Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction

 , , , , , , ,
, , , , , , , 
Abstract
:1. Introduction
2. Materials and Methods
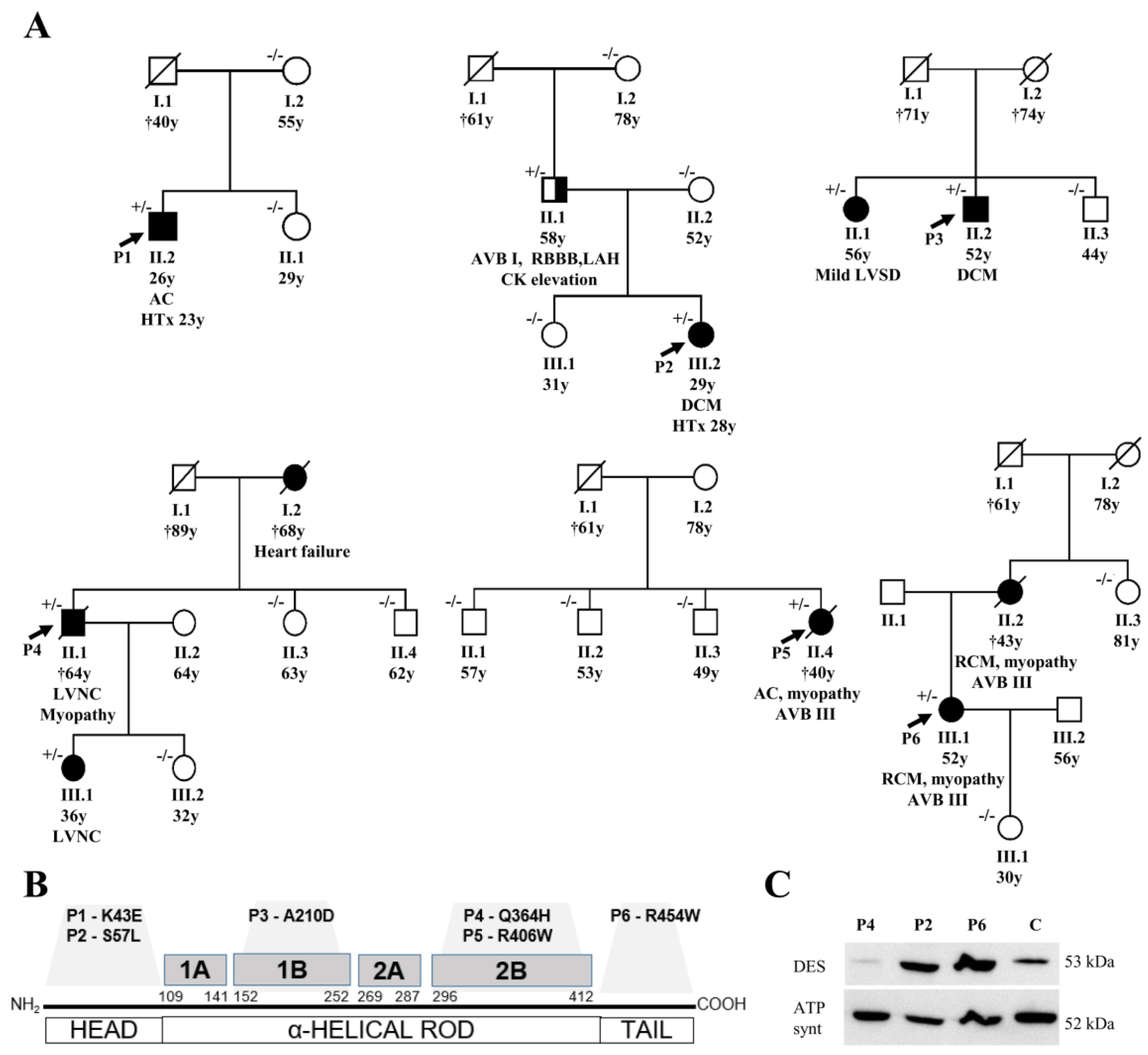
2.1. Clinical Description of Studied Patients and of Their Families
2.2. Genetic Analysis and Detection of Variants
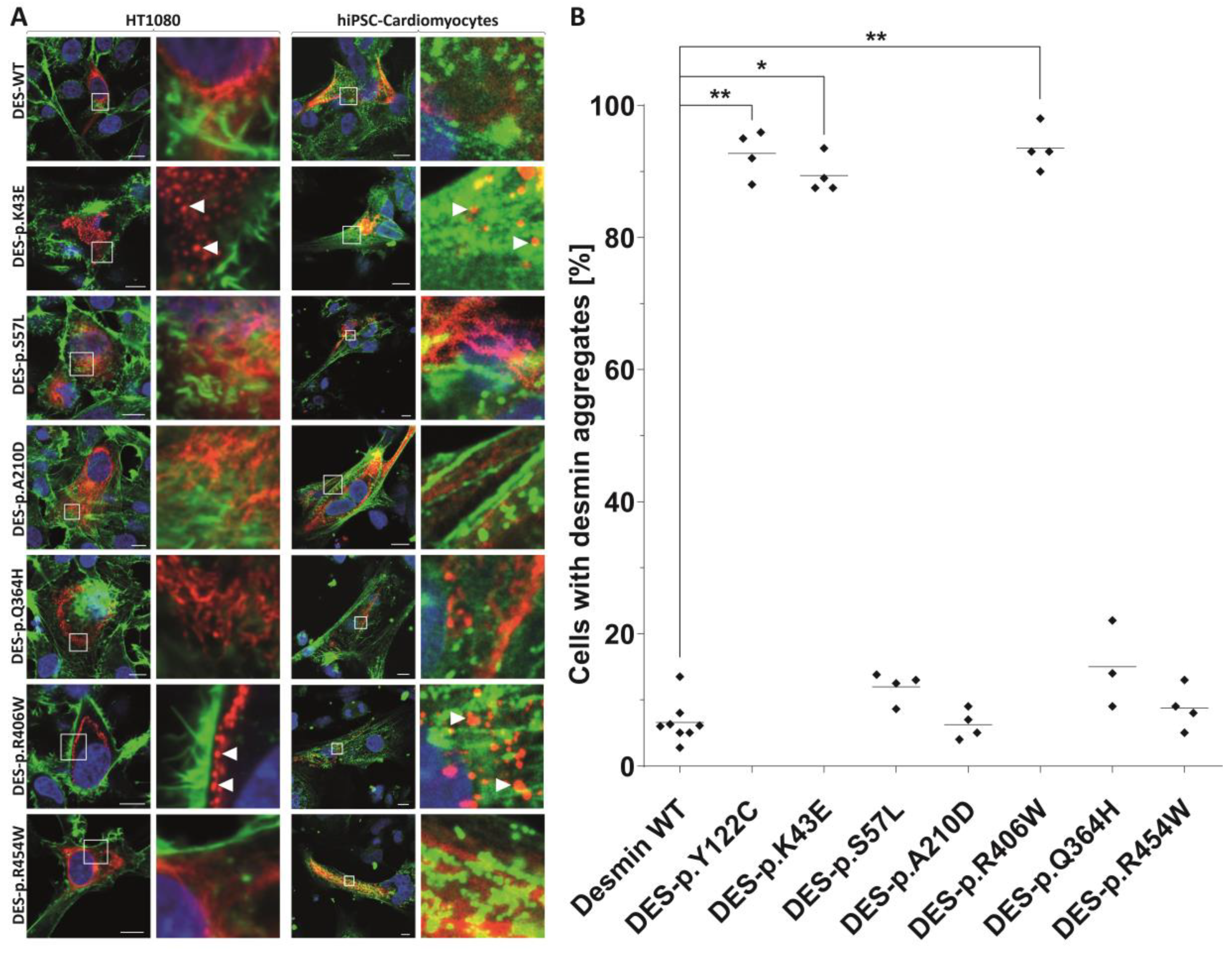
2.3. In Vitro Analysis of DES Variants
2.4. Statistical Analysis of Aggregate Formation
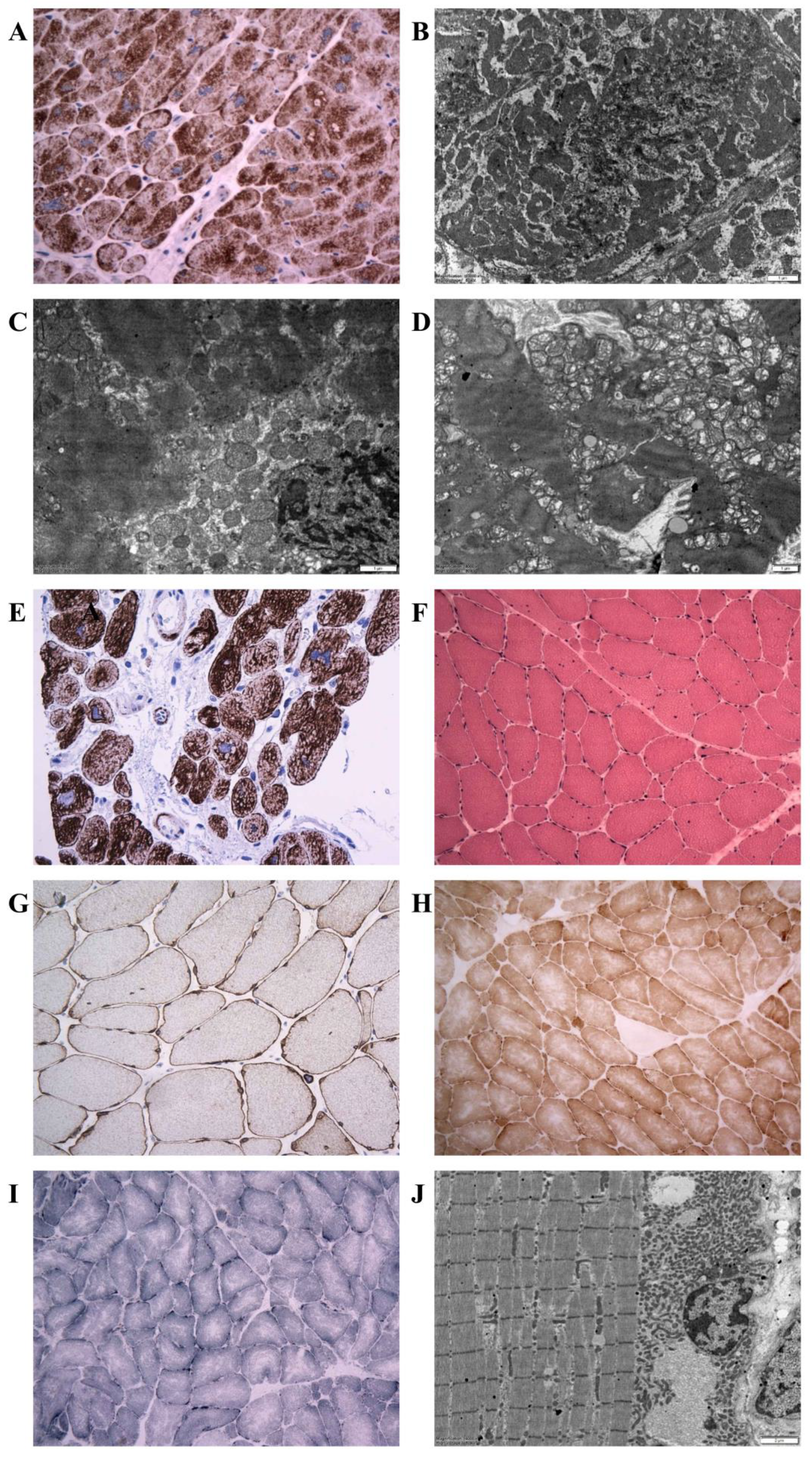
2.5. Histopathology, Immunohistochemistry, Desmin Western Blot, and Electron Microscopy
2.6. Analysis of Mitochondrial Function in Biopsies
3. Results
3.1. Description of DES Variants and Their Segregation in Families
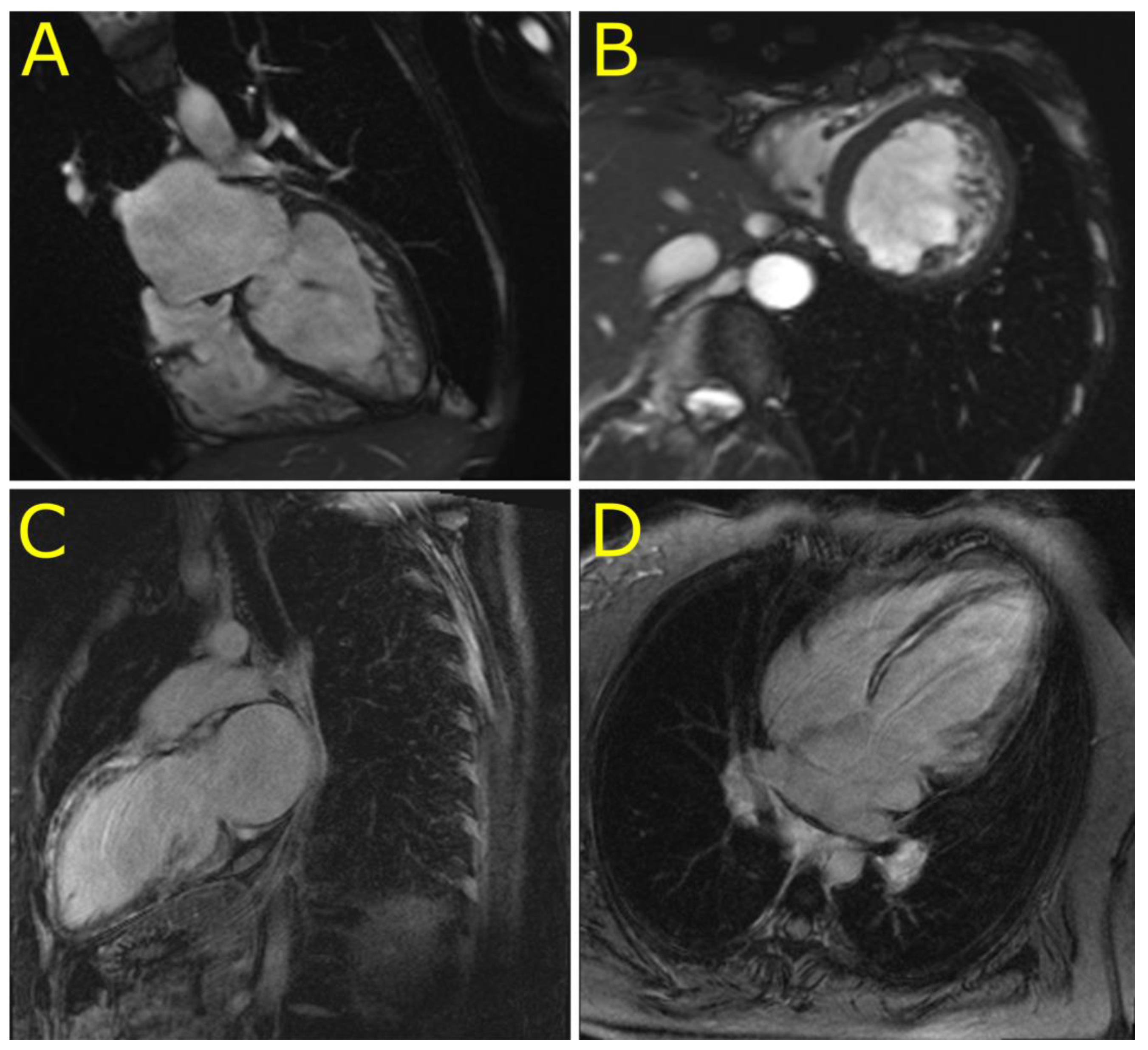
3.2. Phenotypes of Desminopathy
