
Transcriptome Profile Analyses of Head Kidney in Roach (Rutilus rutilus), Common Bream (Abramis brama) and Their Hybrids: Does Infection by Monogenean Parasites in Freshwater Fish Reveal Differences in Fish Vigour among Parental Species and Their Hybrids?
 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Material and Methods
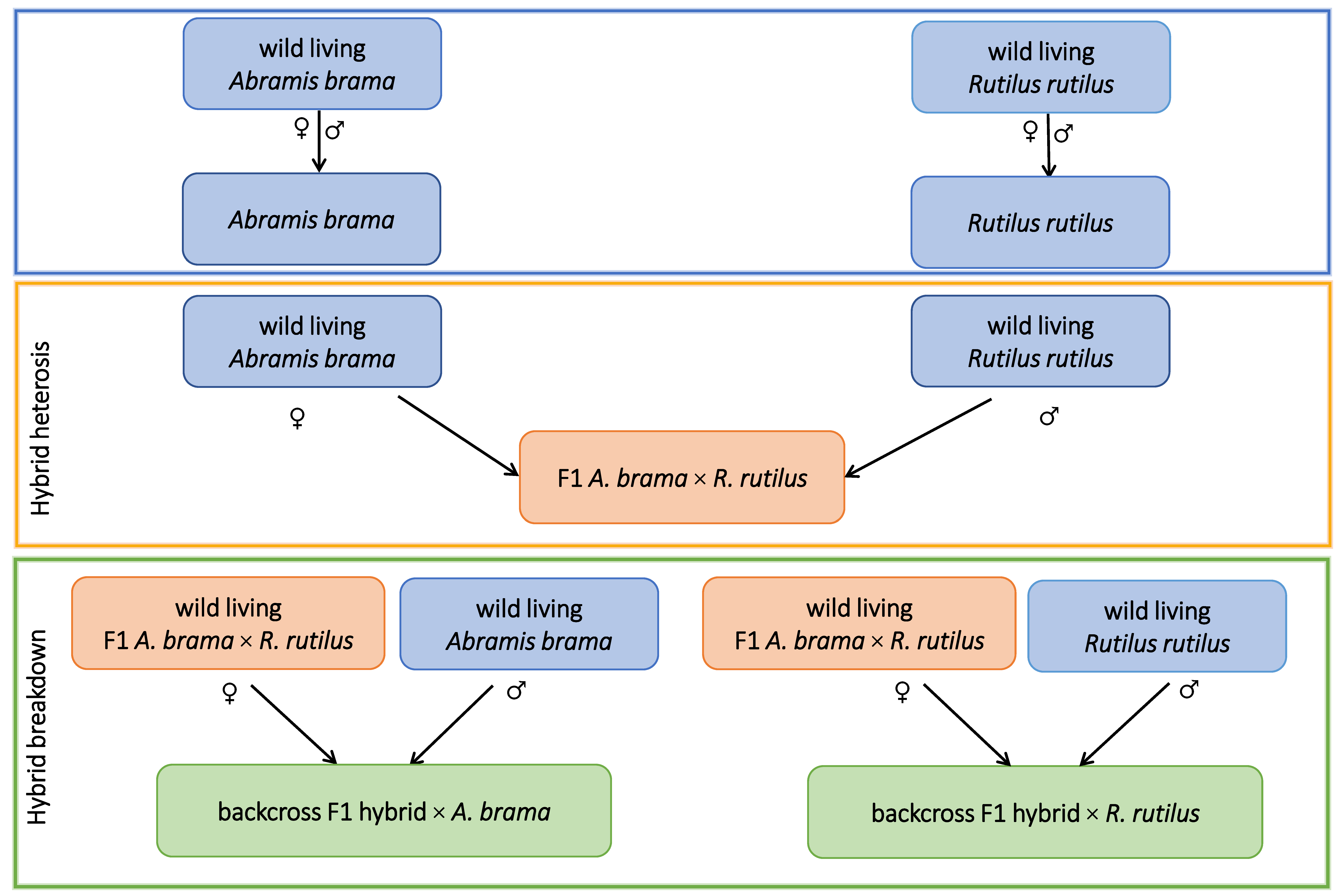
2.1. Experimental Fish Lines
2.2. Monogenean Infection
2.3. RNA Extraction and Library Preparation
2.4. NGS Data Analyses
2.5. Gene Selection and Real-Time Quantitative PCR

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gen Shortcut | Gene Name | Gene Role (References) | Amplicon (bp) | Forward Primer’s Name | Forward Primer (5′-3′) | Reverse Primer’s Name | Reverse Primer (5′-3′) | Primers’ References |
|---|---|---|---|---|---|---|---|---|
| 18S | 18S ribosomal RNA | reference gene (Muduli et al. [88]) | 140 | 18S_F | AACGGCTACCACATCCAAGG | 18S_R | TCGCCCATGGGTTTAGGATAC | Potrok [89] |
| A-tub | tubulin alpha chain-like | reference gene (Zheng & Sun [90]) | 157 | atub_F | TGCCAACAACTACGCCCG | atub_R | AGAGGTGAAACCAGAGCC | Mo et al. [91] |
| RPL8 | ribosomal protein L8 | reference gene (Mo et al. [91]) | 123 | RPL8_F | GCAGCAGAAGGCATCCACAC | RPL8_R | CTCCTCCAGACAGCAGACAAT | Mo et al. [91] |
| CCND1 | Cyclin D1 | target gene (Jirawatnotai et al. [92]; Jiang et al. [93]) | 208 | CCND1_F | AGGAAAATCGTCGCCACATG | CCND1_R | TGCACAACTTCTCTGCTGTT | this study |
| ENTH | ENTH Domain-Containing Protein 1 | target gene (Dodd et al. [94]) | 165 | ENTH_F | CAGACCAGCAGTGCATCTTC | ENTH_R | TTTGCTAGTCTGCTCTGCCT | this study |
| ING | Inhibitor of Growth Family | target gene (He et al. [95]); Dantas et al. [96]) | 182 | ING_F | TCGCAGGAAGAAGCACATTG | ING_R | CCATCTCCCGCTCATGTTTC | this study |
| HBB | Haemoglobin Subunit Beta | target gene (Roesner et al. [97]) | 163 | Hemoglobin_F | CGGAAACATGGTTGAGTGGA | Hemoglobin_R | GGCGTTGTAGAGGTTTCCAA | this study |
| IL34 | Interleukin 34 | target gene (Peixoto et al. [98]) | 161 | IL34_F | GCTTGTGTGCCTCTTGTCC | IL34_R | GGGTGGACGGAGGTATGAAT | this study |
| SCAMP5a | Secretory Carrier-Associated Membrane Protein 5 | target gene (Heo et al. [99]) | 136 | Scamp5a_F2 | AGGCTCAGGAAGAATGGACC | Scamp5a_R2 | TAGTTGTAGGTTGGTGCAGC | this study |
| GSN | Gelsolin | target gene (Azimzadeh & Mohammadisefat [100]) | 154 | GSN_F | GCAAACCTTCCCTCCAGAGA | GSN_R | CCAGGACGGCATAGCATAGA | this study |
| ZF-C2H2 | C2H2 zinc finger domain binding | target gene (Yin et al. [101]) | 172 | ZF-C2H2_F | GGTTCAAGCCAGCAAGAATCA | ZF-C2H2_R | GATTTTCCTCRTTCGATTCCAT | this study |
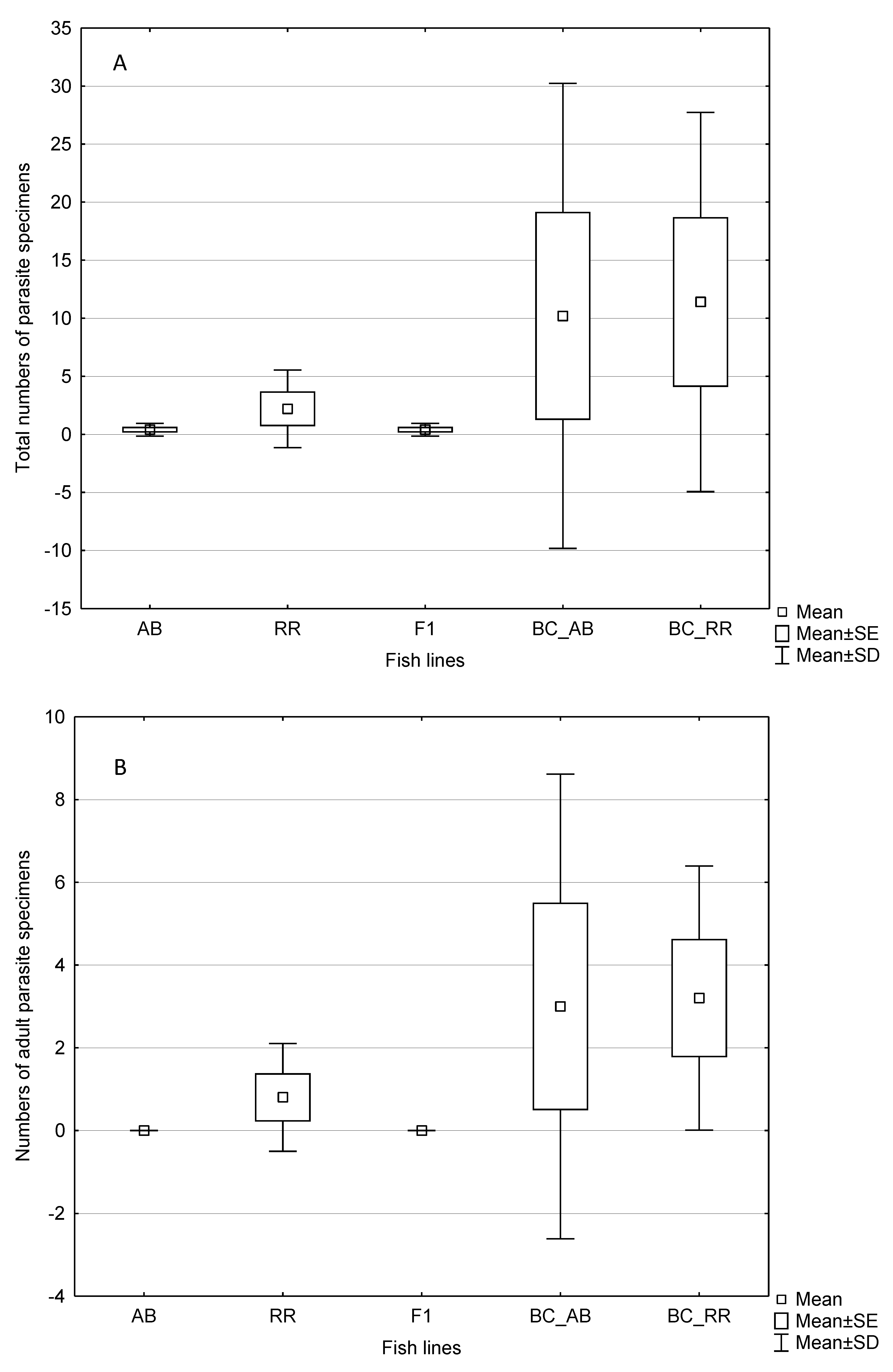
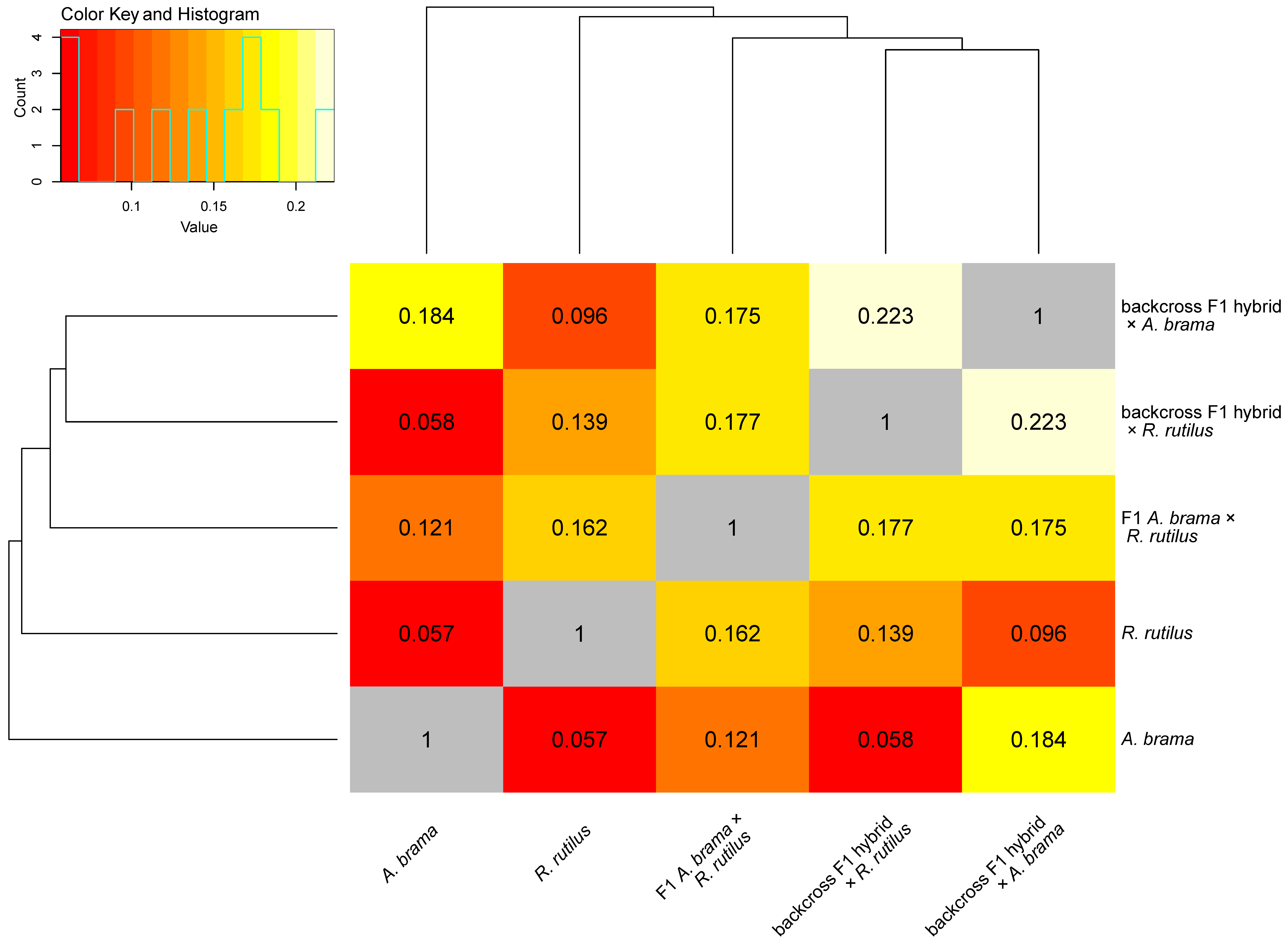
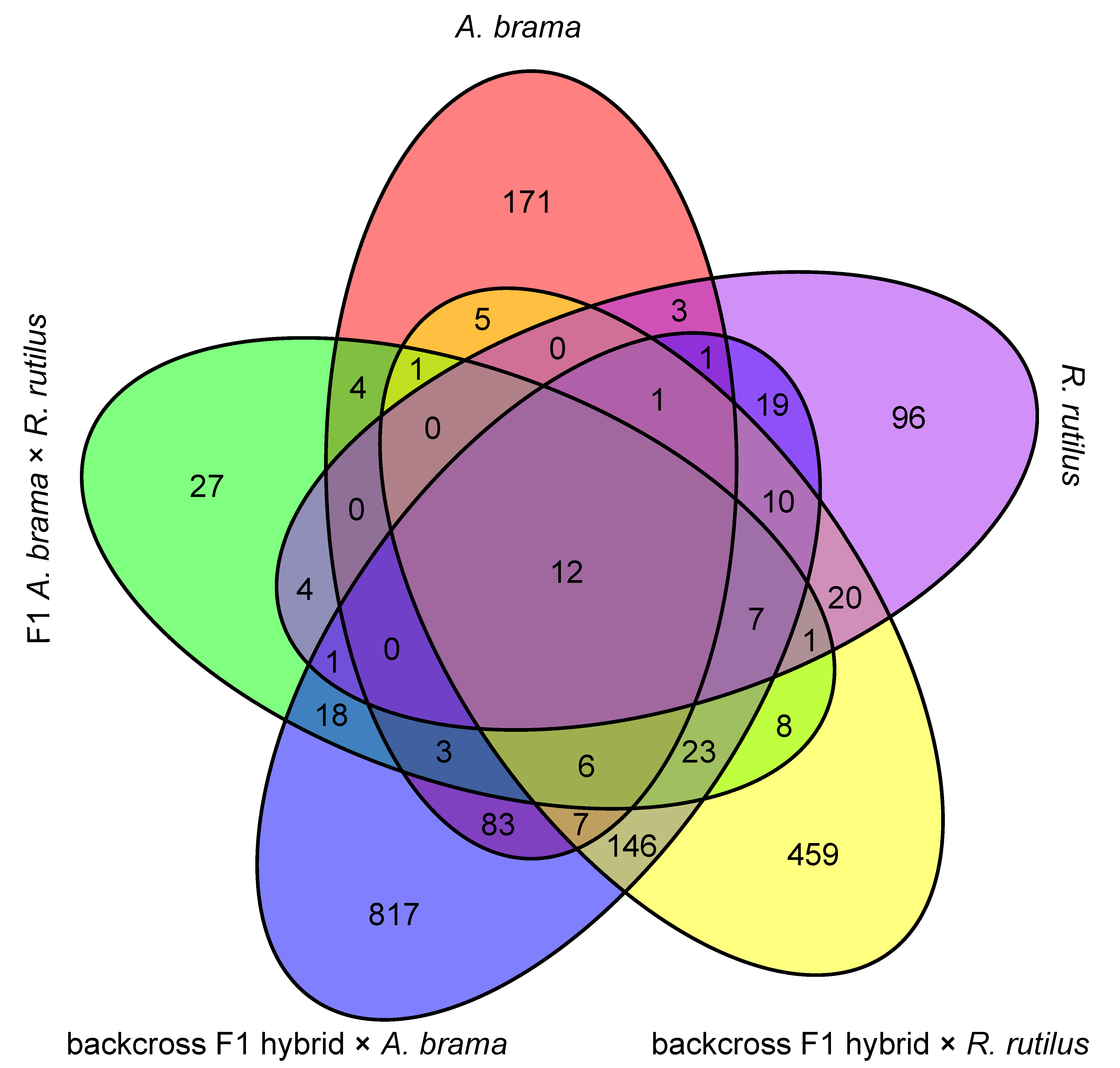
