
Abstract
Repetitive elements have been identified in several amphibian genomes using whole genome sequencing, but few studies have used cytogenetic mapping to visualize these elements in this vertebrate group. Here, we used fluorescence in situ hybridization and genomic data to map the U1 and U2 small nuclear RNAs and histone H3 in six species of African clawed frog (genus Xenopus), including, from subgenus Silurana, the diploid Xenopus tropicalis and its close allotetraploid relative X. calcaratus and, from subgenus Xenopus, the allotetraploid species X. pygmaeus, X. allofraseri, X. laevis, and X. muelleri. Results allowed us to qualitatively evaluate the relative roles of polyploidization and divergence in the evolution of repetitive elements because our focal species include allotetraploid species derived from two independent polyploidization events — one that is relatively young that gave rise to X. calcaratus and another that is older that gave rise to the other (older) allotetraploids. Our results demonstrated conserved loci number and position of signals in the species from subgenus Silurana; allotetraploid X. calcaratus has twice as many signals as diploid X. tropicalis. However, the content of repeats varied among the other allotetraploid species. We detected almost same number of signals in X. muelleri as in X. calcaratus and same number of signals in X. pygmaeus, X. allofraseri, X. laevis as in the diploid X. tropicalis. Overall, these results are consistent with the proposal that allopolyploidization duplicated these tandem repeats and that variation in their copy number was accumulated over time through reduction and expansion in a subset of the older allopolyploids.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Repetitive elements are genomic sequences that are present in multiple copies and are found in all eukaryotic genomes, but with varying abundances and genomic distributions. Content of repetitive DNA ranges from less than 10% in some fishes (e.g., Tetraodon nigroviridis and Cynoglossus semilaevis) and birds (e.g., Phoenicopterus ruber, Struthio camelus, Haliaeetus albicilla) to > 90% in some plants, such as Allium cepa (Chalopin et al. 2015; Chen et al. 2014; Zhang et al. 2014; Canapa et al. 2016; Fu et al. 2019).
Repetitive elements in eukaryotes are categorized into transposable elements (TEs) and tandem repeats. TEs, which are scattered throughout the genome, include retroelements/retrotransposons and DNA transposons. Retrotransposons are reversely transcribed to DNA and replicated using a copy and paste mechanism. Therefore, they may increase genome size (Yampolsky 2016; Clark et al. 2019). Among retrotransposons, the LINEs (Long Interspersed Nuclear Elements) are classified into many subcategories (Valente et al. 2011; Chalopin et al. 2015; Gama et al. 2022). In contrast, tandem repeats are found as tandem arrays or head-to-tail motifs from several to more than a hundred copies (Myers 2007). Tandem repeats are present in clusters within the genome on single chromosomal locus or multiple loci and include microsatellite, minisatellite, satellite DNA, ribosomal genes (minor 5S and major 45S), histone genes, and small nuclear RNA (snRNA) (Symonová et al. 2017b; Knytl et al. 2018a; Sember et al. 2020; Gazoni et al. 2021; Schott et al. 2022). These loci often undergo concerted evolution (Elder and Turner 1995; Liao 1999), causing intraspecific paralogous repetitive elements to be more similar to each other than to their orthologs, even when the origin of repetitive elements precedes speciation (Liao 1999). Repetitive loci on chromosomes show dynamic evolution in terms of where in the genome and how frequently they are found, and they often have a high rate of rearrangement (Bruschi et al. 2014; Liu et al. 2019).
Due to their repetitive nature, short read sequences typically pose challenges to mapping and repeat quantification, including using genomic data. However, localization of repetitive DNA can be achieved using fluorescent in situ hybridization (FISH) cytogenetic techniques. Cytogenetic mapping of repetitive sequences has been studied in teleost fishes (Bishani et al. 2021; Knytl et al. 2022), reptiles (Oliveira et al. 2021; Altmanová et al. 2022), birds (De Oliveira et al. 2017; Kretschmer et al. 2021), and mammals (Milioto et al. 2019; Gerbault-Seureau et al. 2018). In amphibians, cytogenetic mapping of repetitive sequences has identified inter- and intraspecific rearrangements and other features of karyotype evolution (da Silva et al. 2021; Phimphan et al. 2021; Guzmán et al. 2022).
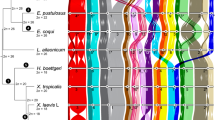
African clawed frogs of the genus Xenopus (Pipidae) include almost 30 species and are divided into two subgenera (Xenopus and Silurana) (Evans et al. 2015) that each is sometimes considered to be a genus (e.g., Dubois et al. 2021). Subgenus Silurana includes four described species, the diploid Xenopus (Silurana) tropicalis and three allotetraploids X. calcaratus, X. epitropicalis, and X. mellotropicalis. For practical reasons, we use the name Silurana throughout this article, but present it at the subgenus level in accordance with Evans et al. (2015). Subgenus Xenopus includes 25 tetraploid, octoploid, or dodecaploid species (Tymowska 1991) that are divided into three species groups: amieti, laevis, and muelleri (Evans et al. 2015). In genus Xenopus, at least eight independent allopolyploidization events occurred, including at least one allotetraploidization event in each subgenus (Fig. 1; for an alternative scenario in Silurana, see Knytl et al. 2023). This variation in independently evolved ploidy levels offers a compelling opportunity to explore how tandem repeats evolve in the context of genome duplications.

Inferred phylogenetic relationships and approximate divergence times among our focal taxa based on Evans et al. (2015), Session et al. (2016), and Feng et al. (2017). Allotetraploidization (depicted as reticulating lineages) occurred independently in each subgenus; inferred diploid lineages with no known extant diploid descendants are indicated with daggers. Time scale is in millions of years ago; chromosome numbers are shown in parentheses
About 34.5% and 40% of the X. tropicalis and X. laevis genomes are repetitive (Hellsten et al. 2010; Session et al. 2016). Ribosomal DNAs (rDNAs) have been cytogenetically mapped on Xenopus chromosomes (Schmid and Steinlein 2015; Knytl et al. 2017, 2023; Roco et al. 2021). In allotetraploid species of subgenus Silurana, rDNA FISH analysis identified nucleolar organizer region (NOR) on chromosome 7a where the letter “a” refers to the “a subgenome” within the allotetraploid genome (Knytl et al. 2017, 2023). That only one pair of homologous chromosomes contains the NOR locus in Xenopus allotetraploids suggests deletion of NOR on chromosome 7b (of the b subgenome) after allotetraploidization (Knytl et al. 2023) because a NOR would have been required in each diploid ancestral species. There is limited cytogenetic information from other repetitive sequences in Xenopus, and thus, it is also not known whether non-rDNA repeats vary among Xenopus species.
