodistribution assessment of a gene therapy product should “have a demonstrated limit of quantification of ≤ 50 copies/µg genomic DNA, so that your assay can detect this limit with 95% confidence” (41). The statement itself is problematic since it refers to a limit of quantification (i.e., LLOQ) yet follows it with a recommendation only for detection with 95% confidence, rather than a demonstration of accuracy and precision criteria appropriate for an LLOQ. Given such ambiguity, it is worthwhile to establish the various components of assay sensitivity and clarify how they should be applied to the development and validation of fit-for-purpose bioanalytical PCR assays. The limit of blank (LOB), limit of detection (LOD), and lower limit of quantification (LLOQ) may all be employed in the characterization and validation of PCR assay sensitivity (46,47,48).
Limit of Blank
LOB is defined as the highest apparent corresponding concentration or signal of the target analyte expected to be detected with a stated probability when a blank sample containing no analyte, such as no template control (NTC), is analyzed. LOB can also be determined in raw analytical platform signals in situations where the platform may report a signal below a certain threshold as having a concentration of 0. For example, in dPCR where the negative samples produce no readout and consequently no standard deviation can be determined, the LOB can be determined in the linear scale using raw partition signals. The LOB is only required to be established in method validation based on the COU (e.g., in dPCR to define the threshold of the blank, or if needed to statistically determine the LOD). If required, the LOB is established in method development and should be confirmed formally in method validation, as it underpins the discrimination of a negative/undetectable response from a positive/detectable response.
Validation Experimental Design
The NTC is prepared as other assay controls and samples are prepared by spiking a mock sample into the PCR reaction mix comprised of primers, probe, dNTPs, and enzymes required for PCR or RT-PCR. The mock sample can be a relevant nucleic acid matrix at an appropriate concentration which is devoid of the analyte. The concentration of any matrix in NTCs should be the same as that which will be used to determine LOD and LLOQ and should ideally be at a concentration relevant for sample analysis. A minimum of 20 replicates of NTC should be run for LOB establishment in validation. LOB determination can be performed in the same runs for the assessment of LOD, but it is also acceptable to determine LOB prior to LOD assessment. LOB may be selected empirically at a 95% confidence (e.g., 19/20 copies are negative in the assay) independent of PCR platform. LOB at 95% confidence limit may also be derived statistically in dPCR if one assumes a Normal/Gaussian distribution of analytical signal response (i.e., positive partitions) from blank samples using the equation below (46, 48):
Validation Acceptance Criteria
-
The LOB is reported as determined.
-
The LOB should be < LOD.
Limit of Detection
Analytical assays can yield a low-level background signal even in the absence of analyte, which is captured by the LOB detailed above. Therefore, analytical methods require the determination of a LOD to establish the lowest analyte concentration that yields an assay response which can be reliably distinguished from the assay response in the absence of analyte (i.e., NTC) but is not required to meet any precision and accuracy criteria for quantification. It is recommended that bioanalytical assays supporting therapeutic development should determine the LOD with a ≥ 95% confidence limit (47). The LOD should be determined in method development and confirmed formally in method validation.
Validation Experimental Design
Different experimental designs may be applied to establishing the LOD, and there is currently no regulatory guidance specifying how LOD should be assessed. However, validated PCR assays have typically determined LOD in one of two ways: empirically or through the application of a statistical model. It is recommended to determine LOD over multiple runs and days, at a minimum, perform ≥ 3 runs, ≥ 2 analysts, ≥ 2 days. The LOD of a validated assay is determined by performing a 1.5–twofold or appropriate serial dilution of the reference template in extracted gDNA or total RNA matrix from the LLOQ concentration (preliminary LLOQ will suffice) down to a sub-LLOQ “low concentration sample.” A “low concentration sample” in this situation may contain as few as 3 nominal copies, which, due to sampling ambiguity, is the theoretical limit of detection at a 95% confidence interval for a sample population following a Poisson distribution (49) Alternatively, a “full process” assessment can be conducted by spiking reference template at each concentration level into 20 replicates of biological matrix (e.g., plasma or tissue homogenate) followed by extraction and PCR analysis. This approach factors in variance introduced by the nucleic acid extraction process.