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thoney, D.A.; Hargis, W.J. Monogenea (Platyhelminthes) as hazards for fish in confinement. Annu. Rev. Fish Dis. 1991, 1, 133–153. [Google Scholar] [CrossRef]
- Bakke, T.A.; Cable, J.; Harris, P.D. The biology of gyrodactylid monogeneans: The “Russian-doll killers“. Adv. Parasitol. 2007, 64, 161–376. [Google Scholar] [PubMed]
- Tu, X.; Ling, F.; Huang, A.; Wang, G. An infection of Gyrodactylus kobayashii Hukuda, 1940 (Monogenea) associated with the mortality of goldfish (Carassius auratus) from central China. Parasitol. Res. 2015, 114, 737–745. [Google Scholar] [CrossRef]
- Deveney, M.R.; Chisholm, L.A.; Whittington, I.D. First published record of the pathogenic monogenean parasite Neobenedenia melleni (Capsalidae) from Australia. Dis. Aquat. Org. 2001, 46, 79–82. [Google Scholar] [CrossRef]
- Ogawa, K. Diseases of cultured marine fishes caused by Platyhelminthes (Monogenea, Digenea, Cestoda). Parasitology 2014, 142, 178–195. [Google Scholar] [CrossRef]
- Shinn, A.; Pratoomyot, J.; Bron, J.; Paladini, G.; Brooker, E.; Brooker, A. Economic impacts of aquatic parasites on global finfish production. Glob. Aquac. Advocate 2015, 2015, 82–84. [Google Scholar]
- Pugachev, O.N.; Gerasev, P.I.; Gussev, A.V.; Ergens, R.; Khotenowsky, I. Guide to Monogenoidea of Freshwater Fish of Palaearctic and Amur Regions; Ledizione-LediPublishing: Milan, Italy, 2010. [Google Scholar]
- Khotenovsky, I. The Subclass Octomacrinea Khotenovsky; Nauka: Leningrad, Russia, 1985. (In Russian) [Google Scholar]
- Gelnar, M.; Koubková, B.; Pláňková, H.; Jurajda, P. Report on metazoan parasites of fishes of the River Morava with remarks on the effects of water pollution. Helminthologia 1994, 31, 47–56. [Google Scholar]
- Matějusová, I.; Koubková, B.; Gelnar, M.; Cunningham, C.O. Paradiplozoon homoion Bychowsky & Nagibina, 1959 versus P. gracile Reichenbach-Klinke, 1961 (Monogenea): Two species or phenotypic plasticity? Syst. Parasitol. 2002, 53, 39–47. [Google Scholar]
- Nejat, F.; Benovics, M.; Řehulková, E.; Vukić, J.; Šanda, R.; Kaya, C.; Tarkan, A.S.; Asghar Abdoli, A.; Aksu, S.; Šimková, A. Diversity, phylogeny and intraspecific variability of Paradiplozoon species (Monogenea: Diplozoidae) parasitizing endemic cyprinoids in the Middle East. Parasitology 2023, 150, 705–722. [Google Scholar] [CrossRef]
- Konstanzová, V.; Koubková, B.; Kašný, M.; Ilgová, J.; Dzika, E.; Gelnar, M. Ultrastructure of the digestive tract of Paradiplozoon homoion (Monogenea). Parasitol. Res. 2015, 114, 1485–1494. [Google Scholar] [CrossRef]