Small nuclear RNAs are components of the spliceosome that perform pre-mRNA splicing. The spliceosome is composed of five RNA tandem-repeat units (U1, U2, U4, U5, and U6 snRNAs) (Valadkhan 2005). Histones are components of the nucleosome that are present in tandem repeat families of five major genes: H1/H5, H2A, H2B, H3, and H4. Here, we used FISH for mapping U1 and U2 snRNA and histone H3 loci on chromosomes from six species of Xenopus, including members of both subgenera which diverged from one another approximately 45–50 Mya (Session et al. 2016; Feng et al. 2017). The allotetraploidization event in subgenus Xenopus is estimated to have occurred earlier (17–18 Mya) than the one in subgenus Silurana and the onset of the polyploid radiation in subgenus Xenopus at approximately 17 Mya (Session et al. 2016). Specifically, we examined the diploid X. tropicalis and its tetraploid close relative X. calcaratus, which are from subgenus Silurana, and the allotetraploids X. pygmaeus, X. allofraseri (both from the amieti species group), X. laevis (laevis species group), and X. muelleri (muelleri species group) which belong to subgenus Xenopus and arose from the older allotetraploidization. In the subgenus Xenopus, the L and S subgenomes diverged from one another about 30–35 Mya, whereas the subgenomes a and b in the tetraploid Silurana diverged from one another about 10 Mya (Evans et al. 2015; Session et al. 2016). With an overarching goal of exploring the effect of polyploidization and divergence on the evolution of tandem repeats, we used cytogenetic methods and a genome database to test the hypotheses that (i) the number of the repeat loci in diploid X. tropicalis is half that in the allotetraploid species, and (ii) the locations of repeat loci in the studied species are homologous.
Materials and methods
Primary cell cultures and metaphase spread preparations
Primary cell cultures were derived from laboratory strains of X. tropicalis (strain ‘Ivory Coast’), X. laevis, and X. muelleri, and wild-derived stains of X. calcaratus and X. allofraseri from Cameroon (Bakingili, where they occur in syntopy, 4.0684°N, 9.0682°E) and X. pygmaeus from the Democratic Republic of the Congo (Kokolopori, Yalokole, near Luo River, 0.2056°N, 22.8884°E). Xenopus muelleri animals were originally obtained from the Institute of Zoology at the University of Geneva (Switzerland). All species were bred at Charles University, Faculty of Science, Prague, Czech Republic. Briefly, tadpoles were anesthetized, hind limbs removed and homogenized (Sinzelle et al. 2012) in a cultivation medium (Knytl et al. 2017) modified according to Knytl et al. (2023). The explants were then cultivated at 29.5 °C with 5.5% CO2 for five days without disturbance. The media was then changed every day for one week. Passages were performed with trypsin-ethylenediaminetetraacetic acid (Knytl et al. 2017).
Chromosomal suspensions were prepared following Krylov et al. (2010) and stored in fixative solution (methanol/acetic acid, 3:1, v/v) at − 20 °C. For cytogenetic analysis, a chromosome suspension was dropped onto a slide (Courtet et al. 2001). Chromosome preparations were aged at − 20 °C for at least one week. For each experiment, mitotic metaphase spreads were counterstained with ProLong™ Diamond Antifade Mountant with the fluorescent 4′,6-diamidino-2-phenylindole, DAPI stain (Invitrogen by Thermo Fisher Scientific, Waltham, MA, USA). From ten to 20 metaphase spreads were analyzed per probe. Microscopy and processing of metaphase images were conducted using a Leica Microsystem (Wetzlar, Germany) as detailed in Seroussi et al. (2019).
Fluorescent in situ hybridization with repetitive DNA probes
In order to generate probes for FISH, genomic DNA from X. tropicalis was used as a template for amplification of the repetitive small nuclear DNA regions (snDNA) U1 and U2, and histone H3. DNA was extracted from tadpole tail tissue using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) according to manufacturer’s instructions. Primers used for amplification are listed in Table 1. The annealing temperature was 54 °C and the elongation step 30 s for all PCR reactions; other conditions for PCR amplification with PPP Master Mix (Top-Bio, Prague, Czech Republic) followed the manufacturer’s recommendations. Labeling PCR was performed as described in Knytl and Fornaini (2021). Digoxigenin-11-dUTP (Jena Bioscience, Jena, Germany) was used for U2 and H3 labeling, and biotin-16-dUTP (Jena Bioscience) was used for U1 labeling. Xenopus tropicalis U1 and U2 snDNA and H3 probes were then hybridized to chromosome spreads of X. tropicalis, X. calcaratus, X. pygmaeus, X. allofraseri, X. laevis, and X. muelleri. The procedures for hybridization mixture preparation, denaturation, and the subsequent overnight hybridization were described previously for rDNA FISH (Knytl et al. 2023). Post-hybridization washing and blocking reactions were performed as described for painting FISH in Krylov et al. (2010). Probe visualization was performed following Knytl et al. (2017). Slides were then de-stained according to the following protocol: nail polish was removed using xylene (2 min) and benzoin (2 min). Slides with cover slip were incubated in 4 × SSC/0.1% Tween for 10 min with agitation, and then cover slips were manually removed. Slides without cover slip were then incubated in 4 × SSC/0.1% Tween for 30 min with agitation followed by dehydration with methanol series (70, 90, 100% for 3 min each) and then air dried. After slide incubation in fixative solution for 30 min, slides were rinsed with distilled water, air dried, and then aged for 90 min at 60 °C.
Painting FISH
We used whole chromosome painting probes generated by laser microdissection of X. tropicalis chromosomes from previous study by Knytl et al. (2023). Whole chromosome painting probes from X. tropicalis chromosomes 1 and 8 were newly labeled with digoxigenin-11-dUTP (Krylov et al. 2010) and biotin-16-dUTP (both Jena Bioscience) (Knytl et al. 2023), respectively. De-stained slides (after FISH with the U1 and U2 probes) of X. tropicalis, X. calcaratus, and X. laevis were used for cross-species painting FISH (Zoo-FISH) with a digoxigenin-labeled probe according to the protocols described in Krylov et al. (2010) and modified in Knytl et al. (2017). Subsequently, slides were de-stained again and used for Zoo-FISH with biotin-labeled probe. Detection of signal was carried as detailed for double-color painting in Knytl et al. (2023). The X. tropicalis FISH experiments were performed in the reverse order from the other species (i.e., painting FISH first followed by de-staining and snDNA FISH).
