Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes

 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. CZMELAC Sequence Capture Panel
2.3. Targeted NGS Analysis
2.4. Bioinformatics
2.5. Variant Filtration and Prioritization
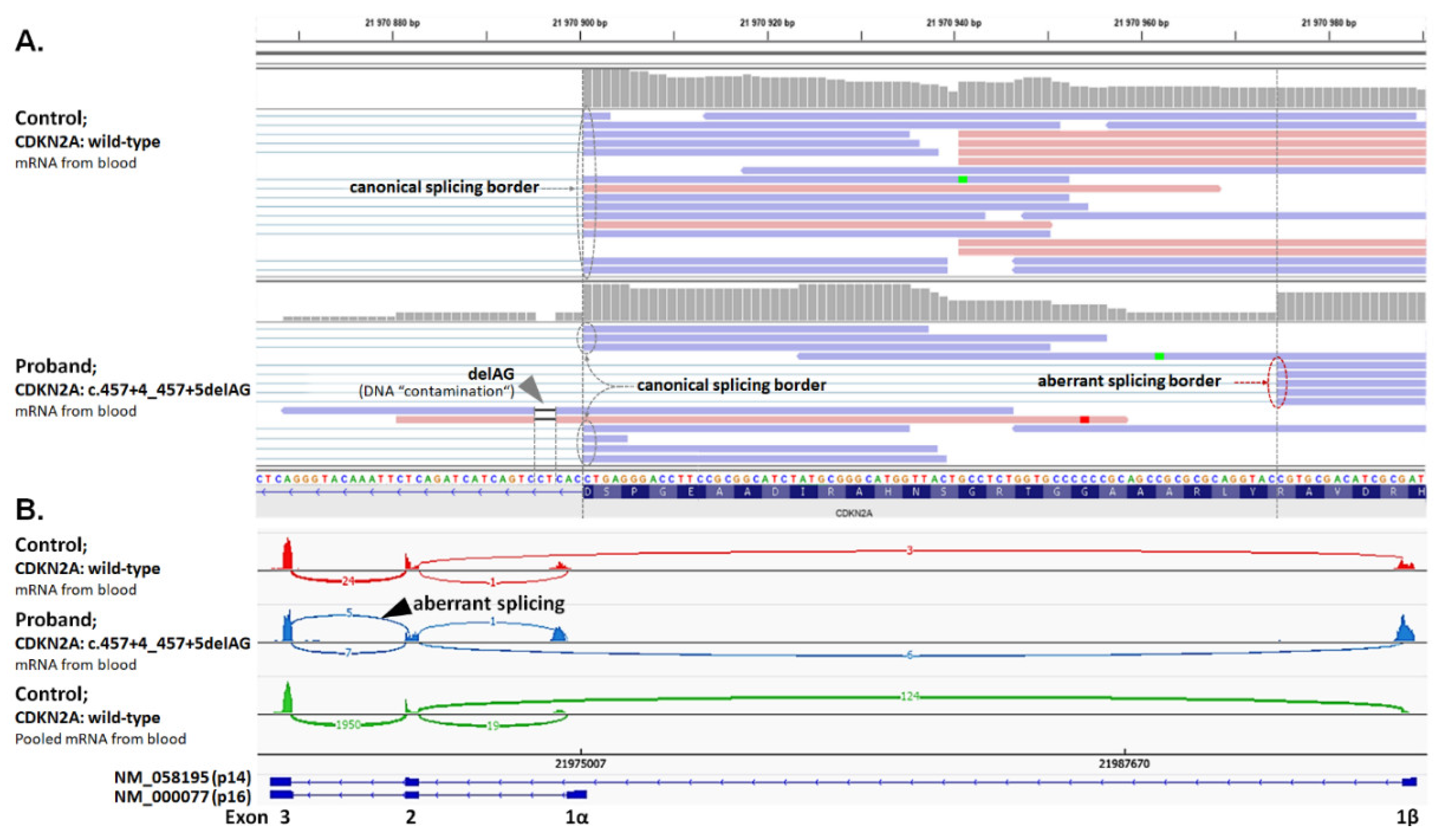
2.6. Analysis of Splicing Alterations
2.7. Statistical Analysis
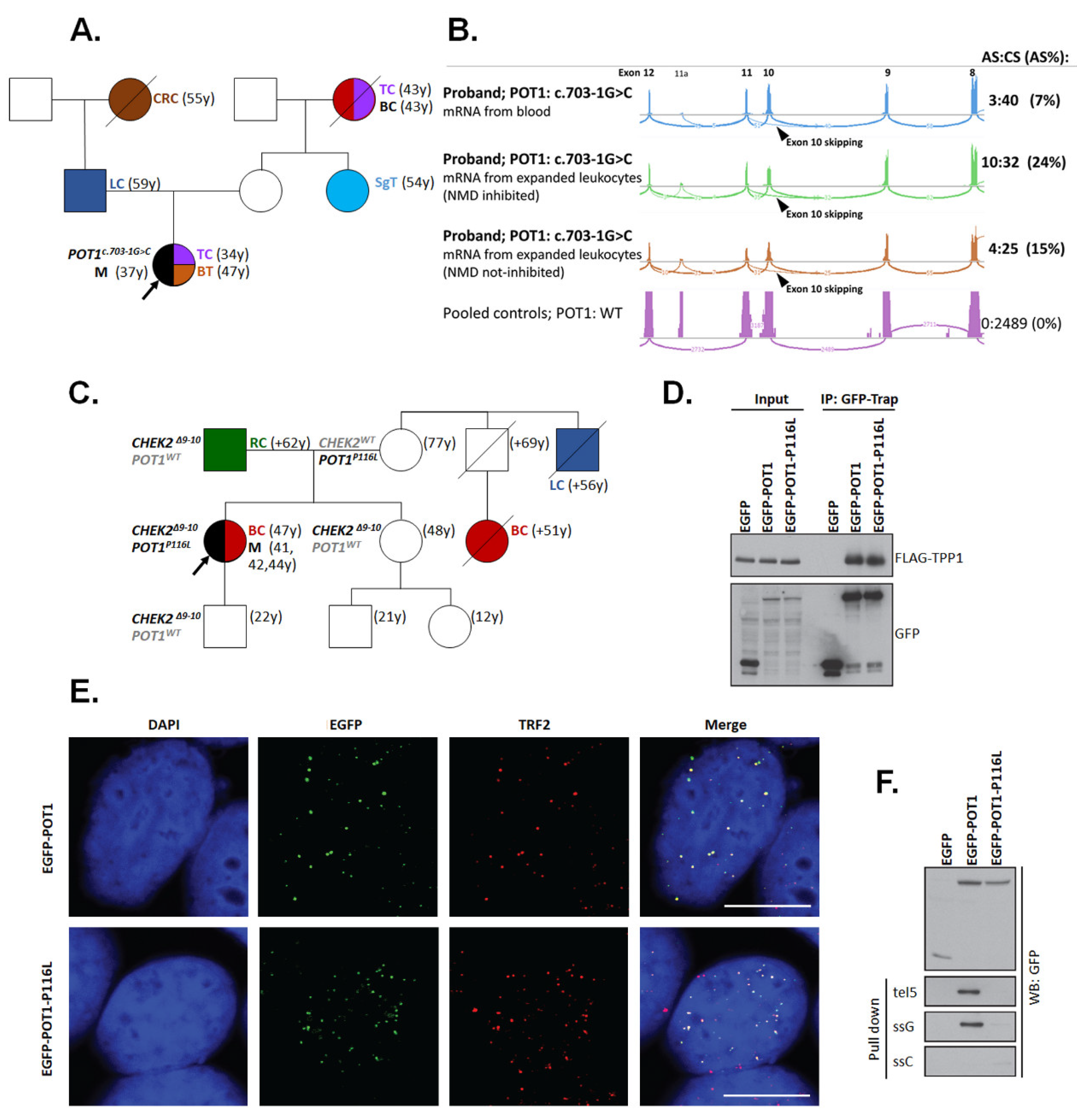
2.8. Functional Assays for Selected Germline Variants
2.8.1. CHEK2 Functional Analysis
2.8.2. POT1 Functional Analysis
3. Results
3.1. Germline Variants in Analyzed Genes
3.1.1. Mutations in High-to-Moderate Melanoma Risk Genes
3.1.2. Mutations in Low-Risk Melanoma Genes
3.1.3. Mutations in Genes Associated with Hereditary Cancer Syndromes
3.1.4. Mutations in Other Genes with Unknown Familial Melanoma Risk
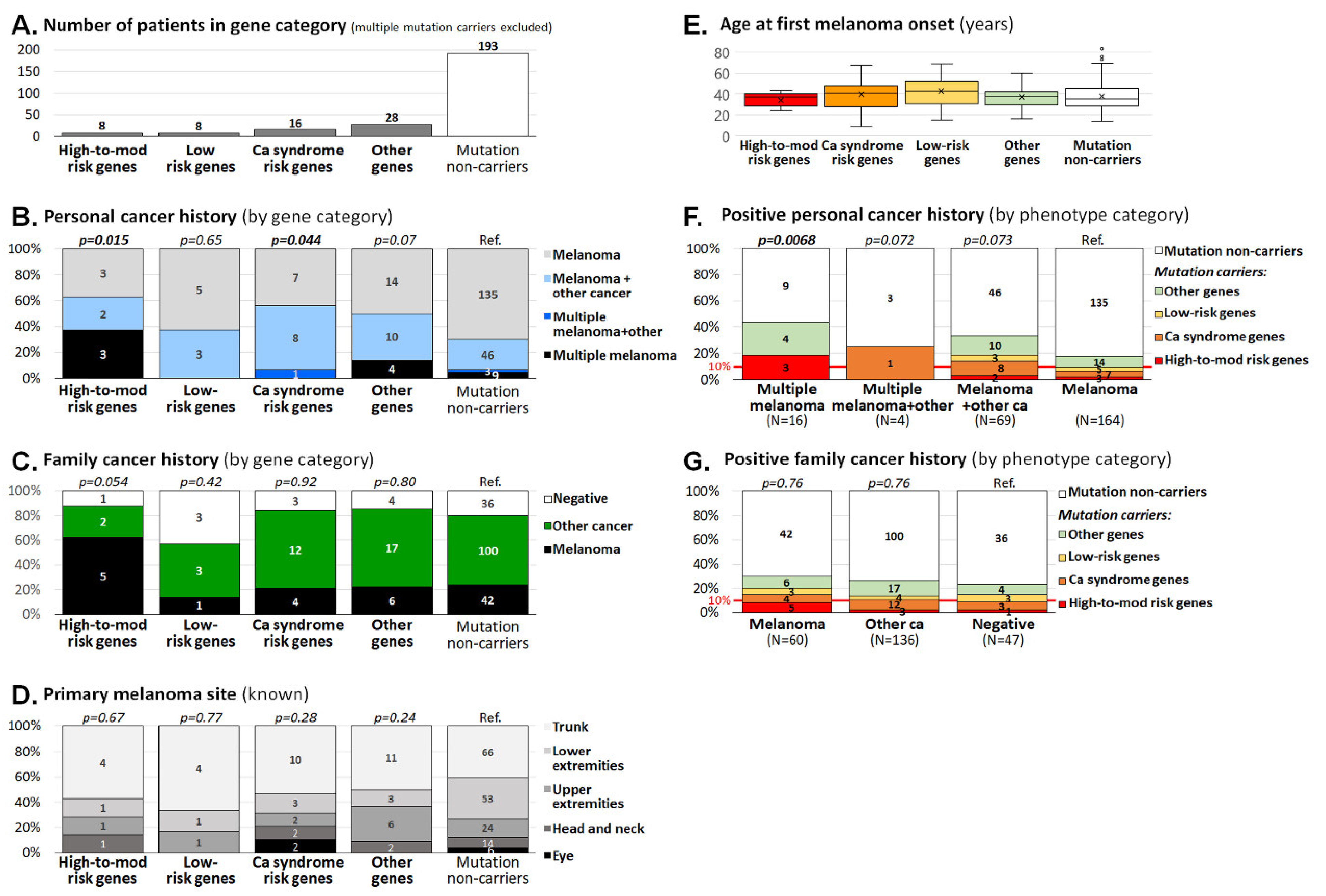
3.2. Clinicopathological Characteristics of Melanoma Patients Carrying Germline Mutations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2018, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narayanan, D.L.; Saladi, R.N.; Fox, J.L. Review: Ultraviolet radiation and skin cancer. Int. J. Dermatol. 2010, 49, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Shain, A.H.; Bastian, B.C. From melanocytes to melanomas. Nat. Rev. Cancer 2016, 16, 345–358. [Google Scholar] [CrossRef]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Möller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer Among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hawkes, J.E.; Truong, A.; Meyer, L.J. Genetic predisposition to melanoma. Semin. Oncol. 2016, 43, 591–597. [Google Scholar] [CrossRef]
- Maggi, L.B.; Winkeler, C.L.; Miceli, A.P.; Apicelli, A.J.; Brady, S.N.; Kuchenreuther, M.J.; Weber, J.D. ARF tumor suppression in the nucleolus. Biochim. Biophys. Acta 2014, 1842, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Gruis, N.A.; Van Der Velden, P.A.; Sandkuijl, L.A.; Prins, D.E.; Weaver-Feldhaus, J.; Kamb, A.; Bergman, W.; Frants, R.R. Homozygotes for CDKN2 (p16) germline mutation in Dutch familial melanoma kindreds. Nat. Genet. 1995, 10, 351–353. [Google Scholar] [CrossRef]
- Hill, V.K.; Gartner, J.J.; Samuels, Y.; Goldstein, A.M. The Genetics of Melanoma: Recent Advances. Annu. Rev. Genom. Hum. Genet. 2013, 14, 257–279. [Google Scholar] [CrossRef] [Green Version]
- Sargen, M.R.; Pfeiffer, R.; Yang, X.R.; Tucker, M.A.; Goldstein, A.M. Variation in Cutaneous Patterns of Melanomagenesis According to Germline CDKN2A/CDK4 Status in Melanoma-Prone Families. J. Investig. Dermatol. 2020, 140, 174–181.E3. [Google Scholar] [CrossRef] [Green Version]