3.3. Morphology of Desminopathy in Myocardial and Skeletal Muscle Samples
3.4. Indications for the Pathogenicity of the Novel Desmin Variants
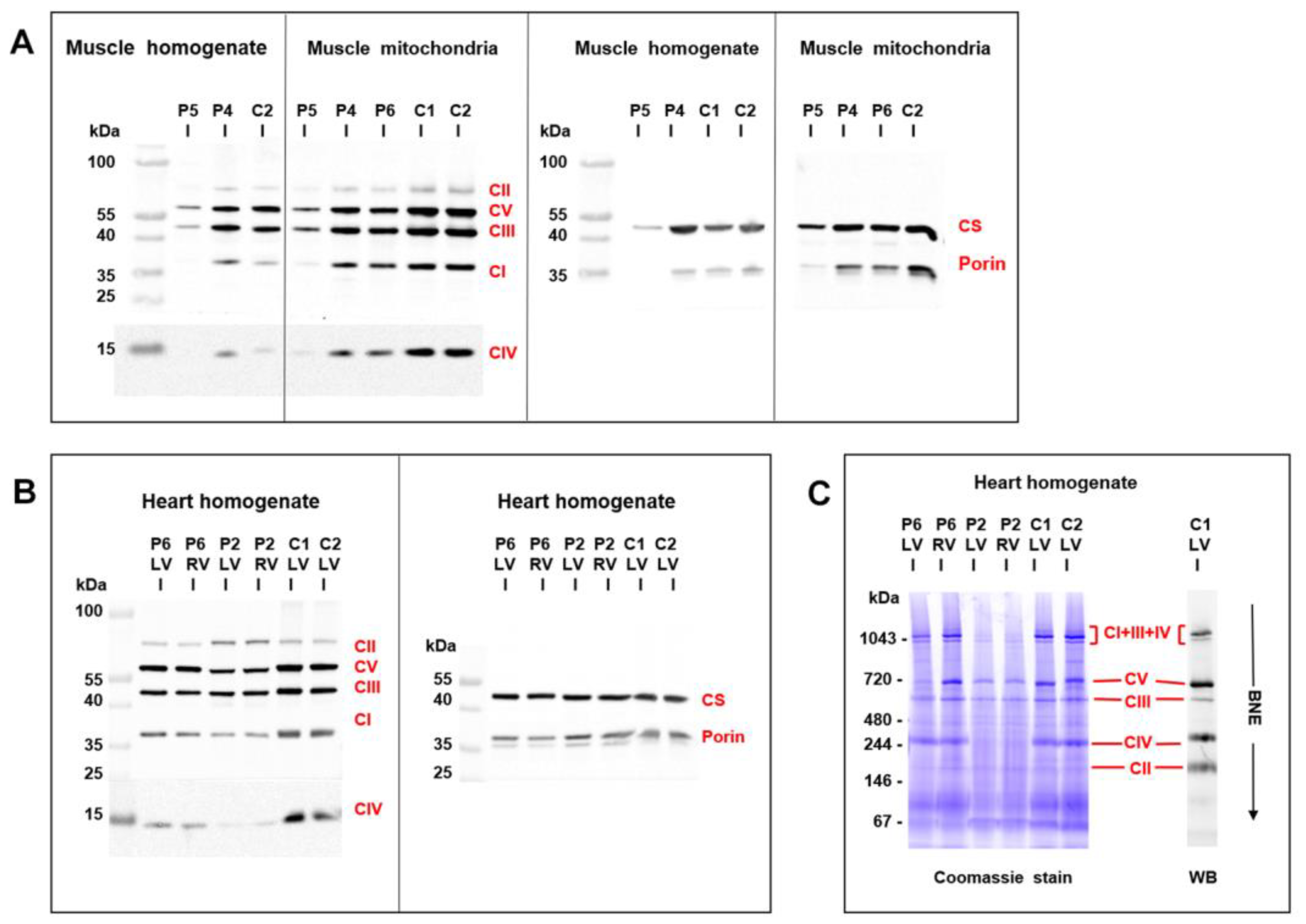
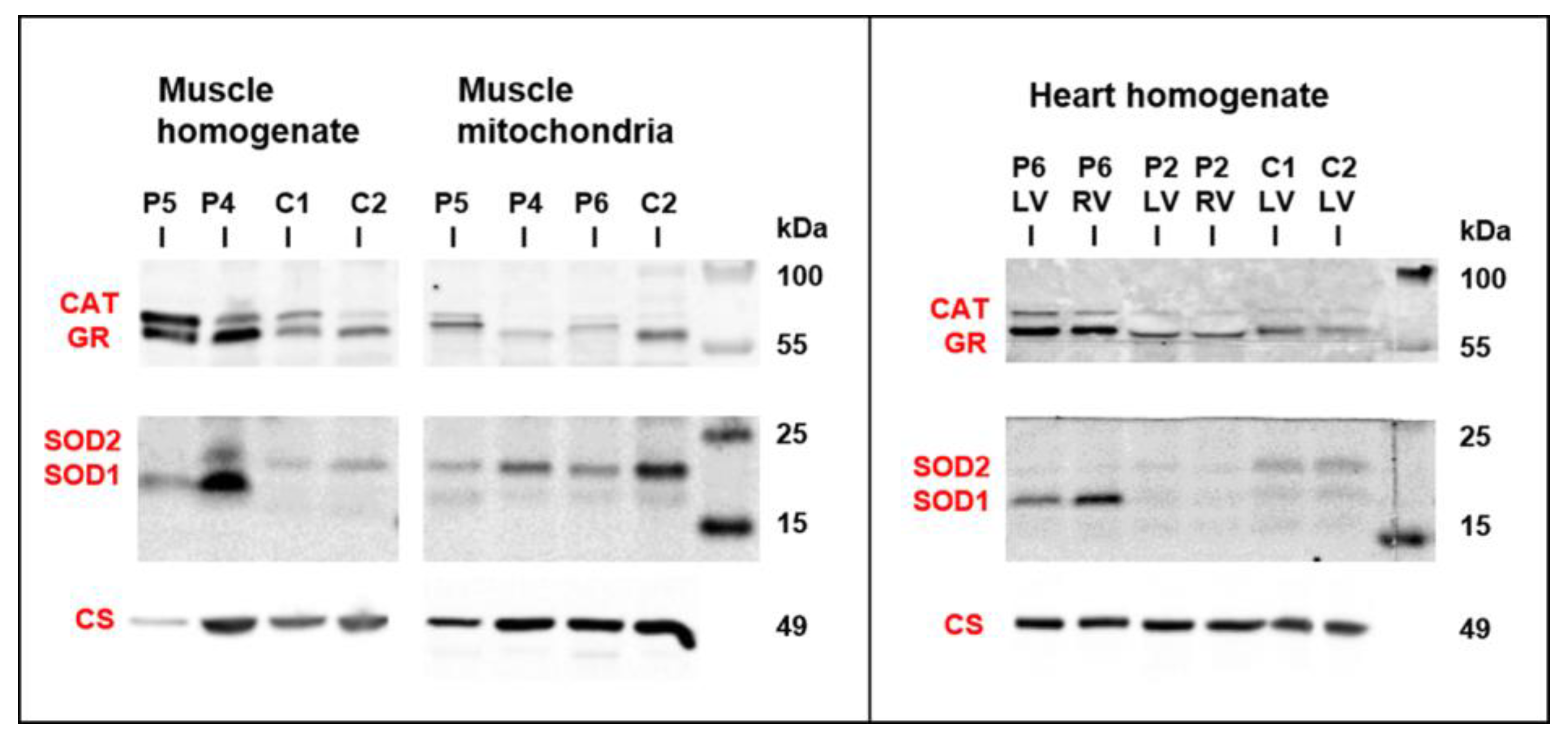
3.5. Mitochondrial Function and Content in Skeletal Muscle and Heart
4. Discussion
4.1. Clinical and Histopathological Correlates of Desminopathy
4.2. Novel Cardiac Phenotypes of Desminopathy
4.3. Mitochondrial Dysfunction in Desminopathy
4.4. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations and Acronyms
| ACM | arrhythmogenic cardiomyopathy |
| CAT | catalase |
| COX | cytochrome c oxidase |
| DCM | dilated cardiomyopathy |
| DES | desmin gene |
| GADPH | glyceraldehyde 3-phosphate dehydrogenase |
| GR | glutathione reductase |