Results
Sanger sequencing and BLAST searching
Amplification of the U1 and U2 snDNA and H3 nuclear genes yielded 119 and 177 and 364 bp long amplicons, respectively. Based on BLASTn searches of the X. tropicalis “Nigeria” strain genome, the U1 amplicon had 98.3% identity (query cover 100%) with the sequence of U1 spliceosomal RNA, LOC116408489 (accession number XR_004220992.1); U2 amplicon had 98.3% identity (query cover 100%) with U2 spliceosomal RNA, LOC116407440 (accession number XR_004220346.1); and the H3 amplicon had 97.2% identity (query cover 98%) with histone H3, LOC100497127, mRNA (accession number XM_012953339.3). Our sequences were deposited to the NCBI GenBank database: U1 snDNA accession number OQ714817, U2 snDNA accession number OQ714818, H3 accession number OQ714819.
FISH with U1 and U2 snDNA probes and their genomic locations in available Xenopus genome assemblies
When hybridized on the diploid species X. tropicalis, the U1 and U2 snDNA probes had intense signals on the distal region of the long (q) arm of chromosomes 1 and 8 (Fig. 2(a)), respectively. The number of copies was determined based on matches at least 50% of the query sequence length and 85% of the query sequence identity. BLAST results using the U1 sequence as a query to the X. tropicalis genome identified ~ 20 copies of U1 snDNA on chromosome 1. Using the U2 sequence as a query, we identified ~ 40 copies on X. tropicalis chromosome 8. In the allotetraploid species X. calcaratus, the U1 and U2 snDNA probes hybridized to the q arms of the chromosomes 1a, 1b and 8a, 8b (Fig. 2(b)), respectively. Chromosomes 1a and 1b are homoeologous to each other, and they are orthologous to X. tropicalis chromosome 1, and the mapped U1 region in X. calcaratus is thus homologous to the orthologous U1 region of X. tropicalis (both species have U1 gene on the distal part of the same chromosome). The U2 snRNA locus also was detected on both homoeologous chromosomes of X. calcaratus (chromosomes 8a and 8b) and in a homologous location to the orthologous U2 gene of X. tropicalis. For detailed definitions of homologous, homoeologous, and orthologous genes and chromosomes in Xenopus, see Song et al. (2021).

Double-color FISH with U1 and U2 snDNA probes. The U1 probe (red) reveals one clear signal (= a pair of homologous chromosomes) in a X. tropicalis, c X. pygmaeus, d X. allofraseri, and e X. laevis, while the same FISH shows two signals, each within homoeologous chromosomes in b X. calcaratus and f X. muelleri. The U2 (green) probe shows one signal in a X. tropicalis, c X. pygmaeus, d X. allofraseri, and e X. laevis, while the U2 probe shows two signals, each of them within homoeologous chromosomes in b X. calcaratus and f X. muelleri. Green and red arrows correspond to the U2 and U1 repeat loci, respectively. Painting probes were used for identification of chromosomes 1 (green) and 8 (red) in a X. tropicalis. Chromosomes were counterstained with DAPI in blue/gray. Scale bars represent 10 µM
These U1 and U2 snDNA probes thus each hybridized to both homoeologous chromosomes in X. calcaratus as expected if copy number of each repeat was doubled by allopolyploidization. Similarly, in X. muelleri, U1 and U2 snDNA probes mapped to the expected homoeologous chromosomes (U1 on 1Lq and 1Sq; U2 on 8Lq and 8Sq; Fig. 2(f)).
In the allotetraploid species X. pygmaeus, X. allofraseri, and X. laevis only one signal was detected for U1 and U2 snDNAs. For the more closely related pair — X. pygmaeus and X. allofraseri — the U1 snDNA probe hybridized most conspicuously to the q arms of chromosome 1L and the U2 snDNA probe hybridized to the q arms of chromosome 8S (Fig. 2(c, d)). In X. laevis, the signal of the U1 and U2 snDNA probes was most conspicuous on the q arms of chromosomes 1S and 8L, respectively (Fig. 2(e)). Both U1 and U2 snRNA signals in X. laevis were located on chromosomes (1S and 8L) that are homoeologous to the chromosomes that bear U1 and U2 snRNA signals in X. pygmaeus and X. allofraseri (1L and 8S) and vice versa. The number of loci did not support the expectation that all tetraploid species should have twice as many snRNA loci as the diploid X. tropicalis, a result that could indicate copy number reduction or loss. Consistent with a scenario of reduction in copy number as opposed to complete loss, a BLAST search using U1 snDNA sequence as a query to the X. laevis genome recovered 11 copies on chromosome 1L and ~ 35 copies on chromosome 1S. Using the U2 sequence as a query against the X. laevis genome sequence, we identified ~ 40 copies on X. laevis chromosome 8L and 12 copies on chromosome 8S.
Painting FISH
To identify chromosomes bearing U1 and U2 loci, we employed FISH with the whole chromosome painting probes from X. tropicalis chromosomes 1 and 8. We successfully identified chromosomes 1 and 8 in X. tropicalis (Fig. 2(a)); chromosomes 1a, 1b, 8a, and 8b in X. calcaratus (Fig. 3); and chromosomes 1L, 1S, 8L, and 8S in X. laevis (Fig. 4). Homoeologous chromosomes of X. calcaratus and X. laevis were identified based on the intensity of the fluorescence signal (Knytl et al. 2023) and in the other studied species in subgenus Xenopus in which the painting FISH approach was not conducted; instead, homoeologous chromosomes were distinguished based on chromosome length (the L homoeologous chromosomes are longer than the S chromosomes; Matsuda et al. 2015; Session et al. 2016).

Sequential FISH in X. calcaratus chromosomes. Cross-species painting FISH experiments with the whole chromosome painting probes from X. tropicalis a chromosome 1 (XTR 1) and b XTR 8 highlight chromosome-bearing U1 and U2 snRNA loci, respectively. c snDNA FISH illustrates that the U1 locus (in red) is localized on X. calcaratus chromosomes 1 (XCA 1a and 1b) and the U2 locus (in green) on XCA 8a and 8b. a and b are shown in the red channel, c is merged in the red-green-blue (RGB) channels. c chromosomes were counterstained with DAPI in gray. Scale bars represent 10 µM

Sequential FISH in X. laevis chromosomes. Cross-species painting FISH experiments with whole chromosome painting probes from a XTR 1 (in red) and XTR 8 (in green) illustrate that b the U1 locus (in red) is localized on X. laevis chromosomes 1S (XLA 1S), and the U2 locus (in green) on XLA 8L. Both a and b are merged in the RGB channels. Chromosomes were counterstained with DAPI in blue. Scale bars represent 10 µM
FISH with histone H3 probe and its genomic locations in available Xenopus genome assemblies
We then hybridized H3 probe to chromosome spreads of X. tropicalis, X. calcaratus, X. pygmaeus, X. allofraseri, X. laevis, and X. muelleri. In all species, signals were present in small patches on multiple chromosomes (Fig. 5). We found signals in all chromosomes in X. tropicalis, X. calcaratus, and X. muelleri. However, X. pygmaeus, X. allofraseri, and X. laevis had signals on about half of chromosomes. The H3 probes mapped to telomeric and pericentromeric regions.