- Betti, M.; Aspesi, A.; Biasi, A.; Casalone, E.; Ferrante, D.; Ogliara, P.; Gironi, L.C.; Giorgione, R.; Farinelli, P.; Grosso, F.; et al. CDKN2A and BAP1 germline mutations predispose to melanoma and mesothelioma. Cancer Lett. 2016, 378, 120–130. [Google Scholar] [CrossRef]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and Cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Xiao, Y.; Sampson, J.; Rotunno, M.; Bennett, H.; Wen, Y.; Jones, K.; Vogt, A.; Burdette, L.; Luo, W.; et al. Rare germline variants in known melanoma susceptibility genes in familial melanoma. Hum. Mol. Genet. 2017, 26, 4886–4895. [Google Scholar] [CrossRef] [PubMed]
- Harland, M.; Petljak, M.; Robles-Espinoza, C.D.; Ding, Z.; Gruis, N.A.; Van Doorn, R.; Pooley, K.A.; Dunning, A.M.; Aoude, L.G.; Wadt, K.A.W.; et al. Germline TERT promoter mutations are rare in familial melanoma. Fam. Cancer 2015, 15, 139–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leachman, S.A.; Lucero, O.M.; Sampson, J.E.; Cassidy, P.; Bruno, W.; Queirolo, P.; Ghiorzo, P. Identification, genetic testing, and management of hereditary melanoma. Cancer Metastasis Rev. 2017, 36, 77–90. [Google Scholar] [CrossRef] [Green Version]
- Chatzinasiou, F.; Lill, C.M.; Kypreou, K.; Stefanaki, I.; Nicolaou, V.; Spyrou, G.; Evangelou, E.; Roehr, J.T.; Kodela, E.; Katsambas, A.; et al. Comprehensive Field Synopsis and Systematic Meta-analyses of Genetic Association Studies in Cutaneous Melanoma. J. Natl. Cancer Inst. 2011, 103, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; GenoMEL, consortium; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.K.; Song, F.; Barrett, J.H.; Kumar, R.; et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 2015, 47, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Roberts, M.; Asgari, M.; Toland, A. Genome-wide association studies and polygenic risk scores for skin cancer: Clinically useful yet? Br. J. Dermatol. 2019, 181, 1146–1155. [Google Scholar] [CrossRef]
- Dušek, L.; Mužík, J.; Malúšková, D.; Májek, O.; Pavlík, T.; Koptíková, J.; Melichar, B.; Buchler, T.; Fínek, J.; Cibula, D.; et al. Cancer incidence and mortality in the Czech Republic. Klin. Onkol. 2014, 27, 406–423. [Google Scholar] [CrossRef]
- Krejci, D.; Zapletalova, M.; Svobodova, I.; Pehalova, L.; Muzik, J.; Klimes, D.; Snajdrova, L.; Bajciova, V.; Mudry, P.; Kodytkova, D.; et al. Epidemiological Trends for Childhood and Adolescent Cancers in the Period 1994–2016 in the Czech Republic. Klin. Onkol. 2019, 32, 10. [Google Scholar] [CrossRef]