In the empirical approach, the LOD is determined to be the target template concentration at which 95% of replicates have assay response values exceeding those of the previously determined LOB (i.e., lower Cq or greater positive partitions). While analyzing a high number of replicates per concentration would better power this determination, it is acceptable to use 20 replicates.
In the statistical model approach, continuous PCR data (e.g., Cq or positive partitions) is converted into a binary scale (detected or non-detected) based on LOB, and a LOD is calculated with a 95% confidence interval via Probit regression analysis with statistical analysis software. When taking this approach, it is critical that the LOB has been characterized for your specific assay, so that data transformation into a binary scale is accurate. In the case of multi-matrix validations supporting biodistribution or vector shedding studies, it is acceptable to demonstrate LOD in a representative or target tissue/biofluid matrix only, unless there is a scientific need to establish LOD in other tissues/biofluid matrices. When starting with single-stranded DNA, like cDNA, the first cycle of PCR produces the complement and amplification starts in cycle 2. Hence, in qPCR, one cycle shall be subtracted if interpolating against a dsDNA reference material.
An alternative statistical approach may be applied for linear-scaled dPCR LOD determinations, much like with the determination of LOB, with the following formula (46, 48):
This approach assumes normal/Gaussian distribution of signal response and applies a 95% confidence limit in the variance of response observed with a “low concentration sample” to the LOB.
Validation Acceptance Criteria
-
LOD must be greater than LOB.
-
LOD must be less than or equal to LLOQ.
-
LOD should be reported as determined with 95% confidence interval.
Lower Limit of Quantification
The LLOQ is the lowest concentration at which the target analyte can be quantified with demonstrated precision and accuracy within established acceptance criteria, in contrast to the LOD which has no quantitative requirements. The LLOQ should be determined in method development and confirmed in precision and accuracy (P&A) runs in validation. Sample results which fall below the LLOQ but above the LOD of the assay are reported as BLQ. This contrast results which fall below the assay LOD are reported as below the limit of detection (BLD).
As previously mentioned, the FDA guidance recommendation stating that PCR assays which support preclinical biodistribution of gene therapy products should “have a demonstrated limit of quantification of ≤ 50 copies/µg genomic DNA, so that your assay can detect this limit with 95% confidence” (41) is commonly used as a PCR assay sensitivity target. Although this sensitivity may be achieved, a less sensitive method may be acceptable depending on the COU, if justified scientifically. It is worth noting that the recent ICH S12 (33) does not recommend a sensitivity target for biodistribution assays. Additionally, analyses supporting bioanalysis in regulated studies may be limited to biological matrices which yield relatively little nucleic acid (e.g., urine) and therefore preclude the loading of 1 µg of nucleic acid into PCR reactions. In these situations, an extrapolation of the sensitivity limit is the only available option if reporting per a specific mass of nucleic acid is required. Sensitivity considerations can become especially problematic for PCR assays supporting cell therapy persistence or cellular kinetics which require a duplex assay to include a reference gene, which may exist at a different abundance than the target gene.
Validation Experimental Design
Validation samples at LLOQ level are prepared by spiking the reference material at the target number of copies in a relevant mass of gDNA or total RNA background per PCR reaction. The use of alternative matrix (e.g., salmon sperm DNA) may be justified depending on fit-for-purpose considerations and assay COU. The mass of gDNA/total RNA loaded per PCR reaction (e.g., 1 µg gDNA or total RNA) should allow for the measurement of the target analyte without assay interference incurred from exceeding platform limitations. Beyond the previously mentioned nucleic acid concentration limitations incurred with some biological matrices, some existing dPCR platforms may prohibit loading of 1 µg gDNA without additional sample restriction digest to improve partitioning and target template accessibility.
Since loading 1 µg of extracted gDNA or total RNA per PCR reaction can be unattainable or limited by the PCR platform, it is acceptable to use a lower concentration of background nucleic acid. However, it is recommended that the LLOQ copy number per reaction decrease commensurately to allow for an extrapolated sensitivity of 50 copies per 1 µg of nucleic acid (e.g., 20 copies per 400 ng of gDNA). An alternative to reporting copy results normalized to sample reaction input mass is to evaluate and report LLOQ on a per reaction basis, although it is recommended to report the LLOQ in the same reportable units of the assay.