FISH with histone H3 probe. The probe (green) shows multiple signals in a X. tropicalis, b X. calcaratus, c X. pygmaeus, d X. allofraseri, e X. laevis, and f X. muelleri. Chromosomes were counterstained with DAPI in blue/gray. Scale bars represent 10 µM
Based on BLAST searches, the H3 sequence occurs on X. tropicalis chromosomes 3 (11 hits), 6 (11 hits), and 9 (27 hits), and on chromosomes 5, 8, and 10, the matches were less than 50% of the query length. In Xenopus laevis, the H3 sequence occurs on chromosomes 3S (12 hits), 5L (6 hits), 5S (3 hits), 6L (2 hits), 6S (1 hit), 9_10L (15 hits), and 9_10S (5 hits). Xenopus laevis chromosomes 1L and 8L show some hits being less than 50% of the query sequence. All U1 and U1 snRNA and H3 loci mapped by FISH and BLAST are shown in Table 2.
Discussion
Quantification and localization of repetitive sequences using high-quality genome sequencing and assembly is an expensive and challenging undertaking as compared to using multiple cytogenetic approaches for gene mapping (Knytl et al. 2018b; Symonová et al. 2017a). As an alternative, we cytogenetically mapped non-rDNA tandem repeats (U1 and U2 snRNA and H3 histone) to one diploid and five allotetraploid Xenopus species with the aim of testing how repetitive elements were affected by genome duplication and divergence. At least two allotetraploidization events occurred in genus Xenopus with respect to our focal species and both — including studied species and their diploid extinct or yet undescribed predecessors — are depicted in Fig. 1.
Barring loss or movement of tandem repeats, we expected allotetraploid species to have twice as many tandem repeats as the diploid X. tropicalis, and that the locations of these elements would be unchanged (i.e., on the same region of both homoeologous chromosomes). In general, the cytogenetic results recovered no evidence for long-range movement of the U1, U2 snRNA or H3 loci. In the allotetraploids X. calcaratus and X. muelleri, the observed number of signals for each of the U1 and U2 snRNA loci matched our expectation based on allopolyploidization. However, for the three other allotetraploids (X. pygmaeus, X. allofraseri, and X. laevis), this expectation was not supported because, although in homologous locations, we found the same number of these tandem repeats as in the diploid X. tropicalis (Table 2). Furthermore, the snRNA signals in X. pygmaeus and X. allofraseri were on a different subgenome (U1 on L, U2 on S) than in X. laevis (U1 on S, U2 on L). Overall, this is consistent with an increased variation in copy number over time following allopolyploidization. BLAST searches of the X. laevis genome recovered each of these loci in both subgenomes, and it appears that the lower-than-expected number of cytogenetic signals is thus a consequence of differences in copy number, at least in this species. That the signals in X. pygmaeus and X. allofraseri are on different subgenomes than in X. laevis suggests that changes in copy number have been an ongoing phenomenon during Xenopus diversification. During diversification of African clawed frogs, the ancestor of X. muelleri diverged from the common ancestor of X. pygmaeus, X. allofraseri, and X. laevis soon after allotetraploidization in subgenus Xenopus, around 17 Mya (Fig. 1; Evans et al. 2015; Session et al. 2016). Thus, a parsimonious interpretation of these results posits that changes in copy number arose independently in an ancestor of X. laevis and again in the most recent common ancestor of X. pygmaeus and X. allofraseri.
In amphibians, U2 snRNA tandem repeats have also been localized in the cycloramphid genus Thoropa (Cholak et al. 2020) and the leptodactylid genus Leptodactylus (Gazoni et al. 2021), and the H3 gene has been localized in the pipid genus Pipa (Zattera et al. 2020). To our knowledge, there are no other cytogenetic studies that localize the U1, U2 snRNA, or H3 gene loci in this vertebrate group.
In frog genus Thoropa, U2 snRNA was detected cytogenetically on chromosomes 6 and 7 in some species, but others had U2 locus only on chromosome 6 or only on chromosome 7 (Cholak et al. 2020). The explanation for this variation might be that U2 loci are universally situated on both chromosomes 6 and 7 in all investigated Thoropa species but with different copy numbers on each of these chromosomes. We found similar variability in the location and number of FISH signals. Using BLAST, we identified locations where the sequence of our probe matched the X. laevis genome sequence and found that the number of U1 and U2 snRNAs detected by BLAST did not match the number of snRNAs detected by FISH. This inconsistency may be due to variation in the copy number of tandem repeats in each X. laevis subgenome and the lack of sensitivity of FISH to detect the locus with a low copy number of repeats. The U1 and U2 snRNA FISH signals were cytogenetically detected on those X. laevis chromosomes that contain ~ 15 and more copies of U1 and U2 repeats. In Leptodactylus, the U2 loci were identified on chromosome 6 in eight species, but one of them had signals on chromosomes 4, 6, 9, and 10 (Gazoni et al. 2021). These authors proposed that the additional signals on chromosomes 4, 9, and 10 can be the result of transposition by TEs followed amplification of the gene. We did not map TEs on Xenopus chromosomes and are thus unable to evaluate the possibility of TE transposition in this genus.
We also evaluated the effects of allotetraploidization and divergence on the evolution of H3 repeats. As expected, due to allotetraploidization, the allotetraploids X. calcaratus and X. muelleri possessed H3 repeats on twice as many chromosomes as the diploid X. tropicalis. However, in the allotetraploid X. pygmaeus, X. allofraseri, and X. laevis, we found signals on eight homologous pairs, which are fewer than expected based on allotetraploidization. This presumably is due to a decreased copy number or deletion of H3 repeats on several chromosomes, possibly in an ancestor of X. pygmaeus, X. allofraseri, and X. laevis based on phylogenetic relationships among these species. Despite the variability associated with allopolyploidization followed by divergence in evolution of tandem repeats, the relative dosages of signals of U1, U2, and H3 with respect to X. tropicalis are similar in each allotetraploid species we examined.