- Soura, E.; Eliades, P.J.; Shannon, K.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management. J. Am. Acad. Dermatol. 2016, 74, 395–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soura, E.; Eliades, P.J.; Shannon, K.M.; Stratigos, A.J.; Tsao, H. Hereditary melanoma: Update on syndromes and management: Emerging melanoma cancer complexes and genetic counseling. J. Am. Acad. Dermatol. 2016, 74, 411–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Read, J.; Wadt, K.; Hayward, N.K. Melanoma genetics. J. Med Genet. 2015, 53, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, W.; Clyne, M.; Khoury, M.J.; Gwinn, M. Phenopedia and Genopedia: Disease-centered and gene-centered views of the evolving knowledge of human genetic associations. Bioinformatics 2009, 26, 145–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhota, F.; Zemankova, P.; Kleiblova, P.; Soukupova, J.; Vocka, M.; Stranecky, V.; Janatova, M.; Hartmannová, H.; Hodaňová, K.; Kmoch, S.; et al. Hereditary truncating mutations of DNA repair and other genes in BRCA1/BRCA2/PALB2 -negatively tested breast cancer patients. Clin. Genet. 2016, 90, 324–333. [Google Scholar] [CrossRef]
- Soukupova, J.; Zemankova, P.; Lhotova, K.; Janatova, M.; Borecka, M.; Stolarova, L.; Lhota, F.; Foretova, L.; Machackova, E.; Stranecky, V.; et al. Validation of CZECANCA (CZEch CAncer paNel for Clinical Application) for targeted NGS-based analysis of hereditary cancer syndromes. PLoS ONE 2018, 13, e0195761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Walker, L.C.; Lattimore, V.L.; Kvist, A.; Kleiblova, P.; Zemankova, P.; De Jong, L.; Wiggins, G.A.R.; Hakkaart, C.; Cree, S.L.; Behar, R.; et al. Comprehensive Assessment of BARD1 Messenger Ribonucleic Acid Splicing with Implications for Variant Classification. Front. Genet. 2019, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Kleiblova, P.; Stolarova, L.; Krizova, K.; Lhota, F.; Hojny, J.; Zemankova, P.; Havranek, O.; Vocka, M.; Cerna, M.; Lhotova, K.; et al. Identification of deleterious germline CHEK2 mutations and their association with breast and ovarian cancer. Int. J. Cancer 2019, 145, 1782–1797. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chan, J.; Lambelé, M.; Yusufzai, T.; Stumpff, J.; Opresko, P.L.; Thali, M.; Wallace, S.S. NEIL3 Repairs Telomere Damage during S Phase to Secure Chromosome Segregation at Mitosis. Cell Rep. 2017, 20, 2044–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loayza, D.; De Lange, T. POT1 as a terminal transducer of TRF1 telomere length control. Nat. Cell Biol. 2003, 423, 1013–1018. [Google Scholar] [CrossRef] [PubMed]
- Calvete, O.; Lázaro, C.; Domínguez, F.; Bougeard, G.; Kunze, K.; Braeuninger, A.; Teule, A.; Lasa, A.; Cajal, T.R.Y.; Llort, G.; et al. The wide spectrum of POT1 gene variants correlates with multiple cancer types. Eur. J. Hum. Genet. 2017, 25, 1278–1281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, M.; Podell, E.R.; Cech, T.R. Structure of human POT1 bound to telomeric single-stranded DNA provides a model for chromosome end-protection. Nat. Struct. Mol. Biol. 2004, 11, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.G.; Wadt, K.; Pritchard, A.L.; Hayward, N.K. Genetics of familial melanoma: 20 years after CDKN2A. Pigment. Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Bianchi-Scarrà, G.; Bruno, W.; et al. Features associated with germline CDKN2A mutations: A GenoMEL study of melanoma-prone families from three continents. J. Med Genet. 2006, 44, 99–106. [Google Scholar] [CrossRef]