The assessment of LLOQ occurs in the runs designated for precision and accuracy assessment (≥ 6 runs, ≥ 2 analysts, ≥ 2 days). Successful demonstration of LLOQ performance meeting the acceptance criteria below validates the LLOQ concentration and establishes it for use in sample analysis. In qPCR, the LLOQ is not typically included in system suitability QCs; however, it should be a calibration point on the calibration curve. Consider running LLOQ on routine sample analysis runs in dPCR, since no calibration curve is run in every run, as is done for qPCR.
Validation Acceptance Criteria
-
LLOQ samples in each P&A run must meet the inter- and intra-assay criteria of ≤ 50%CV for either interpolated qPCR or absolute dPCR copy results.
-
Intra and inter-assay accuracy of − 50 to 100%RE on interpolated LLOQ copies for qPCR. For dPCR, the inter-assay accuracy for LLOQ absolute copies measured should be |%RE|≤ 50.
-
LLOQ must be equal to or greater than LOD.
Specificity and Selectivity
Specificity is intended to assess the ability of the primer probe set to “specifically” detect the analyte/target of interest in the presence of gDNA/total RNA or other interfering substances across various tissue types. Selectivity is the ability of an analytical method to measure the analyte in the presence of interfering substances in representative individual samples from the biological matrix of interest. During method development, specificity is determined primarily to select the ideal primer and probe set. It can be demonstrated either in silico using BLAST or experimentally through the evaluation of an NTC which contains a relevant nucleic acid background matrix. Expansion of the specificity parameter to include evaluation in individuals provides for the additional assessment of selectivity.
Validation Experimental Design
At least 10 individual naïve gDNA or total RNA samples are analyzed unspiked and spiked at the level of the LLOQ, at minimum. Higher concentration spikes (e.g., QC levels) may also be included if necessary. In the case of rare matrices and/or a study with multiple disease matrix types, assessing with a lower number of individuals (e.g., 3 or 4 individual lots from each disease/matrix type) is acceptable.
Samples for specificity and selectivity should contain the same amount of background nucleic acid used to determine LLOQ precision and accuracy. Specificity and selectivity may alternatively or additionally be demonstrated through spiking naïve samples with a non-specific template to demonstrate no non-specific amplification. Performing this assessment of a non-specific template spike should be based on the COU.
Validation Acceptance Criteria
-
100% of unspiked individual lots must demonstrate measured (dPCR) or interpolated results (qPCR) below the LOD.
-
At least 8 out of 10 spiked individual lots or 67% (e.g., 2/3 individuals) should meet the P&A acceptance criteria for LLOQ, or QCs if used.
-
If interfering material is used in validation to determine specificity to the target of interest in the presence of a similar target (e.g., different virus), 100% of samples spiked with the non-relevant target must demonstrate measured (dPCR) or interpolated results (qPCR) below the LOD.
Robustness and Ruggedness
An assay should be robust and rugged enough to allow for variations in operator, equipment, and reagents without significant impact to the generated data. Performance characteristics meeting validation acceptance criteria should be maintained across assay executions, even with alterations to critical reagent lots, instruments employed, and the day of assay execution. Assay ruggedness evaluates the impact of deliberate variations in assay execution (e.g., lab, analyst, instrument, day) to assay performance (i.e., P&A). Assay robustness is a parameter used to evaluate the stability of the method when internal factors of the assay method are varied (e.g., annealing temperatures, deliberate pipetting errors, delayed droplet digital read time). For the purposes of this section, robustness and ruggedness are used synonymously since any PCR assay which is unable to demonstrate robustness will typically fail to demonstrate ruggedness.
When an assay is expected to support sample analysis spanning years, it can be valuable to demonstrate the ruggedness of the assay in validation with different reagent lots or reagent manufacturers, since the chance to incur reagent variation increases with longer study durations. If a formal assessment is not performed in validation, it is recommended to keep aliquots of both controls and reagents reserved for future in-study bridging experiments.
Validation Experimental Design
Capturing the full variation incurred by possible internal and external method alterations is beyond the scope of a typical bioanalytical method validation. However, a reasonable assessment of ruggedness should be conducted during PCR method validation by evaluating the assay’s precision and accuracy across all PCR runs which contained deliberate variations in equipment, and days (i.e., by at least 2 operators, using at least 2 instruments, and performed on at least 2 different days).