Until recently, there was only one study in which the H3 gene was mapped on amphibian chromosomes (Zattera et al. 2020). In Pipa carvalhoi, a representative of the same family as Xenopus but deeply phylogenetically divergent by about 115–120 My (Feng et al. 2017; Hime et al. 2021), H3 signals were detected on chromosomes 1, 5, 6, 8, and 9, and less intense signals were visible on other chromosomes. Multiple distributions of H3 locus on all chromosomes have been observed, for example, in insect species of the family Acrididae (Oliveira et al. 2011; Bueno et al. 2013) where the distribution of the H3 locus may be related to the transposition of 5S ribosomal RNA (rRNA). In Xenopus, 5S rRNA is situated on telomeres of multiple chromosomes (Knytl et al. 2017, 2023), and therefore, the observed distribution of the H3 locus on multiple Xenopus chromosomes may also be associated to the distribution of 5S rRNA.
Co-localization of repetitive elements
An interesting finding that emerged from this study is a genomic association of various repetitive elements with rRNA; this can potentially provide evolutionary insights into genome organization. Synteny exists between As51 satellite DNA (originating from TEs) and NORs (Vicari et al. 2008) or between U1 snRNA and 5S rRNA in the characid fish genus Astyanax (Silva et al. 2015). One of possible roles of a co-localization can be a silencing of ribosomal genes by TEs (Vicari et al. 2008). All Xenopus species have NOR on a single homologous pair (Tymowska 1991). Xenopus tropicalis has NOR on chromosome 7, tetraploids from subgenus Silurana on 7a, and tetraploids from subgenus Xenopus on 3L (Tymowska 1991; Session et al. 2016; Roco et al. 2021; Knytl et al. 2023). We have not identified any snRNA loci on chromosomes 7, 7a, or 3L, and this finding argues against a genomic association between distinct NOR and snRNA repeats. The 5S ribosomal genes have been detected at the distal regions of the X. mellotropicalis and X. calcaratus (Silurana tetraploids) chromosomes 8bq (Knytl et al. 2017, 2023) which is the same position as the U2 snRNA locus in X. calcaratus (this study), supporting a genomic association between 5S rRNA and U2 snRNA. The NOR locus presumably was deleted on chromosome 7b in tetraploids of subgenus Silurana and on chromosome 3S in tetraploids of subgenus Xenopus (Session et al. 2016; Knytl et al. 2023). However, U1 and U2 loci map to both homoeologs in some tetraploids, and thus, deletion of snRNA in an ancestor of Xenopus tetraploids did not take place. There is the possibility that all our investigated Xenopus tetraploids have twice as many snRNA repeat loci as the diploid X. tropicalis but that these loci have unequal copy number within individual subgenomes, meaning that copy number of snRNA repeats was reduced in one subgenome (11 copies of U1 snRNA on X. laevis chromosome 1L is less than 20 copies on X. tropicalis chromosome 1; thus, the copy number of U1 snRNA on X. laevis chromosome 1L was reduced) but expanded in the other subgenome (35 copies of U1 snRNA on X. laevis chromosome 1S versus 20 copies in X. tropicalis). Moreover, some tandem repeat loci maintain a stable number of copies, for example, 40 copies of U2 snRNA on X. tropicalis chromosome 8 and X. laevis chromosomes 8L). These stable copy numbers within the U2 gene are consistent with studies that observed a higher evolutionary conservation of the X. laevis subgenome L than subgenome S, compared to X. tropicalis genome (Session et al. 2016). Reduction, expansion, and stability in the copy number of non-rDNA tandem repeats and complete loss of rRNA locus are the possible events that occur after polyploidization (Session et al. 2016; Knytl et al. 2023). Genes retained as duplicates have distinct evolution leading to loss of function, subfunctionalization, or neofunctionalization and are under strong purifying selection (Force et al. 1999; Lynch et al. 2001), often followed by intra- and/or interchromosomal translocations, inversions, deletions, or degenerations (Evans 2007; Sémon and Wolfe 2007; Session et al. 2016; Knytl et al. 2017). Our study highlighted that copy number reduction and expansion of tandem repeats can be an important driver of evolution following allopolyploidization such as translocation, inversion, deletion, and degeneration.
Conclusion
Repetitive elements are integral components of a genome, and their evolution can be affected by mechanism of polyploidization or divergence among species. We used cytogenetic approaches to study repetitive elements in six species of Xenopus frogs (one diploid, five allotetraploids) with an aim of localizing and quantifying tandem repeats in their genomes. For U1 and U2 snRNA, and H3 tandem repeats, the locations were generally homologous, but we detected variation in copy numbers that resulted from reduction and expansion after allotetraploidization. The dynamic evolution of tandem repeats was most apparent in allotetraploid species that arose from the older allotetraploidization event.
Data availability
The Sanger sequences that support the findings of this study are openly available in GenBank (accession numbers OQ714817-9).