- Kibe, T.; Zimmermann, M.; De Lange, T. TPP1 Blocks an ATR-Mediated Resection Mechanism at Telomeres. Mol. Cell 2016, 61, 236–246. [Google Scholar] [CrossRef] [Green Version]
- Kendellen, M.F.; Barrientos, K.S.; Counter, C.M. POT1 Association with TRF2 Regulates Telomere Length. Mol. Cell. Biol. 2009, 29, 5611–5619. [Google Scholar] [CrossRef] [Green Version]
- Calvete, O.; Martínez, P.; García-Pavía, P.; Benitez-Buelga, C.; Paumard, B.; Fernandez, V.; Domínguez, F.; Salas, C.; Romero-Laorden, N.; García-Donás, J.; et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li–Fraumeni-like families. Nat. Commun. 2015, 6, 8383. [Google Scholar] [CrossRef] [Green Version]
- Richard, M.A.; Lupo, P.J.; Morton, L.M.; Yasui, Y.A.; Sapkota, Y.A.; Arnold, M.A.; Aubert, G.; Neglia, J.P.; Turcotte, L.M.; Leisenring, W.M.; et al. Genetic variation in POT1 and risk of thyroid subsequent malignant neoplasm: A report from the Childhood Cancer Survivor Study. PLoS ONE 2020, 15, e0228887. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, A.; Miao, B.; Skopelitou, D.; Kumar, V.; Kumar, A.; Paramasivam, N.; Bonora, E.; Hemminki, K.; Försti, A.; Bandapalli, O.R. A Germline Mutation in the POT1 Gene Is a Candidate for Familial Non-Medullary Thyroid Cancer. Cancers 2020, 12, 1441. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.L.-S.; Hattangady, N.; Lerario, A.M.; Williams, C.; Koeppe, E.; Quinonez, S.; Osborne, J.; Else, T.; Cha, K.B. A new POT1 germline mutation—Expanding the spectrum of POT1-associated cancers. Fam. Cancer 2017, 53, 1–566. [Google Scholar] [CrossRef] [PubMed]
- He, H.; Li, W.; Comiskey, J.D.F.; Liyanarachchi, S.; Nieminen, T.T.; Wang, Y.; DeLap, M.K.E.; Brock, P.; De La Chapelle, A. A Truncating Germline Mutation of TINF2 in Individuals with Thyroid Cancer or Melanoma Results in Longer Telomeres. Thyroid 2020, 30, 204–213. [Google Scholar] [CrossRef]
- Aoude, L.G.; Pritchard, A.L.; Robles-Espinoza, C.D.; Wadt, K.; Harland, M.; Choi, J.; Gartside, M.; Quesada, V.; A Johansson, P.; Palmer, J.M.; et al. Nonsense Mutations in the Shelterin Complex Genes ACD and TERF2IP in Familial Melanoma. J. Natl. Cancer Inst. 2015, 107, dju408. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Andreotti, V.; Dalmasso, B.; Vanni, I.; Ciccarese, G.; Mandalà, M.; Spadola, G.; Pizzichetta, M.A.; Ponti, G.; Tibiletti, M.G.; et al. Insights into Genetic Susceptibility to Melanoma by Gene Panel Testing: Potential Pathogenic Variants in ACD, ATM, BAP1, and POT1. Cancers 2020, 12, 1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macháčková, E.; Házová, J.; Hrabincová, E.S.; Vašíčková, P.; Navrátilová, M.; Svoboda, M.; Foretová, L. Retrospective NGS Study in High-risk Hereditary Cancer Patients at Masaryk Memorial Cancer Institute. Klin. Onkol. 2016, 29 (Suppl. S1), S35–S45. [Google Scholar] [CrossRef]