To assess the comparability of PCR reagent manufacturer or lots, a clear experimental design and acceptance criteria for demonstration of reagent equivalency should be established. One approach is to prepare two sets of master mix on the day of the assay which only differ in the single PCR reagent under evaluation. Calibration curves (if qPCR) and QCs are then assessed on the same plate with both sets of master mixes and evaluated for performance within P&A acceptance criteria. In cases where original reagents are not available, historical data can be used to determine acceptance.
Validation Acceptance Criteria
-
P&A of results for runs with varied analysts, instruments, and days should demonstrate P&A conforming to that established in the validation P&A assessment.
-
For reagent comparability, in the case of qPCR, each calibrator curve assessed should demonstrate linearity as detailed above in the linearity section. In both qPCR and dPCR, QCs prepared in each respective master reaction mix should yield PCR results within established precision and accuracy criteria.
Stability
Stability is an integral part of validation as it enables planning for downstream studies. At minimum, freeze/thaw, short-term (room temperature, 4°C, and − 20°C) and long-term (− 70/ − 80°C) stability should be assessed.
There is an overwhelming body of literature that supports long-term stability of extracted nucleic acids for 2 years or more. Accelerated stability studies have also shown stability of frozen nucleic acids for 12 years for RNA and 60 years for DNA (50). Although extracted nucleic acids have demonstrated stability for long periods of time under frozen conditions, the gold standard for storage of biospecimens and tissues for DNA/RNA extraction is storage at ultra-low temperatures. Demonstrating the stability of a nucleic acid target of interest in a frozen biological matrix is technically challenging — particularly with solid tissues. When the use of incurred study samples is not possible or desirable, it is possible to perform this assessment in a contrived fashion by spiking a reference material into tissue homogenate pre-extraction, but the relevance of such a contrived sample is unclear. Even when it is possible to use incurred study samples in long-term stability assessments, the contribution of extraction efficiency to results at a given stability time-point adds significant analytical complexity which would require deconvolution. Given the historical evidence of extracted nucleic acid stability (51,52,53), it is not currently recommended to assess the stability of extracted nucleic acid. The need for assessing long-term stability in a contrived or incurred sample should be evaluated on a case-by-case basis. Specific considerations for assessing long-term stability should include assay context of use, study duration, sample storage duration, sample type, drug product characteristics, as well as historical data. For example, a clinical study where sample matrices (e.g., urine for a shedding assay) are stored for a long period of time before extraction and batch PCR analysis.
Validation Experimental Design
To perform the stability assessment, a sufficient number of high and low QCs should be prepared at the beginning of the study to cover assessment runs at each planned time-point, including the baseline time-point.
Freeze/thaw stability should be assessed for a minimum of 3 cycles. During testing, samples should be at frozen storage temperature for a minimum of 10 h and allowed to completely come to room temperature during the thawing cycle, unless required to stay on ice (e.g., RNA).
Room temperature stability can be assessed up to a minimum of 48 hours (i.e., tested at 1, 4, 8, 24, 48 hours), but 72 hours would be preferred. Short-term stability of 4°C and − 20°C can be included but are only recommended if sample handling conditions dictate inclusion (i.e., the inability to store at − 80°C prior to analysis). If included 4°C stability should be tested for up to 7 days (days 1, 3, 5, and 7 are recommended) and − 20°C stability should be tested for up to 30 days (can be tested weekly).
Long-term stability at − 70/ − 80°C should also use high and low QCs as with other stability assessments. Long-term storage at − 70/ − 80°C should be assessed up to a minimum of 12 months (1-, 3-, 6-, and 12-month timepoints are recommended). Eighteen- and 24-month timepoints can be added based on sample analysis testing schedule and follow-up assessment requirements.
Validation Acceptance Criteria
-
Samples would be considered stable in these studies if a stability QC sample yields a mean result which is 70–130% of the baseline QC sample result and < 30% CV between replicates.
-
If assessing long-term stability using a contrived or incurred sample, it is recommended to set acceptance criteria based on the demonstrated extraction efficiency variability of the developed method.