- Krasnovyd, V.; Vetešník, L.; Gettová, L.; Civáňová, K.; Šimková, A. Patterns of parasite distribution in the hybrids of non-congeneric cyprinid fish species: Is asymmetry in parasite infection the result of limited coadaptation? Int. J. Parasitol. 2017, 47, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Kvach, Y.; Ondračková, M.; Bryjová, A.; Jurajda, P. Parasites as biological tags of divergence in Central European gudgeon populations (Actinopterygii: Cyprinidae: Gobioninae). Biologia 2017, 72, 671–679. [Google Scholar] [CrossRef]
- Aydogdu, N.; Avenant-Oldewage, A.; Dos Santos, Q.M.; Aydogdu, A. Prevalence and intensity of Paradiplozoon homoion (Monogenea: Diplozoidae) from Manyas spirlin, Alburnoides manyasensis, an endemic fish of Turkey: New host and geographical record. Iran. J. Fish. Sci. 2022, 19, 3301–3309. [Google Scholar]
- Dedic, N.; Vetešník, L.; Šimková, A. Monogeneans in intergeneric hybrids of leuciscid fish: Is parasite infection driven by hybrid heterosis, genetic incompatibilities, or host-parasite coevolutionary interactions? Front. Zool. 2023, 20, 5. [Google Scholar] [CrossRef]
- Kawatsu, H. Studies on the anemia of fish-IX. Nippon Suisan Gakkaishi 1978, 44, 1315–1319. [Google Scholar] [CrossRef]
- Rohlenová, K.; Morand, S.; Hyršl, P.; Tolarová, S.; Flajšhans, M.; Šimková, A. Are fish immune systems really affected by parasites? An immunoecological study of common carp (Cyprinus carpio). Parasites Vectors 2011, 4, 120. [Google Scholar] [CrossRef]
- Hayden, B.; Pulcini, D.; Kelly-Quinn, M.; O’Grady, M.; Caffrey, J.; McGrath, A.; Mariani, S. Hybridisation between two cyprinid fishes in a novel habitat: Genetics, morphology and life-history traits. BMC Evol. Biol. 2010, 10, 169. [Google Scholar] [CrossRef]
- Toscano, B.J.; Pulcini, D.; Hayden, B.; Russo, T.; Kelly-Quinn, M.; Mariani, S. An ecomorphological framework for the coexistence of two cyprinid fish and their hybrids in a novel environment. Biol. J. Linn. Soc. 2010, 99, 768–783. [Google Scholar] [CrossRef]
- Kuparinen, A.; Vinni, M.; Teacher, A.G.F.; Kähkönen, K.; Merilä, J. Mechanism of hybridization between bream Abramis brama and roach Rutilus rutilus in their native range. J. Fish Biol. 2014, 84, 237–242. [Google Scholar] [CrossRef]
- Konopinski, M.K.; Amirowicz, A. Genetic composition of a population of natural common bream Abramis brama x roach Rutilus rutilus hybrids and their morphological characteristics in comparison with parent species. J. Fish Biol. 2018, 92, 365–385. [Google Scholar] [CrossRef]
- Bartley, D.M.; Rana, K.; Immink, A.J. The use of inter-species hybrids in aquaculture and fisheries. Rev. Fish Biol. Fish. 2001, 10, 325–337. [Google Scholar] [CrossRef]
- Šimková, A.; Dávidová, M.; Papoušek, I.; Vetešník, L. Does interspecies hybridization affect the host specificity of parasites in cyprinid fish? Parasites Vectors 2013, 6, 95. [Google Scholar] [CrossRef] [PubMed]