- Foretová, L.; Navrátilová, M.; Svoboda, M.; Házová, J.; Vašíčková, P.; Sťahlová, E.H.; Fabian, P.; Schneiderová, M.; Macháčková, E.; Hrabincová, E.S. BAP1 Syndrome—Predisposition to Malignant Mesothelioma, Skin and Uveal Melanoma, Renal and Other Cancers. Klin. Onkol. 2019, 32, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, A.; Bellanger, D.; Zahed, L.; Burglen, L.; Derrien, A.; D’Enghien, C.D.; Lespinasse, J.; Parfait, B.; Pedespan, J.; Rieunier, G.; et al. DNA repair functional analyses of NBN hypomorphic variants associated with NBN-related infertility. Hum. Mutat. 2020, 41, 608–618. [Google Scholar] [CrossRef]
- Lhotova, K.; Stolarova, L.; Zemankova, P.; Vočka, M.; Janatova, M.; Borecka, M.; Cerna, M.; Jelinkova, S.; Kral, J.; Volkova, Z.; et al. Multigene Panel Germline Testing of 1333 Czech Patients with Ovarian Cancer. Cancers 2020, 12, 956. [Google Scholar] [CrossRef]
- Dębniak, T.; Górski, B.; Cybulski, C.; Jakubowska, A.; Kurzawski, G.; Lener, M.; Mierzejewski, M.; Masojć, B.; Mędrek, K.; Kładny, J.; et al. Germline 657del5 mutation in the NBS1 gene in patients with malignant melanoma of the skin. Melanoma Res. 2003, 13, 365–370. [Google Scholar] [CrossRef]
- Steffen, J.; Varon, R.; Mosor, M.; Maneva, G.; Maurer, M.; Stumm, M.; Nowakowska, D.; Rubach, M.; Kosakowska, E.; Ruka, W.; et al. Increased cancer risk of heterozygotes withNBS1 germline mutations in poland. Int. J. Cancer 2004, 111, 67–71. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Stapelmann, H.; Frank, B.; Varon, R.; Burwinkel, B.; Schmitt, C.; Boettger, M.B.; Klaes, R.; Sperling, K.; Hemminki, K.; et al. Molecular genetic analysis of NBS1 in German melanoma patients. Melanoma Res. 2007, 17, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Gass, J.M.; Jackson, J.; Macklin, S.; Blackburn, P.; Hines, S.; Atwal, P.S. A case of contralateral breast cancer and skin cancer associated with NBN heterozygous pathogenic variant c.698_701delAACA. Fam. Cancer 2017, 2013, 551–553. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.J.; Bishop, D.T.; Robles-Espinoza, C.D. Melanoma predisposition—A limited role for germline BRCA1 and BRCA2 variants. Pigment. Cell Melanoma Res. 2019, 33, 6–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, A.-T.N.; Leboeuf, N.R.; Nambudiri, V.E. Skin cancer risk in CHEK2 mutation carriers. J. Eur. Acad. Dermatol. Venereol. 2020. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Criteria | Posit. FCH incl. M. | Posit. FCH incl. Other Cancers | Negative FCH | Unknown FCH | Patients; N (%) | Mean Age; yrs (Range) |