| HCM | hypertrophic cardiomyopathy |
| LVNC | left ventricular non-compaction cardiomyopathy |
| NADH | reduced form of nicotinamide adenine dinucleotide |
| MPS | massively parallel sequencing |
| RCM | restrictive cardiomyopathy |
| SDH | succinate dehydrogenase |
| SOD | superoxide dismutase |
| WES | whole exome sequencing |
References
- van Spaendonck-Zwarts, K.Y.; van Hessem, L.; Jongbloed, J.D.; de Walle, H.E.; Capetanaki, Y.; van der Kooi, A.J.; van Langen, I.M.; van den Berg, M.P.; van Tintelen, J.P. Desmin-related myopathy. Clin. Genet. 2011, 80, 354–366. [Google Scholar] [CrossRef]
- Li, D.; Tapscoft, T.; Gonzalez, O.; Burch, P.E.; Quiñones, M.A.; Zoghbi, W.A.; Hill, R.; Bachinski, L.L.; Mann, D.L.; Roberts, R. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999, 100, 461–464. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R.; Slavov, D.; Ku, L.; Di Lenarda, A.; Sinagra, G.; Carniel, E.; Haubold, K.; Boucek, M.M.; Ferguson, D.; Graw, S.L.; et al. Prevalence of desmin mutations in dilated cardiomyopathy. Circulation 2007, 115, 1244–1251. [Google Scholar] [CrossRef] [Green Version]
- Goldfarb, L.G.; Park, K.Y.; Cervenakova, L.; Gorokhova, S.; Lee, H.S.; Vasconcelos, O.; Nagle, J.W.; Semino-Mora, C.; Sivakumar, K.; Dalakas, M.C. Missence mutations in desmin associated with familial cardiac and skeletal myopathy. Nat. Genet. 1998, 19, 402–403. [Google Scholar] [CrossRef]