Extraction Efficiency
To assess reproducibility and efficiency of extraction method(s), a matrix extraction evaluation should be performed. Extraction efficiency may be determined in development and verified in validation, and acceptance criteria should be based on the assay’s COU.
Validation Experimental Design
In validation, spike a known amount of drug product and/or reference material (if no drug product availability or not applicable) at a minimum of one QC level that is within the standard curve range (e.g., 10 times LLOQ, or MQC or HQC levels) in biofluids and representative tissue homogenates and perform at least three independent extractions in each matrix (as applicable, depending on assay COU). For large biodistribution studies, a small subset of biodistribution tissues of interest can be selected to test in this parameter based on the therapeutic indication (target tissue) and safety considerations (liver and gonads). An unspiked replicate should be included for each matrix tested. The inclusion of positive and negative extraction controls is also recommended in extraction efficiency experiments. Note that if the assay uses a reference gene, this should also be included in this evaluation.
Validation Acceptance Criteria
-
Extraction efficiency is reported and generally acceptable with a spike recovery between > 20% and < 120%. However, values outside of this range may be acceptable based on data obtained during method development optimization.
-
Consider the COU if the performance exceeds this acceptance criteria.
-
If a reference gene is used, ensure extraction efficiency meets acceptance for how that reference gene will be used.
Summary and Conclusions
The purpose of this publication is to improve consistency, clarity, and sound scientific validations for qPCR and dPCR assays that are used to support gene and cell therapies. The information presented herein is built on experience from molecular and bioanalytical scientists in the industry who have experience in developing and validating these assays for regulatory submissions. The recommendations are intended to present general considerations and examples of experimental design and suitable acceptance criteria for assays used to support cell and gene therapies by qPCR and dPCR, especially in the absence of regulatory guidance for analytical validation of these assays.
Data Availability
Information herein are consensus recommendations from subject matter experts within the AAPS Bioanalytical Community PCR Working Group. The recommendations are based on experience and expert opinions and not associated with specific data from repositories or other.
References
FDA. What is Gene therapy? https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/what-gene-therapy. 25 July 2018. Accessed 25 June 2023.
Sayed N, Allawadhi P, Khurana A, Singh V, Navik U, Pasumarthi SK, et al. Gene therapy: comprehensive overview and therapeutic applications. Life Sci. 2022;294: 120375. https://doi.org/10.1016/j.lfs.2022.120375.
Jahangiri S, Rahimnejad M, Boroujeni NN, Ahmadi Z, Fath PM, Ahmadi S, et al. Viral and non-viral gene therapy using 3D (bio)printing. J Gene Med. 2022;24(12): e3458. https://doi.org/10.1002/jgm.3458.
American Society of Gene and Cell Therapy. Gene & Cell Therapy FAQs. 2021. https://asgct.org/education/more-resources/gene-and-cell-therapy-faqs. Accessed 27 June 2023.
Lin H, Cheng J, Mu W, Zhou J, Zhu L. Advances in universal CAR-T cell therapy. Front Immunol. 2021;12: 744823. https://doi.org/10.3389/fimmu.2021.744823.
Labanieh L, Majzner RG, Mackall CL. Programming CAR-T cells to kill cancer. Nay Biomed Eng. 2018;6:377–91. https://doi.org/10.1038/s41551-018-0235-9.
Ma M, Balasubramanian N, Dodge R, Zhang Y. Challenges and opportunities in bioanalytical support for gene therapy medicinal product development. Bioanalysis. 2017;9(18):1423–30. https://doi.org/10.4155/bio-2017-0116.
Bernt KM, Ni S, Gaggar A, Li ZY, Shayakhmetov DM, Lieber A. The effect of sequestration by nontarget tissues on anti-tumor efficacy of systemically applied, conditionally replicating adenovirus vectors. Mol Ther. 2003;8(5):746–55. https://doi.org/10.1016/j.ymthe.2003.07.006.
Gonin P, Gaillard C. Gene transfer vector biodistribution: pivotal safety studies in clinical gene therapy development. Gene Ther. 2004;11(Suppl 1):S98–108. https://doi.org/10.1038/sj.gt.3302378.