|---|---|---|---|---|---|---|
| Multiple primary M. & other cancer | 0 | 4 | 0 | 2 | 6 (2.3) | 45.0 (38–58) |
| Multiple primary M. | 5 | 8 | 3 | 1 | 17 (6.4) | 37.3 (24–75) |
| M. & other cancer | 9 | 45 | 9 | 8 | 71 (26.9) | 47.3 (14–83) |
| M. only, dg at < 25 yrs | 5 | 17 | 11 | 3 | 36 (13.6) | 20.0 (9–24) |
| M. only, dg at ≥ 25 yrs | 41 | 62 | 24 | 7 | 134 (50.8) | 37.1 (25–69) |
| Patients; N (% of all) | 60 (22.7) | 136 (51.5) | 47 (17.8) | 21 (8.0) | 264 (100) | 37.7 (9–83) |
| Mean age; yrs (range) | 38.9 (9–69) | 37.8 (14–83) | 33.0 (15–66) | 44.2 (14–75) | - | - |
| High-to-moderate melanoma risk genes | ACD, BAP1, CDK4, CDKN2A, MITF, POT1, TERF2IP, TERT |
| Low melanoma risk genes | AGR3, ARNT, ASIP, CASP8, CCND1, CDKN2B, CLPTM1L, FTO, HERC2, IRF4, MC1R, MGMT, MTAP, MX2, OBFC1, OCA2, PARP1, PLA2G6, SETDB1, SLC24A4, SLC45A2, TERF1, TERF2, TINF2, TYR, TYRP1 |
| Hereditary cancer syndrome genes with uncertain melanoma risk | APC, ATM, BARD1, BMPR1A, BRCA1, BRCA2, BRIP1, CDH1, FH, CHEK2, KIT, MET, MSH2, MSH3, MSH6, NBN, NF1, NF2, PALB2, PMS2, POLD1, POLE, PTEN, RAD51C, RAD51D, RB1, RET, SDHA, SDHB, SDHC, SDHD, SMAD4, STK11, TP53, VHL, WRN, WT1 |
| Genes with unknown impact on hereditary melanoma development | ABLIM1, APEX1, ATRN, AURKA, BBC3, BLM, BRAF, BRMS1, CASP10, CBL, CCAR2, CCNH, CDK10, CDK7, CDKN1A, CDKN1B, CDKN1C, CEBPA, COX8A, CTLA4, CTNNB1, CYP11A1, CYP17A1, CYP19A1, CYP1A1, CYP1A2, CYP3A5, DAB2IP, DCAF4, DDB1, DDB2, EDNRB, EGF, EGFR, EIF1AX, EPCAM, ERBB2, ERBB4, ERCC1, ERCC2, ERCC3, ERCC4, ERCC5, ERCC6, ERCC8, EXOC2, EZH2, FANCC, FANCL, FANCM, FAS, FASLG, FGFR2, FGFR4, FLCN, FLT1, FOXP3, GATA2, GATA4, GC, GNA11, GNAQ, GPC3, GSTM1, GSTM3, GSTP1, GSTT1, H2AFY, HRAS, IDH1, IDH2, IFIH1, IFNA1, IFNG, IL10, IL2RA, IL4, IL6, IL8, ING4, KAT6A, KIAA1967, KMT2A, KRAS, LRIG1, MAP2K1, MDM2, MLH1, MLH3, MMP1, MMP3, MUTYH, MYH7B, NCOA6, NFKB1, NFKBIE, NOD2, NOTCH3, NRAS, PAX5, PDGFRA, PIGU, PIK3CA, PIK3R1, PIK3R4, PMAIP1, PMS1, POLH, POMC, PPM1D, PPP6C, PRF1, PTGS2, PTCH1, PTPN11, PTPN22, RAC1, RAD23A, RAD23B, RASEF, RECQL, RECQL4, RHOBTB2, RUNX1, SBDS, SF3B1, SH2B3, SLX4, SMARCB1, SNX31, STAG2, STK19, SUZ12, TACC1, TERC, TLR3, TRPM1, TSC1, TSC2, VDR, XAB2, XPA, XPC, XRCC1, XRCC3, ZNF365 |
| (a) | Gene: Coding Sequence (Protein) Change - Concomitant Mutation | Mel Site (Age) (b) | Other Tumors in Proband (Age) | Family Cancer History Tumor Type (N) (c) |
|---|---|---|---|---|
| High-to-moderate risk genes | ||||
| F | CDKN2A: c.16_20del5 (p.G6Qfs*7) | TR (38) | none | BC (1), Leu (1), Mel (1), other 3 relatives with unknown tumors |