References
Altmanová M, Doležálková-Kaštánková M, Jablonski D, Strachinis I, Vergilov V, Vacheva E et al (2022) Karyotype stasis but species-specific repetitive DNA patterns in Anguis lizards (Anguidae), in the evolutionary framework of Anguiformes, PREPRINT (Version 1) available at Research Square. https://doi.org/10.21203/rs.3.rs-2413537/v1
Bishani A, Prokopov DY, Romanenko SA, Molodtseva AS, Perelman PL, Interesova EA et al (2021) Evolution of tandemly arranged repetitive DNAs in three species of Cyprinoidei with different ploidy levels. Cytogenet Genome Res 161(1–2):32–42. https://doi.org/10.1159/000513274
Bruschi DP, Rivera M, Lima AP, Zúñiga AB, Recco-Pimentel SM (2014) Interstitial telomeric sequences (ITS) and major rDNA mapping reveal insights into the karyotypical evolution of Neotropical leaf frogs species (Phyllomedusa, Hylidae, Anura). Mol Cytogenet 7(1):1–12. https://doi.org/10.1186/1755-8166-7-22
Bueno D, Palacios-Gimenez OM, Cabral-de-Mello DC (2013) Chromosomal mapping of repetitive DNAs in the grasshopper Abracris flavolineata reveal possible ancestry of the B chromosome and H3 histone spreading. PLoS One 8(6). https://doi.org/10.1371/journal.pone.0066532
Canapa A, Barucca M, Biscotti MA, Forconi M, Olmo E (2016) Transposons, genome size, and evolutionary insights in animals. Cytogenet Genome Res 147(4):217–239. https://doi.org/10.1159/000444429
Chalopin D, Naville M, Plard F, Galiana D, Volff JN (2015) Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol Evol 7(2):567–580. https://doi.org/10.1093/gbe/evv005
Chen S, Zhang G, Shao C, Huang Q, Liu G, Zhang P et al (2014) Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat Genet 46:253–260. https://doi.org/10.1038/ng.2890
Cholak LR, Haddad CFB, Parise-Maltempi PP (2020) Cytogenetic analysis of the genus Thoropa Cope, 1865 (Anura-Cycloramphidae) with evolutionary inferences based on repetitive sequences. Genet Mol Biol 43(3):e20190364. https://doi.org/10.1590/1678-4685-GMB-2019-0364
Clark DP, Pazdernik NJ, McGehee MR (2019) Molecular biology. Elsevier, Academic Cell. https://doi.org/10.1016/C2015-0-06229-3
Colgan DJ, McLauchlan A, Wilson GDF, Livingston SP, Edgecombe GD, Macaranas J et al (1998) Histone H3 and U2 snRNA DNA sequences and arthropod molecular evolution. Aust J Zool 46:419–437. https://doi.org/10.1071/ZO98048
Courtet M, Flajnik M, Du Pasquier L (2001) Major histocompatibility complex and immunoglobulin loci visualized by in situ hybridization on Xenopus chromosomes. Dev Comp Immunol 25(2):149–157. https://doi.org/10.1016/S0145-305X(00)00045-8
Da Silva DS, Da Silva Filho HF, Cioffi MB, De Oliveira EHC, Gomes AJB (2021) Comparative cytogenetics in four leptodactylus species (Amphibia, Anura, Leptodactylidae): evidence of inner chromosomal diversification in highly conserved karyotypes. Cytogenet Genome Res 161(1–2):52–62. https://doi.org/10.1159/000515831
De Oliveira FI, Kretschmer R, Dos Santos MS, De Lima Carvalho CA, Gunski RJ, O’Brien PCM et al (2017) Chromosomal mapping of repetitive DNAs in Myiopsitta monachus and Amazona aestiva (Psittaciformes, Psittacidae) with emphasis on the sex chromosomes. Cytogenet Genome Res 151(3):151–160. https://doi.org/10.1159/000464458
Dubois A, Ohler A, Pyron RA (2021) New concepts and methods for phylogenetic taxonomy and nomenclature in zoology, exemplified by a new ranked cladonomy of recent amphibians (Lissamphibia). Megataxa 5(1):1–738. https://doi.org/10.1159/000464458
Elder JF, Turner BJ (1995) Concerted evolution of repetitive DNA sequences in eukaryotes. Q Rev Biol 70(3):297–320. https://doi.org/10.1086/419073
Evans BJ (2007) Ancestry influences the fate of duplicated genes millions of years after polyploidization of clawed frogs (Xenopus). Genetics 176(2):1119–1130. https://doi.org/10.1534/genetics.106.069690
Evans BJ, Carter TF, Greenbaum E, Gvoždík V, Kelley DB, McLaughlin PJ et al (2015) Genetics, morphology, advertisement calls, and historical records distinguish six new polyploid species of African clawed frog (Xenopus, Pipidae) from West and Central Africa. PLoS One 10(12):e0142823. https://doi.org/10.1371/journal.pone.0142823
Feng YJ, Blackburn DC, Liang D, Hillis DM, Wake DB, Cannatella DC et al (2017) Phylogenomics reveals rapid, simultaneous diversification of three major clades of Gondwanan frogs at the Cretaceous-Paleogene boundary. Proc Natl Acad Sci USA 114(29):E5864–E5870. https://doi.org/10.1073/pnas.1704632114
Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J (1999) Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151(4):1531–1545. https://doi.org/10.1093/genetics/151.4.1531
Fu J, Zhang H, Guo F, Ma L, Wu J, Yue M et al (2019) Identification and characterization of abundant repetitive sequences in Allium cepa. Sci Rep 9(1):1–7. https://doi.org/10.1038/s41598-019-52995-9
Gama JM, Ludwig A, Gazolla AB, Guizelini D, Recco-Pimentel SM, Bruschi DP (2022) A genomic survey of LINE elements in Pipidae aquatic frogs shed light on Rex-elements evolution in these genomes. Mol Phylogenet Evol 168:107393. https://doi.org/10.1016/j.ympev.2022.107393
Gazoni T, Dorigon NS, Da Silva MJ, Cholak LR, Haddad CFB, Parise-Maltempi PP (2021) Chromosome mapping of U2 snDNA in species of Leptodactylus (Anura, Leptodactylidae). Cytogenet Genome Res 161(1–2):63–69. https://doi.org/10.1159/000515047
Gerbault-Seureau M, Cacheux L, Dutrillaux B (2018) The relationship between the (in-)stability of NORs and their chromosomal location: the example of cercopithecidae and a short review of other primates. Cytogenet Genome Res 153(3):138–146. https://doi.org/10.1159/000486441
Guzmán K, Roco ÁS, Stöck M, Ruiz-García A, García-Muñoz E, Bullejos M (2022) Identification and characterization of a new family of long satellite DNA, specific of true toads (Anura, Amphibia, Bufonidae). Sci Rep 12(1). https://doi.org/10.1038/s41598-022-18051-9
Hellsten U, Harland RM, Gilchrist MJ, Hendrix D, Jurka J, Kapitonov V et al (2010) The genome of the Western clawed frog Xenopus tropicalis. Science (80- ) 328(5978):633–6. https://doi.org/10.1126/science.1183670