- Šimková, A.; Civáňová, K.; Vetešník, L. Heterosis versus breakdown in fish hybrids revealed by one-parental species-associated viral infection. Aquaculture 2022, 546, 737406. [Google Scholar] [CrossRef]
- Krasnovyd, V.; Vetešník, L.; Šimková, A. Distribution of host-specific parasites in hybrids of phylogenetically related fish: The effects of genotype frequency and maternal ancestry? Parasites Vectors 2020, 13, 402. [Google Scholar] [CrossRef] [PubMed]
- Hayden, B.; Massa-Gallucci, A.; Caffrey, J.; Harrod, C.; Mariani, S.; O’Grady, M.; Kelly-Quinn, M. Trophic dynamics within a hybrid zone—Interactions between an abundant cyprinid hybrid and sympatric parental species. Freshw. Biol. 2011, 56, 1723–1735. [Google Scholar] [CrossRef]
- Nzau Matondo, B.; Ovidio, M.; Poncin, P.; Kakesa, T.A.; Wamuini, L.S.; Philippart, J.C. Hybridization success of three common European cyprinid species, Rutilus rutilus, Blicca bjoerkna and Abramis brama and larval resistance to stress tests. Fish. Sci. 2007, 73, 1137–1146. [Google Scholar] [CrossRef]
- Hayden, B.; McLoone, P.; Coyne, J.; Cafrey, J.M. Extensive hybridization between roach, Rutilus rutilus L., and common bream, Abramis brama L. in Irish lakes and rivers. Biol. Environ. 2014, 114, 35–39. [Google Scholar] [CrossRef]
- Šimková, A.; Gettová, L.; Civáňová, K.; Seifertová, M.; Janáč, M.; Vetešník, L. Diversity of MHC IIB genes and parasitism in hybrids of evolutionarily divergent cyprinoid species indicate heterosis advantage. Sci. Rep. 2021, 11, 16860. [Google Scholar] [CrossRef]
- Dobzhansky, T. Genetics and the Origin of Species; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Muller, H.J. Isolating mechanisms, evolution, and temperature. Biol. Symp. 1942, 6, 71–125. [Google Scholar]
- Rand, D.M.; Haney, R.A.; Fry, A.J. Cytonuclear coevolution: The genomics of cooperation. Trends Evol. Ecol. 2004, 19, 645–653. [Google Scholar] [CrossRef]
- Renaut, S.; Nolte, A.W.; Bernatchez, I. Gene expression divergence and hybrid misexpression between lake whitefish species pairs (Coregonus spp. Salmonidae). Mol. Biol. Evol. 2009, 26, 925–936. [Google Scholar] [CrossRef] [PubMed]
- Renaut, S.; Bernatchez, L. Transcriptome-wide signature of hybrid breakdown associated with intrinsic reproductive isolation in lake whitefish species pairs (Coregonus spp. Salmonidae). Heredity 2011, 106, 1003–1011. [Google Scholar] [CrossRef]
- Stelkens, R.B.; Schmid, C.; Seehausen, O. Hybrid breakdown in cichlid fish. PLoS ONE 2015, 10, e0127207. [Google Scholar] [CrossRef] [PubMed]
- Tichopád, T.; Vetešník, L.; Šimková, A.; Rodina, M.; Franěk, R.; Pšenička, M. Spermatozoa morphology and reproductive potential in F1 hybrids of common carp (Cyprinus carpio) and gibel carp (Carassius gibelio). Aquaculture 2020, 521, 735092. [Google Scholar] [CrossRef]