| F | CDKN2A: c.71G>C (p.R24P) | TR (24) | Mel (35) | CRC (1), Mel (1), UrC(1) |
| F | CDKN2A: c.71G>C (p.R24P) | TR (28) | Mel (38) | Mel (2) |
| F | CDKN2A: c.95_112del (p.L32_L37del) | LE (28) | GC (48) | BC (2), CRC(1), GC (1), LC (1), Mel (2) |
| M | CDKN2A: c.334C>G (p.R112G) | HE (43) | none | Mel (1), PaC(1) |
| F | CDKN2A: c.457+4_457+5delAG (p.Y129Hfs*11) | TR (29) | Mel (34) | BT (1) |
| F | POT1: c.347C>T (p.P116L); - CHEK2: c.909-2028_1095+330del5395 (p.M304Lfs*15) | UE (41) | Mel (41,42,44); BC (47) | RC (1) |
| F | POT1: c.703-1G>C (p.V235Gfs*22) | n.a. (37) | TC (34); BT (47) | BC (1), CRC (1), LC(1), SgT (1), TC (1) |
| M | ACD: c.755delA (p.D255Afs*9) | UE (39) | none | negative |
| Low-risk genes | ||||
| F | OCA2: c.1211C>T (p.T404M); - KAT6A: c.1138G>T (p.E380*) | n.a. (29) | none | Mel(1) |
| M | OCA2: c.1327G>A (p.V443I) | TR (15) | none | negative |
| F | OCA2: c.1327G>A (p.V443I) | TR (43) | none | BC (3), CRC (3), PaC (1) |
| F | OCA2: c.1327G>A (p.V443I) | LE (52) | Ly (38); SkC (49) | Leu (1), Unknown (1) |
| M | OCA2: c.2037G>C (p.W679C) | n.a. (50) | none | negative |
| M | OCA2: c.2037G>C (p.W679C) | n.a. (68) | SkC (68) | n.a. |
| M | TYRP1: c.1054_1057del4 (p.N353Vfs*31); - TRPM1: Δe2-7 (p.?) | TR (36) | none | Mel (2) |
| M | SLC45A2: Δe1-2 (p.?); - GSTM3: c.393C>A (p.Y131*) | EY (25) | none | n.a. |
| F | SLC45A2: Δe1-4 (p.?) | TR (42) | BC (41) | PrC (1) |
| M | TYR: c.650G>A (p.R217Q) | TR (37) | none | negative |
| F | TYR: c.1037-7T>A (p.?); - FANCC: c.455dupA (p.N152Kfs*9) | HE (66) | BC (52); CRC (66) | BC (2), HCC (1), |
| F | TINF2: c.796C>T (p.R266*) | UE (48) | none | CRC (2), GbC (1), Mel (1), PrC (2), RC (1), Sarcoma (1) |
| Hereditary cancer syndrome genes | ||||
| F | NBN: c.657_661del5 (p.K219Nfs*16) | TR (24) | none | BC (1), BT (1), Mel (1) |
| F | NBN: c.657_661del5 (p.K219Nfs*16) | EY (25) | none | negative |
| M | NBN: c.657_661del5 (p.K219Nfs*16) | TR (37) | none | n.a. |
| F | NBN: c.657_661del5 (p.K219Nfs*16) | HE (45) | Mel (68); OC (56) | n.a. |
| F | NBN: c.657_661del5 (p.K219Nfs*16) | TR (65) | OC (67) | negative |
| M | NBN: c.1126delG (p.D376Ifs*2) | n.a. (47) | none | LC (2), Mel (1), |
| F | NBN: c.1723G>T (p.E575*); - NFKBIE: c.165_169dup5 (p.E57Afs*51) | LE (9) | none | Mel (1) |
| M | BRCA2: c.475G>A (p.V159M) | UE (45) | RC (46) | HL (1) |
| F | BRCA2: c.1389_1390delAG (p.V464Gfs*3) | LE (47) | BC (59,59) | GC (2) |
| F | BRCA2: c.5682C>G (p.Y1894*) | n.a. (67) | BT (59); BC (56) | 3 sisters with gynecological tumors, LC (1), retinoblastoma (1) |
| M | BRCA2: c.7007G>A (p.R2336H); - IFIH1: c.2464C>T (p.R822*) | HE (22) | none | BT (1), PrC (2), TC (1) |
| M | BRCA2: c.8168_8172ins4 (p.Y2726Mfs*10); - TYRP1: c.1254C>A (p.Y418*) | n.a. (40) | Mel (36); NHL (38) | LC (2) |