US FDA. Guidance for industry: design and analysis of shedding studies for virus or bacteria-based gene therapy and oncolytic products. 2015. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/design-and-analysis-shedding-studies-virus-or-bacteria-based-gene-therapy-and-oncolytic-products. Accessed 10 Feb 2023.
Hays A, Durham J, Gullick B, Rudemiller N, Schneider T. Bioanalytical assay strategies and considerations for measuring cellular kinetics. Int J Mol Sci. 2022;24(1):695. https://doi.org/10.3390/ijms24010695.
Yang TY, Doddareddy R. Considerations in the development and validation of real-time quantitative polymerase chain reaction and its application in regulated bioanalysis to characterize the cellular kinetics of CAR-T products in clinical studies. Bioanalysis. 2021;13(2):115–28. https://doi.org/10.4155/bio-2020-0221.
Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009;55(4):611–22. https://doi.org/10.1373/clinchem.2008.112797.
Huggett J, Whale AS, Spiegeaere WD, Trypsteen W, Abdel Nour A, Bae YK, et al. The digital MIQE Guidelines update: minimum information for publication of quantitative digital PCR experiments for 2020. Clin Chem. 2020;66(8):1012–29. https://doi.org/10.1093/clinchem/hvaa125.
Ma H, Bell KN, Loker RN. qPCR and qRT-PCR analysis: regulatory points to consider when conducting biodistribution and vector shedding studies. Mol Ther Methods Clin Dev. 2020;20:152–68. https://doi.org/10.1016/j.omtm.2020.11.007.
Corsaro B, Yang TY, Murphy R, Sonderegger I, Exley A, Bertholet S, et al. 2020 White Paper on Recent Issues in Bioanalysis: Vaccine Assay Validation, qPCR Assay Validation, QC for CAR-T Flow Cytometry, NAb Assay Harmonization and ELISpot Validation ( Part 3 - Recommendations on Immunogenicity Assay Strategies, NAb Assays, Biosimilars and FDA/EMA Immunogenicity Guidance/Guideline, Gene & Cell Therapy and Vaccine Assays). Bioanalysis. 2021;6:415–63. https://doi.org/10.4155/bio-2021-0007.
Hays A, Islam R, Matys K, Williams D. Best practices in qPCR and dPCR validation in regulated bioanalytical laboratories. AAPS J. 2022;24(2):36. https://doi.org/10.1208/s12248-022-00686-1.
Loo L, Harris S, Milton M, Meena A, Lembke W, Berisha F, et al. 2021 White Paper on Recent Issues in Bioanalysis: TAb/NAb, Viral Vector CDx, Shedding Assays; CRISPR/Cas9 & CAR-T Immunogenicity; PCR & Vaccine Assay Performance; ADA Assay Comparability & Cut Point Appropriateness (Part 3 - Recommendations on Gene Therapy, Cell Therapy, Vaccine Assays; Immunogenicity of Biotherapeutics and Novel Modalities; Integrated Summary of Immunogenicity Harmonization). Bioanalysis. 2021;14(11):737–93. https://doi.org/10.4155/bio-2022-0081.
Lauren A, Braun M, Pybre P, Cazzin C, Colletti K, Cox C, et al. Applying context of use to quantitative polymerase chain reaction method validation and analysis: a recommendation from the European Bioanalysis Forum. Bioanalysis. 2021;23:1723–9. https://doi.org/10.4155/bio-2021-0218.
Lauren A, Braun M, Cazzin C, Colletti K, Cox C, Dietz L, et al. Quantitative polymerase chain reaction in the bioanalytical laboratory and technical and scientific considerations for nonclinical and clinical assay characterization, validation and sample analysis. Bioanalysis. 2022;16:1085–93. https://doi.org/10.4155/bio-2022-0170.
Wissel M, Pourier M, Satterwhite C, Lin J, Islam R, et al. Recommendations on qPCR/ddPCR assay validation by GCC. Bioanalysis. 2022;12:853–63. https://doi.org/10.4155/bio-2022-0109.
Uchiyama A, Naritomi Y, Hashimoto Y, Hanada T, Watanabe K, Kitta K, et al. Understanding quantitative polymerase chain reaction bioanalysis issues before validation planning: Japan Bioanalysis Forum discussion group. Bioanalysis. 2022;21:1391–405. https://doi.org/10.4155/bio-2022-0190.