- Stolbunova, V.V.; Pavlova, V.V.; Kodukhova, Y.V. Asymmetric hybridization of roach Rutilus rutilus and common bream Abramis brama in controlled reverse crosses: Genetic and morphological patterns. Biosyst. Divers. 2020, 28, 376–383. [Google Scholar] [CrossRef]
- Zhi, T.; Huang, C.; Sun, R.; Zheng, Y.; Chen, J.; Xu, X.; Brown, C.L.; Yang, T. Mucosal immune response of Nile tilapia Oreochromis niloticus during Gyrodactylus cichlidarum infection. Fish Shellfish Immunol. 2020, 106, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ling, F.; Ji, J.; Kang, Y.-J.; Wang, G.-X. Expression of immune related genes in goldfish gills induced by Dactylogyrus intermedius infections. Fish Shellfish Immunol. 2013, 34, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Tu, X.; Qi, X.; Huang, A.; Ling, F.; Wang, G. Cytokine gene expression profiles in goldfish (Carassius auratus) during Gyrodactylus kobayashii infection. Fish Shellfish Immunol. 2019, 86, 116–124. [Google Scholar] [CrossRef]
- Zhou, S.; Liu, Y.T.; Dong, J.; Yang, Q.H.; Xu, N.; Yang, Y.B.; Gu, Z.M.; Ai, X.H. Transcriptome analysis of goldfish (Carassius auratus) in response to Gyrodactylus kobayashii infection. Parasitol. Res. 2021, 120, 161–171. [Google Scholar] [CrossRef]
- Zhou, S.; Li, W.X.; Zou, H.; Zhang, J.; Wu, S.G.; Li, M.; Wang, G.T. Expression analysis of immune genes in goldfish (Carassius auratus) infected with the monogenean parasite Gyrodactylus kobayashii. Fish Shellfish Immunol. 2018, 77, 40–45. [Google Scholar] [CrossRef]
- Zhi, T.; Xu, X.; Chen, J.; Zheng, Y.; Zhang, S.; Peng, J.; Brown, C.L.; Yang, T. Expression of immune related genes of Nile tilapia Oreochromis niloticus after Gyrodactylus cichlidarum and Cichlidogyrus sclerosus infections demonstrating immunesuppression in coinfection. Fish Shellfish Immunol. 2018, 80, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Gela, D.; Kocour, M.; Rodina, M.; Flajšhans, M.; Beránková, P.; Linhart, O. Controlled Reproduction Technology of Common Carp (Cyprinus carpio L.); Methodology Editon; University of South Bohemia in České Budějovice, Faculty of Fisheries and Protection of Waters: Vodňany, Czech Republic, 2009; Volume 99. [Google Scholar]
- Linhart, O.; Rodina, M.; Bastl, J.; Cosson, J. Urinary bladder, ionic composition of seminal fluid and urine with characterization of sperm motility in tench (Tinca tinca L.). J. Appl. Ichthyol. 2003, 19, 177–181. [Google Scholar] [CrossRef]
- Pečínková, M.; Matějusová, I.; Koubková, B.; Gelnar, M. Investigation of Paradiplozoon homoion (Monogenea, Diplozoidae) life cycle under experimental conditions. Parasitol. Int. 2007, 56, 179–183. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 1 May 2023).