| F | BRCA1: c.68_69delAG (p.E23Vfs*17) | TR (47) | UrC (56); OC (57) | n.a. |
| F | BRCA1: c.1687C>T (p.Q563*) | EY (54) | BC (46) | OC (1) |
| F | BRCA1: c.4214delT (p.I1405Kfs*10); - ATM: c.7630-2A>C (p.?); - MUTYH c.1187G>A (p.G396D) | LE (46) | OC (46); BC (49) | BC (3), OC (2) |
| F | BRCA1: c.5266dupC (p.Q1756Pfs*74) | TR (53) | BC (54) | negative |
| M | CHEK2:c.909-2028_1095+330del5395 (p.M304Lfs*15) | UE (28) | none | CRC(1), Ly (1), Mel (1), MMT (1) |
| M | CHEK2: c.846+4_846+7del4 (p.D265-H282del) | TR (38) | none | BC (1), CRC (2) |
| F | ATM: c.381delA (p.V128*) - WRN: c.1105C>T (p.R369*) | TR (41) | Mel (50) | BC (2), PaC (1) |
| F | ATM: c.5932G>T (p.E1978*) | TR (35) | none | LC (1), UrC (1) |
| F | RAD51D: c.405+2T>C (p.?); - CHEK2: c.917G>C (p.G306A) | TR (26) | none | CC (1) |
| F | RB1: c.608-1G>T (p.?) | TR (32) | BC (45) | GbC (1), LC (1) |
| Melanoma Susceptibility Class | P/LP Variants; N (%) | OR (95%CI); p | |
|---|---|---|---|
| 264 Patients | 1479 Controls | ||
| Multiple Mutation Carriers INCLUDED * | |||
| High-to-moderate risk melanoma genes | 9 (3.4) | 1 (0.1) | 52.2 (6.6–413.1); 3.2 × 10-7 |
| Low-risk melanoma genes | 12 (4.5) | 35 (2.4) | 1.9 (1.0–3.8); 0.06 |
| Hereditary cancer syndrome genes | 22 (8.3) | 57 (3.9) | 2.3 (1.4–3.8); 0.003 |
| Genes with unknown familial melanoma risk | 28 (10.6) | 132 (8.9) | 1.2 (0.8–1.8); 0.4 |
| Multiple Mutation Carriers EXCLUDED | |||
| High-to-moderate risk melanoma genes | 8 (3.2) | 1 (0.1) | 48.1 (6.4–2116.9); 1.5 × 10-6 |
| Low-risk melanoma genes | 8 (3.2) | 35 (2.4) | 1.3 (0.5–3.0); 0.51 |
| Hereditary cancer syndrome genes | 16 (6.3) | 57 (3.9) | 1.7 (0.9–3.0); 0.09 |
| Genes with unknown familial melanoma risk | 28 (10.6) | 132 (8.9) | 1.2 (0.8–1.8); 0.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stolarova, L.; Jelinkova, S.; Storchova, R.; Machackova, E.; Zemankova, P.; Vocka, M.; Kodet, O.; Kral, J.; Cerna, M.; Volkova, Z.; et al. Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes. Biomedicines 2020, 8, 404. https://doi.org/10.3390/biomedicines8100404
Stolarova L, Jelinkova S, Storchova R, Machackova E, Zemankova P, Vocka M, Kodet O, Kral J, Cerna M, Volkova Z, et al. Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes. Biomedicines. 2020; 8(10):404. https://doi.org/10.3390/biomedicines8100404
Chicago/Turabian StyleStolarova, Lenka, Sandra Jelinkova, Radka Storchova, Eva Machackova, Petra Zemankova, Michal Vocka, Ondrej Kodet, Jan Kral, Marta Cerna, Zuzana Volkova, and et al. 2020. "Identification of Germline Mutations in Melanoma Patients with Early Onset, Double Primary Tumors, or Family Cancer History by NGS Analysis of 217 Genes" Biomedicines 8, no. 10: 404. https://doi.org/10.3390/biomedicines8100404