International Organization of Standardization. Biotechnology — Requirements for evaluating the performance of quantification methods for nucleic acid target sequences — qPCR and dPCR. (ISO Standard No. 20395:2019). https://www.iso.org/standard/67893.html.
US FDA. Guidance for Industry: Bioanalytical Method Validation. 2018. https://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf.
European Medicines Agency. Guideline on Bioanalytical Method Validation. 2011. https://www.ema.europa.eu/en/bioanalytical-method-validation.
US FDA. Guidance for Industry. M10 Bioanalytical Method Validation and Study Sample Analysis. 2022. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/m10-bioanalytical-method-validation.
Bustin SA, Mueller R, Nolan T. Parameters for successful PCR primer design. Method Mol Biol. 2020;2065:5–22. https://doi.org/10.1007/978-1-4939-9833-3_2.
Latorra D, Arar K, Hurley JM. Design considerations and effects of LNA in PCR primers. Mol Cell Probes. 2003;17(5):253–9. https://doi.org/10.1016/s0890-8508(03)00062-8.
Peters IR, Helps CR, Hall EJ, Day MJ. Real-time RT-PCR: considerations for efficient and sensitive assay design. J Immunol Methods. 2004;286(1–2):203–17. https://doi.org/10.1016/j.jim.2004.01.003.
Lind K, Ståhlberg A, Zoric N, Kubista M. Combining sequence-specific probes and DNA binding dyes in real-time PCR for specific nucleic acid quantification and melting curve analysis. Biotechniques. 2006;40(3):315–9. https://doi.org/10.2144/000112101.
Sedlak RH, Kuypers J, Jerome KR. A multiplexed droplet digital PCR assay performs better than qPCR on inhibition prone samples. Diagn Microbiol Infect Dis. 2014;80(4):285–6. https://doi.org/10.1016/j.diagmicrobio.2014.09.004.
Verheul RC, van Deutekom JCT, Datson NA. Digital droplet PCR for the absolute quantification of exon skipping induced by antisense oligonucleotides in (pre-)clinical development for Duchenne muscular dystrophy. PLoS ONE. 2016;11(9): e0162467. https://doi.org/10.1371/journal.pone.0162467.
US FDA. Guidance for Industry. S12 Nonclinical Biodistribution considerations for Gene Therapy Products. 2023. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/s12-nonclinical-biodistribution-considerations-gene-therapy-products.
Zhang H, Korenkova V, Sjoback R, Svec D, Bjorkman J, Kruhoffer M, et al. Biomarkers for monitoring pre-analytical quality variation of mRNA in blood samples. PLoS One. 2014;9(11):e111644. https://doi.org/10.1371/journal.pone.0111644. (eCollection 2014).
Malentacchi F, Pizzamiglio S, Verderio P, Pazzagli M, Orlando C, Ciniselli CM, et al. Influence of storage conditions and extraction methods on the quantity and quality of circulating cell-free DNA (ccfDNA): the SPIDIA-DNAplas External Quality Assessment experience. Clin Chem Lab Med. 2015;53(12):1935–42. https://doi.org/10.1515/cclm-2014-1161.
Shroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, et al. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Bol Biol. 2006;7:3. https://doi.org/10.1186/1471-2199-7-3.
Fleige S, Pfaffl M. RNA integrity and the effect on the real-time qRT-PCR performance. Mol Aspects Med. 2006;27(2–3):126–39. https://doi.org/10.1016/j.mam.2005.12.003.
Hiramatsu K, Matsuda C, Masago K, Toriyama K, Sasaki E, Fujita Y, et al. Diagnostic utility of DNA integrity number as an indicator of sufficient DNA quality in next-generation sequencing-based genomic profiling. Am J Clin Pathol. 2023; aqad046. https://doi.org/10.1093/ajcp/aqad046.
Stahlberg A, Kubista M, Pfaffl M. Comparison of reverse transcriptase in gene expression analysis. Clin Chem. 2004;50(9):1678–80. https://doi.org/10.1373/clinchem.2004.035469.
Stahlberg A, Hakansson J, Xian X, Semb H, Kubista K. Properties of the reverse transcription reaction in mRNA quantification. Clin Chem. 2004;50(3):509–15. https://doi.org/10.1373/clinchem.2003.026161.