- Chu, J.; Sadeghi, S.; Raymond, A.; Jackman, S.D.; Nip, K.M.; Mar, R.; Mohamadi, H.; Butterfield, Y.S.; Robertson, A.G.; Birol, I. BioBloom tools: Fast, accurate and memory-efficient host species sequence screening using bloom filters. Bioinformatics 2014, 30, 3402–3404. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute. Picard Toolkit. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 15 June 2023).
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. and 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Liao, Y.; Smyth, G.K.; Shi, W. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Gilbert, D. EvidentialGene: tr2aacds, mRNA Transcript Assembly Software. 2013. Available online: https://arthropods.eugenes.org/EvidentialGene (accessed on 1 May 2023).
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Suvorov, V.; Prjibelski, A.D. rnaQUAST: A quality assessment tool for de novo transcriptome assemblies. Bioinformatics 2016, 32, 2210–2212. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef]
- Haas, B.J. TransDecoder. 2015. Available online: https://github.com/TransDecoder/TransDecoder (accessed on 1 May 2023).
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.D.; Kuo, T.H.; Davis, F.G.; et al. A tissue-mapped axolotl de novo transcriptome enables identification of limb regeneration factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Nucleotide [Internet]; National Library of Medicine (US), National Center for Biotechnology Information: Bethesda, MD, USA, 1988. Available online: https://www.ncbi.nlm.nih.gov/nuccore (accessed on 15 June 2023).
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated profile HMM searches. PLoS Comp. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef]
- Nielsen, H. Predicting Secretory Proteins with SignalP. In Protein Function Prediction: Methods and Protocols; Humana: New York, NY, USA, 2017; pp. 59–73. [Google Scholar]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO Hub and Web Presence Working Group. AmiGO: Online access to ontology and annotation data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef]
- Suzuki, S.; Kakuta, M.; Ishida, T.; Akiyama, Y. Faster sequence homology searches by clustering subsequences. Bioinformatics 2015, 31, 1183–1190. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. Cluster Profiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar]
- Dowle, M.; Srinivasan, A. data.table: Extension of ‘data.frame’. 2023. Available online: https://Rdatatable.gitlab.io/data.table (accessed on 15 June 2023).
- Kolde, R.; Kolde, M.R. Package ‘pheatmap’. 2023. Available online: https://cran.r-project.org/web/packages/pheatmap/index.html (accessed on 15 June 2023).
- Wickham, H. ggplot2. Wiley Interdiscip. Rev. Comput. Stat. 2011, 3, 180–185. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, 2002–2007. [Google Scholar] [CrossRef]
- Vandesompele, J.; De Preter, K.; Pattyn, F.; Poppe, B.; Van Roy, N.; De Paepe, A.; Speleman, F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002, 3, research0034.1. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔCq) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Muduli, C.; Rathore, G.; Singh, A.; Srivastava, R. Identification of reference genes for quantitative expression analysis in Indian catfish, Clarias magur, under physiological and pathological conditions. Aquac. Res. 2022, 53, 2785–2795. [Google Scholar] [CrossRef]
- Potrok, A. The Effect of Single-Species Infection by Dactylogyrus Parasites on Expression of Selected Immune Genes in Goldfish (Carassius auratus gibelio). Master’s Thesis, Faculty of Science, Masaryk University, Brno, Czech Republic, 2020. Available online: https://is.muni.cz/th/gjmad/?studium=246504;lang=cs (accessed on 1 March 2023).