Hime PM, Lemmon AR, Lemmon ECM, Prendini E, Brown JM, Thomson RC et al (2021) Phylogenomics reveals ancient gene tree discordance in the Amphibian Tree of Life. Syst Biol 70:49–66. https://doi.org/10.1093/sysbio/syaa034
Karimi K, Vize PD (2014) The Virtual Xenbase: transitioning an online bioinformatics resource to a private cloud. Database (Oxford) 2014:bau108. https://doi.org/10.1093/database/bau108
Knytl M, Fornaini NR (2021) Measurement of chromosomal arms and FISH reveal complex Genome architecture and standardized karyotype of model fish, genus Carassius. Cells 10(9):2343. https://doi.org/10.3390/cells10092343
Knytl M, Smolík O, Kubíčková S, Tlapáková T, Evans BJ, Krylov V (2017) Chromosome divergence during evolution of the tetraploid clawed frogs, Xenopus mellotropicalis and Xenopus epitropicalis as revealed by Zoo-FISH. PLoS One 12(5):e0177087. https://doi.org/10.1371/journal.pone.0177087
Knytl M, Kalous L, Rylková K, Choleva L, Merilä J, Ráb P (2018a) Morphologically indistinguishable hybrid Carassius female with 156 chromosomes: a threat for the threatened crucian carp, C. carassius, L. PLoS One 13(1):e0190924. https://doi.org/10.1371/journal.pone.0190924
Knytl M, Tlapakova T, Vankova T, Krylov V (2018b) Silurana chromosomal evolution: a new piece to the puzzle. Cytogenet Genome Res 156(4):223–228. https://doi.org/10.1159/000494708
Knytl M, Forsythe A, Kalous L (2022) A fish of multiple faces, which show us enigmatic and incredible phenomena in nature: biology and cytogenetics of the genus Carassius. Int J Mol Sci 23(15):8095. https://doi.org/10.3390/ijms23158095
Knytl M, Fornaini NR, Bergelová B, Gvoždík V, Černohorská H, Kubíčková S et al (2023) Divergent subgenome evolution in the allotetraploid frog Xenopus calcaratus. Gene 851:146974. https://doi.org/10.1016/j.gene.2022.146974
Kretschmer R, Rodrigues BS, Barcellos SA, Costa AL, de Cioffi M, B, Garnero ADV, et al (2021) Karyotype evolution and genomic organization of repetitive DNAs in the saffron finch, sicalis flaveola (Passeriformes, aves). Animals 11(5):1456. https://doi.org/10.3390/ani11051456
Krylov V, Kubickova S, Rubes J, Macha J, Tlapakova T, Seifertova E et al (2010) Preparation of Xenopus tropicalis whole chromosome painting probes using laser microdissection and reconstruction of X. laevis tetraploid karyotype by Zoo-FISH. Chromosom Res 18(4):431–439. https://doi.org/10.1007/s10577-010-9127-x
Liao D (1999) Concerted evolution: molecular mechanism and biological implications. Am J Hum Genet 64(1):24–30. https://doi.org/10.1086/302221
Liu Y, Song M, Luo W, Xia Y, Zeng X (2019) Chromosomal evolution in the Amolops mantzorum species group (Ranidae; Anura) narrated by repetitive DNAs. Cytogenet Genome Res 157(3):172–178. https://doi.org/10.1159/000499416
Lynch M, O’Hely M, Walsh B, Force A (2001) The probability of preservation of a newly arisen gene duplicate. Genetics 159(4):1789–1804. https://doi.org/10.1093/genetics/159.4.1789
Matsuda Y, Uno Y, Kondo M, Gilchrist MJ, Zorn AM, Rokhsar DS et al (2015) A new nomenclature of Xenopus laevis chromosomes based on the phylogenetic relationship to Silurana/Xenopus tropicalis. Cytogenet Genome Res 145(3–4):187–191. https://doi.org/10.1159/000381292
Milioto V, Vlah S, Mazzoleni S, Rovatsos M, Dumas F (2019) Chromosomal localization of 18S–28S rDNA and (TTAGGG)n sequences in wo South African dormice of the genus Graphiurus (Rodentia: Gliridae). Cytogenet Genome Res 158(3):145–151. https://doi.org/10.1159/000500985
Myers PZ (2007) Tandem repeats and morphological variation. Nat Educ 1(1):1. https://www.nature.com/scitable/topicpage/tandem-repeats-and-morphological-variation-40690/
Oliveira VCS, Altmanová M, Viana PF, Ezaz T, Bertollo LAC, Ráb P et al (2021) Revisiting the karyotypes of alligators and caimans (Crocodylia, Alligatoridae) after a half-century delay: bridging the gap in the chromosomal evolution of reptiles. Cells 10(6):1397. https://doi.org/10.1186/1755-8166-4-24
Oliveira NL, Cabral-de-Mello DC, Rocha MF, Loreto V, Martins C, Moura RC (2011) Chromosomal mapping of rDNAs and H3 histone sequences in the grasshopper rhammatocerus brasiliensis (acrididae, gomphocerinae): extensive chromosomal dispersion and co-localization of 5S rDNA/H3 histone clusters in the A complement and B chromosome. Mol Cytogenet 4:24. https://doi.org/10.1186/1755-8166-4-24
Phimphan S, Aiumsumang S, Tanomtong A (2021) Characterization of chromosomal and repetitive elements in the genome of “Rana nigrovittata” (Anura, Ranidae): revealed by classical and molecular techniques. Cytol Genet 55(6):583–589. https://doi.org/10.3103/S0095452721060104
Roco ÁS, Liehr T, Ruiz-García A, Guzmán K, Bullejos M (2021) Comparative distribution of repetitive sequences in the karyotypes of Xenopus tropicalis and Xenopus laevis (Anura, Pipidae). Genes (basel) 12(5):617. https://doi.org/10.3390/genes12050617
Schmid M, Steinlein C (2015) Chromosome banding in Amphibia. XXXII. The genus Xenopus (Anura, Pipidae). Cytogenet Genome Res 145:201–217. https://doi.org/10.1159/000433481
Schott SCQ, Glugoski L, Azambuja M, Moreira-Filho O, Vicari MR, Nogaroto V (2022) Comparative cytogenetic and sequence analysis of U small nuclear RNA genes in three Ancistrus species (Siluriformes: Loricariidae). Zebrafish 19(5):200–209. https://doi.org/10.1089/zeb.2022.0040
Sember A, Pelikánová Š, de Bello CM, Šlechtová V, Hatanaka T, Do Doan H et al (2020) Taxonomic diversity not associated with gross karyotype differentiation: the case of bighead carps, genus Hypophthalmichthys (Teleostei, Cypriniformes, Xenocyprididae). Genes (basel) 11(5):479. https://doi.org/10.3390/genes11050479
Sémon M, Wolfe KH (2007) Consequences of genome duplication. Curr Opin Genet Dev 17(6):505–512. https://doi.org/10.1016/j.gde.2007.09.007
Seroussi E, Knytl M, Pitel F, Elleder D, Krylov V, Leroux S et al (2019) Avian expression patterns and genomic mapping implicate leptin in digestion and TNF in immunity, suggesting that their interacting adipokine role has been acquired only in mammals. Int J Mol Sci 20(18):4489. https://doi.org/10.3390/ijms20184489