US FDA. Guidance for Industry: Long Term Follow-up After Administration of Human Gene Therapy Products. 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/long-term-follow-after-administration-human-gene-therapy-products.
European Medicines Agency. Follow-up of Patients Administered with Gene Therapy Medicinal Products. 2009. https://www.ema.europa.eu/en/follow-patients-administered-gene-therapy-medicinal-products.
Kubista M, Andrade JM, Bengtsson M, Forootan A, Jonak J, Lind K, et al. The real-time polymerase chain reaction. Mol Aspects Med. 2006;27(2–3):95–125. https://doi.org/10.1016/j.mam.2005.12.007.
Svec D, Tichopad A, Novosadvoa V, Pfaffl MW, Kubista M. How good is a PCR efficiency estimate: recommendations for precise and robust qPCR efficiency assessments. Biomol Detect Quantif. 2015;3:9–16. https://doi.org/10.1016/j.bdq.2015.01.005.
CLSI. Evaluation of Linearity of Quantitative Measurement Procedures, 2nd edn. CLSI Guideline EP06; Clinical and Laboratory Standards Institute; 2021.
Ambruster DA, Pry T. Limit of blank, limit of detection and limit of quantitation. Clin Biochem Rev. 2008;29(Suppl 1):S49-52.
Forootan A, Sjoback R, Bjorkman J, Sjogreen B, Linz L, Kubista M. Methods to determine limit of detection and limit of quantification in quantitative real-time PCR (qPCR). Biomol Detect Quantif. 2017;2017(12):1–6. https://doi.org/10.1016/j.bdq.2017.04.001.eCollection.
CLSI. EP17: Protocols for Determination of Limits of Detection and Limits of Quantitation, Approved Guideline, 2nd edn. CLSI Guideline EP17-A2. Clinical and Laboratory Standards Institute; 2012.
Stahlberg A, Kubista M. The workflow of single-cell expression profiling using quantitative real-time PCR. Expert Rev Mol Diagn. 2014;14(3):323–31. https://doi.org/10.1586/14737159.2014.901154.
Lou JJ, Mirsadraei L, Sanchez D, Wilson RW, Shabihkhani M, Lucey GM, et al. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin Biochem. 2014;47(4–5):267–73. https://doi.org/10.1016/j.clinbiochem.2013.12.011.
Hartman C, Lennartz K, Ibrahim H, Coz A, Kasper Y, Lenz C, et al. Application note: stable 16-year storage of DNA purified with the QIAamp® DNA blood mini kit. https://www.qiagen.com/us/resources. October 2016. Accessed 24 July 2023.
Walther W, Schmeer M, Kobelt D, Baier R, Harder A, Walhorn V, Anselmetti D, Aumann J, Fichtner I, Schleef M. A seven-year storage report of good manufacturing practice-grade naked plasmid DNA: stability, topology, and in vitro/in vivo functional analysis. Hum Gene Ther Clin Dev. 2013;24:147–53.
Seelenfreund E, Robinson WA, Amato CM, Tan AC, Kim J, Robinson SE. Long term storage of dry versus frozen RNA for next generation molecular studies. PLoS ONE. 2014;9: e111827. https://doi.org/10.1371/journal.pone.0111827.
Author information
Authors and Affiliations
Contributions
The authors are members of the AAPS Bioanalytical Community PCR Working Group. All authors contributed equally to discussions, writing, reviewing, and editing of this manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors are employed by and receive compensation from their affiliations that are listed in this manuscript. The authors have no other relevant affiliations or financial involvements with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Disclaimer
The views and conclusion presented in this paper are those of the authors and do not necessarily reflect the representative affiliation or company’s position on the subject.
Additional information
Communicated by Vittal Shivva, Yan Ni, Steven Louie and Mario L. Rocci Jr.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
A majority of the work was conducted under Melanie Chalfant’s prior affiliation with Janssen Research & Development, LLC.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hays, A., Wissel, M., Colletti, K. et al. Recommendations for Method Development and Validation of qPCR and dPCR Assays in Support of Cell and Gene Therapy Drug Development. AAPS J 26, 24 (2024). https://doi.org/10.1208/s12248-023-00880-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1208/s12248-023-00880-9