- Zheng, W.J.; Sun, L. Evaluation of housekeeping genes as references for quantitative real time RT-PCR analysis of gene expression in Japanese flounder (Paralichthys olivaceus). Fish Shellfish Imunol. 2011, 30, 638–645. [Google Scholar] [CrossRef]
- Mo, F.; Zhao, J.; Liu, N.; Cao, L.H.; Jiang, S.X. Validation of reference genes for RT-qPCR analysis of CYP4T expression in crucian carp. Genet. Mol. Biol. 2014, 37, 500–507. [Google Scholar] [CrossRef]
- Jirawatnotai, S.; Hu, Y.; Livingston, D.M.; Sicinski, P. Proteomic identification of a direct role for cyclin d1 in DNA damage repair. Cancer Res. 2012, 72, 4289–4293. [Google Scholar] [CrossRef]
- Jiang, W.D.; Hu, K.; Liu, Y.; Jiang, J.; Wu, P.; Zhao, J.; Zhang, Y.A.; Zhou, X.Q.; Feng, L. Dietary myo-inositol modulates immunity through antioxidant activity and the Nrf2 and E2F4/cyclin signalling factors in the head kidney and spleen following infection of juvenile fish with Aeromonas hydrophila. Fish Shellfish Immunol. 2016, 49, 374–386. [Google Scholar] [CrossRef]
- Dodd, M.E.; Hatzold, J.; Mathias, J.R.; Walters, K.B.; Bennin, D.A.; Rhodes, J.; Kanki, J.P.; Look, A.T.; Hammerschmidt, M.; Huttenlocher, A. The ENTH domain protein Clint1 is required for epidermal homeostasis in zebrafish. Development 2009, 136, 2591–2600. [Google Scholar] [CrossRef]
- He, G.H.; Helbing, C.C.; Wagner, M.J.; Sensen, C.W.; Riabowol, K. Phylogenetic analysis of the ING family of PHD finger proteins. Mol. Biol. Evol. 2005, 22, 104–116. [Google Scholar] [CrossRef]
- Dantas, A.; Al Shueili, B.; Yang, Y.; Nabbi, A.; Fink, D.; Riabowol, K. Biological functions of the ING proteins. Cancers 2019, 11, 1817. [Google Scholar] [CrossRef]
- Roesner, A.; Hankeln, T.; Burmester, T. Hypoxia induces a complex response of globin expression in zebrafish (Danio rerio). J. Exp. Biol. 2006, 209, 2129–2137. [Google Scholar] [CrossRef]
- Peixoto, D.; Machado, M.; Azeredo, R.; Costas, B. Chronic Inflammation Modulates Opioid Receptor Gene Expression and Triggers Respiratory Burst in a Teleost Model. Biology 2022, 11, 764. [Google Scholar] [CrossRef]
- Heo, M.J.; Kim, A.; Park, C.I. Data on molecular characterization and gene expression analysis of secretory carrier-associated membrane protein 5 (SCAMP5) from the red sea bream (Pagrus major). Data Brief 2019, 25, 103901. [Google Scholar] [CrossRef]
- Azimzadeh, K.; Mohammadisefat, P. Alterations of Cystatin c, Gelsolin, Hepcidin and Sphingosine 1 phosphate in rainbow trout (Oncorhynchus mykiss) with naturally infected Ichthyophthirius multifiliis in Urmia: Determination of Possible Potential Diagnostic Biomarker. Turk. J. Fish. Aquat. Sci. 2022, 22, TRJFAS19913. [Google Scholar] [CrossRef]
- Yin, Z.; Nie, H.; Jiang, K.; Yan, X. Molecular mechanisms underlying Vibrio tolerance in Ruditapes philippinarum revealed by comparative transcriptome profiling. Front. Immunol. 2022, 13, 879337. [Google Scholar] [CrossRef]
- Fritz, R.S.; Nichols-Orians, C.M.; Brunsfeld, S.J. Interspecific hybridization of plants and resistance to herbivores: Hypotheses, genetics, and variable responses in a diverse herbivore community. Oecologia 1994, 97, 106–117. [Google Scholar] [CrossRef]
- Artamonovaa, V.S.; Makhrova, A.A.; Shulmanb, B.S.; Khaiminac, O.V.; Yurtsevad, A.O.; Lajusd, D.L.; Shirokove, V.A.; Shurov, I.L. Response of the Atlantic Salmon (Salmo salar L.) Population of the Keret River to the Invasion of Parasite Gyrodactylus salaris Malmberg. Russ. J. Biol. Invasions 2011, 2, 73–80. [Google Scholar] [CrossRef]
- Zhigileva, O.N.; Uslamina, I.M. Helminths’ Infestation of Various Mitochondrial Lines of the Sable Martes zibellina and the Pine Marten Martes martes. Russ. J. Genet. Appl. Res. 2017, 7, 648–653. [Google Scholar] [CrossRef]
- Toh, S.Q.; Glanfield, A.; Gobert, G.N.; Jones, M.K. Heme and blood-feeding parasites. Friends or foes? Parasites Vectors 2010, 3, 108. [Google Scholar]
- Martínez-Sernández, V.; Mezo, M.; González-Warleta, M.; Perteguer, M.J.; Muino, L.; Guitián, E.; Gárate, T.; Ubeira, F.M. The MF6p/FhHDM-1 Major Antigen Secreted by the Trematode Parasite Fasciola hepatica Is a Heme-binding Protein. J. Biol. Chem. 2014, 289, 1441–1456. [Google Scholar] [CrossRef]
- Vorel, J.; Cwiklinski, K.; Roudnický, P.; Ilgová, J.; Jedličková, L.; Dalton, J.P.; Mikeš, L.; Gelnar, M.; Kašný, M. Eudiplozoon nipponicum (Monogenea, Diplozoidae) and its adaptation to haematophagy as revealed by transcriptome and secretome profiling. BMC Genom. 2021, 22, 274. [Google Scholar]
- Morgan, W.T.; Smith, A. Binding and transport of iron-porphyrins by hemopexin. Adv. Inorg. Chem. 2001, 51, 205–241. [Google Scholar]
- Vincent, S.H.; Grady, R.W.; Shaklai, N.; Snider, J.M.; Mullereberhard, U. The influence of heme-binding proteins in heme-catalyzed oxidations. Arch. Biochem. Biophys. 1988, 265, 539–550. [Google Scholar] [CrossRef]
- Pleic, I.L.; Buselic, I.; Trumbic, Z.; Bocina, I.; Sprung, M.; Mladineo, I. Expression analysis of the Atlantic bluefin tuna (Thunnus thynnus) pro-inflammatory cytokines, IL-1 beta, TNF alpha 1 and TNF alpha 2 in response to parasites Pseudocycnus appendiculatus (Copepoda) and Didymosulcus katsuwonicola (Digenea). Fish Shellfish Immunol. 2015, 45, 946–954. [Google Scholar] [CrossRef] [PubMed]
- Parida, S.; Mohapatra, A.; Mohanty, J.; Sahoo, P. Labeo rohita and Argulus siamensis infection: Host size, local inflammatory reaction and immunity modulate ectoparasite load on fish. Aquac. Res. 2018, 49, 757–766. [Google Scholar] [CrossRef]