Session AM, Uno Y, Kwon T, Chapman JA, Toyoda A, Takahashi S et al (2016) Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538(7625):336–343. https://doi.org/10.1038/nature19840
Silva DMZA, Utsunomia R, Pansonato-Alves JC, Oliveira C, Foresti F (2015) Chromosomal mapping of repetitive DNA sequences in five species of astyanax (Characiformes, Characidae) reveals independent location of U1 and U2 snRNA sites and association of U1 snRNA and 5S rDNA. Cytogenet Genome Res 146(2):144–152. https://doi.org/10.1159/000438813
Sinzelle L, Thuret R, Hwang H-Y, Herszberg B, Paillard E, Bronchain OJ et al (2012) Characterization of a novel Xenopus tropicalis cell line as a model for in vitro studies. Genesis 50(3):316–324. https://doi.org/10.1002/dvg.20822
Song XY, Furman BLS, Premachandra T, Knytl M, Cauret CMS, Wasonga DV et al (2021) Sex chromosome degeneration, turnover, and sex-biased expression of sex-linked transcripts in African clawed frogs (Xenopus). Philos Trans R Soc Lond B Biol Sci 376(1832):20200095. https://doi.org/10.1098/rstb.2020.0095
Symonová R, Havelka M, Amemiya CT, Howell WM, Kořínková T, Flajšhans M et al (2017a) Molecular cytogenetic differentiation of paralogs of Hox paralogs in duplicated and re-diploidized genome of the North American paddlefish (Polyodon spathula). BMC Genet 18(1):19. https://doi.org/10.1186/s12863-017-0484-8
Symonová R, Ocalewicz K, Kirtiklis L, Delmastro GB, Pelikánová Š, Garcia S et al (2017b) Higher-order organisation of extremely amplified, potentially functional and massively methylated 5S rDNA in European pikes (Esox sp.). BMC Genomics 18(1):391. https://doi.org/10.1186/s12864-017-3774-7
Tymowska J (1991) Polyploidy and cytogenetic variation in frogs of the genus Xenopus. In: Green DM, Sessions SK (eds) Amphibian cytogenetics and evolution. Academic Press, San Diego, pp 259–297
Valadkhan S (2005) snRNAs as the catalysts of pre-mRNA splicing. Curr Opin Chem Biol 9(6):603–608. https://doi.org/10.1016/j.cbpa.2005.10.008
Valente GT, Mazzuchelli J, Ferreira IA, Poletto AB, Fantinatti BEA, Martins C (2011) Cytogenetic mapping of the retroelements Rex1, Rex3 and Rex6 among cichlid Fish: new insights on the chromosomal distribution of transposable elements. Cytogenet Genome Res 133(1):34–42. https://doi.org/10.1159/000322888
Vicari MR, Artoni RF, Moreira-Filho O, Bertollo LAC (2008) Colocalization of repetitive DNAs and silencing of major rRNA genes. A case report of the fish Astyanax janeiroensis. Cytogenet Genome Res 122(1):67–72. https://doi.org/10.1159/000151318
Yampolsky LY (2016) Mutation and genome evolution. Encyclop Evol Biol 77–83. Elsevier. https://doi.org/10.1016/B978-0-12-800049-6.00170-0
Zattera ML, Gazolla CB, de Soares A, A, Gazoni T, Pollet N, Recco-Pimentel SM et al (2020) Evolutionary dynamics of the repetitive DNA in the karyotypes of Pipa carvalhoi and Xenopus tropicalis (Anura, Pipidae). Front Genet 11:1–10. https://doi.org/10.3389/fgene.2020.00637
Zhang G, Li B, Li C, Gilbert MTP, Jarvis ED, Wang J (2014) Comparative genomic data of the Avian phylogenomics project. Gigascience 3(1):1–8. https://doi.org/10.1186/2047-217X-3-26
Acknowledgements
We thank two anonymous reviewers for their constructive comments on early versions of the manuscript. Also, we thank Xenbase (Karimi and Vize 2014) for making available the high-quality genome assemblies of X. tropicalis (v10.1) and X. laevis (v9.2).
Funding
Open access publishing supported by the National Technical Library in Prague. The research was supported by the institutional support from the IVB CAS (RVO: 68081766); the Ministry of Culture of the Czech Republic (DKRVO 2019–2023/6.VII.e, National Museum, 00023272) (both VG); institutional support from the Ministry of Agriculture of the Czech Republic (MZE-RO0518) (HČ, SK); the Natural Science and Engineering Research Council of Canada (RGPIN-2017–05770) (BJE); the P JAC project CZ.02.01.01/00/22_010/0002902 MSCA Fellowships CZ – UK (MK).
Author information
Authors and Affiliations
Contributions
Laboratory experiments [Nicola R. Fornaini], [Barbora Bergelová], [Václav Gvoždík] and [Halina Černohorská]; Graphic design [Nicola R. Fornaini] and [Ben J. Evans]; Fieldwork [Václav Gvoždík], [Eric B. Fokam], [Gabriel Badjedjea] and [Ben J. Evans]; Funding acquisition [Václav Gvoždík], [Halina Černohorská], [Svatava Kubíčková], [Ben J. Evans] and [Martin Knytl]; Conceptualization of the study [Václav Gvoždík], [Ben J. Evans], [Martin Knytl]; Visualization [Martin Knytl]; Data curation and validation [Martin Knytl]; Project administration [Martin Knytl]; Writing – original draft preparation [Martin Knytl]; Supervision [Martin Knytl]; All authors reviewed, edited, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Statement of ethics
Charles University has registered experimental breeding facilities for pipid frogs (16OZ12891/2018–17214, 37428/2019-MZE-18134). All experimental procedures involving frogs were approved by the Institutional Animal Care and Use Committee of Charles University, according to the directives from the State Veterinary Administration of the Czech Republic, reference number MSMT-20585/2022–4 issued by the Ministry of Education, Youth and Sport of the Czech Republic (MK is a manager of the experimental project on living Xenopus animals). MK is a holder of the Certificate of professional competence to design experiments according to §15d(3) of the Czech Republic Act No. 246/1992 coll. on the Protection of Animals against Cruelty (Registration number CZ 03973), provided by the Ministry of Agriculture of the Czech Republic.
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fornaini, N.R., Bergelová, B., Gvoždík, V. et al. Consequences of polyploidy and divergence as revealed by cytogenetic mapping of tandem repeats in African clawed frogs (Xenopus, Pipidae). Eur J Wildl Res 69, 81 (2023). https://doi.org/10.1007/s10344-023-01709-8
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10344-023-01709-8