- Mo, Z.Q.; Wu, H.K.; Hu, Y.T.; Lu, Z.J.; Lai, X.L.; Chen, H.P.; He, Z.K.; Luo, X.C.; Lee, Y.V.; Dan, X.M. Transcriptomic analysis reveals innate immune mechanisms of an underlying parasite-resistant grouper hybrid (Epinephelus fuscogutatus × Epinephelus lanceolatus). Fish Shellfish Immunol. 2021, 119, 67–75. [Google Scholar] [CrossRef]
- Niu, J.J.; Sun, M.M.; Li, Z.Y.; Wang, Z.Y.; Kong, M.; Wang, Y.F.; Song, J.Q.; Zhang, Q.Q.; He, Y.; Qi, J. Whole transcriptome analysis provides new insight on immune response mechanism of golden pompano (Trachinotus ovatus) to Amyloodinium ocellatum infestation. Aquaculture 2022, 560, 738396. [Google Scholar]
- Lindenstrøm, T.; Buchmann, K.; Secombes, C.J. Gyrodactylus derjavini infection elicits IL-1β expression in rainbow trout skin. Fish Shellfish Immunol. 2003, 15, 107–115. [Google Scholar] [PubMed]
- Tu, X.; Liu, L.; Qi, X.; Chen, W.; Wang, G.; Ling, F. Characterization of Toll-like receptor gene expression in goldfish (Carassius auratus) during Dactylogyrus intermedius infection. Dev. Comp. Immunol. 2016, 63, 78–83. [Google Scholar] [PubMed]
- Reyes-Becerril, M.; Alamillo, E.; Trasviña, A.; Hirono, I.; Kondo, H.; Jirapongpairoj, W.; Ascencio-Valle, F.; Angulo, C. In vivo and in vitro studies using larval and adult antigens from Neobenedenia melleni on immune response in yellowtail (Seriola lalandi). J. Fish Dis. 2017, 40, 1497–1509. [Google Scholar]
- Duan, C.; Ma, Z.; Wu, S.; Ding, X.J.; Tu, X.; Ye, J. Functional characterization of complement factor D on the defence against Gyrodactylus kobayashii (Monogenea) infection in goldfish (Carassius auratus). Aquaculture 2021, 545, 737214. [Google Scholar]
- Roudnický, P.; Vorel, J.; Ilgová, J.; Benovics, M.; Norek, A.; Jedličková, L.; Mikeš, L.; Potešil, D.; Zdráhal, Z.; Dvořák, J.; et al. Identification and partial characterization of a novel serpin from Eudiplozoon nipponicum (Monogenea, Polyopisthocotylea). Parasite 2018, 25, 61. [Google Scholar]
- Jedličková, L.; Dvořak, J.; Hrachovinová, I.; Ulrychová, L.; Kašný, M.; Mikeš, L. A novel Kunitz protein with proposed dual function from Eudiplozoon nipponicum (Monogenea) impairs haemostasis and action of complement in vitro. Int. J. Parasitol. 2019, 49, 337–346. [Google Scholar]
- Tan, E.; Low, K.W.; Wong, W.S.F.; Leung, K.Y. Internalization of Aeromonas hydrophila by fish epithelial cells can be inhibited with a tyrosine kinase inhibitor. Microbiology 1998, 144, 299–307. [Google Scholar]
- Li, Z.X.; Li, Y.W.; Xu, S.; Xu, Y.; Mo, Z.Q.; Dan, X.M.; Luo, X.C. Grouper (Epinephelus coioides) TCR signaling pathway was involved in response against Cryptocaryon irritans infection. Fish Shellfish Immunol. 2017, 64, 176–184. [Google Scholar]
- Howe, K.; Matthew, D.C.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Stemple, D.L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar]
- Shin, J.T.; Fishman, M.C. From zebrafish to human: Modular medical models. Annu. Rev. Genom. Hum. Genet. 2002, 3, 311–340. [Google Scholar]
- Giardoglou, P.; Beis, D. On Zebrafish Disease Models and Matters of the Heart. Biomedicines 2019, 7, 15. [Google Scholar] [PubMed]
- Das, B.K.; Chakraborty, H.; Rout, A.K.; Behera, B.K. De novo whole transcriptome profiling of Edwardsiella tarda isolated from infected fish (Labeo catla). Gene 2019, 701, 152–160. [Google Scholar] [PubMed]
- Hung, K.S.; Chen, S.Y.; Hsu, P.H.; Lin, B.A.; Hu, C.H.; Yang, C.H.; Pai, T.W.; Tzou, W.S.; Chung, H.Y. Comparative transcriptome analysis of organ-specific adaptive responses to hypoxia provides insights to human diseases. Genes 2022, 13, 1096. [Google Scholar] [PubMed]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [PubMed]











| qPCR | RNA seq | |||
|---|---|---|---|---|
| Gene Name | R | p | R | p |
| HBB | 0.951 | <0.001 | 0.651 | 0.001 |
| CCND1 | 0.906 | <0.001 | 0.851 | <0.001 |
| ING | 0.709 | 0.007 | 0.845 | <0.001 |
| IL34 | −0.342 | 0.253 | −0.645 | 0.001 |
| ZK-C2H2 | 0.172 | 0.575 | 0.787 | <0.001 |
| GSN | −0.515 | 0.072 | −0.788 | <0.001 |
| SCAMP5a | 0.747 | 0.003 | 0.792 | <0.001 |
| ENTH | 0.806 | 0.001 | 0.781 | <0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Šimková, A.; Civáňová Křížová, K.; Voříšková, K.; Vetešník, L.; Bystrý, V.; Demko, M. Transcriptome Profile Analyses of Head Kidney in Roach (Rutilus rutilus), Common Bream (Abramis brama) and Their Hybrids: Does Infection by Monogenean Parasites in Freshwater Fish Reveal Differences in Fish Vigour among Parental Species and Their Hybrids? Biology 2023, 12, 1199. https://doi.org/10.3390/biology12091199
Šimková A, Civáňová Křížová K, Voříšková K, Vetešník L, Bystrý V, Demko M. Transcriptome Profile Analyses of Head Kidney in Roach (Rutilus rutilus), Common Bream (Abramis brama) and Their Hybrids: Does Infection by Monogenean Parasites in Freshwater Fish Reveal Differences in Fish Vigour among Parental Species and Their Hybrids? Biology. 2023; 12(9):1199. https://doi.org/10.3390/biology12091199
Chicago/Turabian StyleŠimková, Andrea, Kristína Civáňová Křížová, Kristýna Voříšková, Lukáš Vetešník, Vojtěch Bystrý, and Martin Demko. 2023. "Transcriptome Profile Analyses of Head Kidney in Roach (Rutilus rutilus), Common Bream (Abramis brama) and Their Hybrids: Does Infection by Monogenean Parasites in Freshwater Fish Reveal Differences in Fish Vigour among Parental Species and Their Hybrids?" Biology 12, no. 9: 1199. https://doi.org/10.3390/biology12091199