- Arbustini, E.; Morbini, P.; Grasso, M.; Fasani, R.; Verga, L.; Bellini, O.; Dal Bello, B.; Campana, C.; Piccolo, G.; Febo, O.; et al. Restrictive cardiomyopathy, atrioventricular block and mild to subclinical myopathy in patients with desmin-immunoreactive material deposits. J. Am. Coll. Cardiol. 1998, 31, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Park, K.Y.; Dalakas, M.C.; Semino-Mora, C.; Lee, H.S.; Litvak, S.; Takeda, K.; Ferrans, V.J.; Goldfarb, L.G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N. Engl. J. Med. 2000, 342, 770–780. [Google Scholar] [CrossRef]
- Pruszczyk, P.; Kostera-Pruszczyk, A.; Shatunov, A.; Goudeau, B.; Dramiñska, A.; Takeda, K.; Sambuughin, N.; Vicart, P.; Strelkov, S.V.; Goldfarb, L.G.; et al. Restrictive cardiomyopathy with atrioventricular conduction block resulting from a desmin mutation. Int. J. Cardiol. 2007, 117, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Olive, M.; Armstrong, J.; Miralles, F.; Pou, A.; Fardeau, M.; Gonzalez, L.; Martínez, F.; Fischer, D.; Martínez Matos, J.A.; Shatunov, A.; et al. Phenotypic patterns of desminopathy associated with three novel mutations in the desmin gene. Neuromuscul. Disord. 2007, 17, 443–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otten, E.; Asimaki, A.; Maass, A.; van Langen, I.M.; van der Wal, A.; de Jonge, N.; van den Berg, M.P.; Saffitz, J.E.; Wilde, A.A.; Jongbloed, J.D.; et al. Desmin mutations as a cause of right ventricular heart failure affect the intercalated disks. Heart Rhythm 2010, 7, 1058–1064. [Google Scholar] [CrossRef] [PubMed]
- Klauke, B.; Kossmann, S.; Gaertner, A.; Brand, K.; Stork, I.; Brodehl, A.; Dieding, M.; Walhorn, V.; Anselmetti, D.; Gerdes, D.; et al. De novo desmin mutation N116S is associated with arrhythmogenic right ventricular cardiomyopathy. Hum. Mol. Genet. 2010, 19, 4595–4607. [Google Scholar] [CrossRef] [Green Version]
- Lorenzon, A.; Beffagna, G.; Bauce, B.; De Bortoli, M.; Li Mura, I.E.A.; Calore, M.; Dazzo, E.; Basso, C.; Nava, A.; Thiene, G.; et al. Desmin mutations and arrhythmogenic right ventricular cardiomyopathy. Am. J. Cardiol. 2013, 111, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Capetanaki, Y.; Bloch, R.J.; Kouloumenta, A.; Mavroidis, M.; Psarras, S. Muscle intermediate filaments and their links to membranes and membranous organelles. Exp. Cell. Res. 2007, 313, 2063–2076. [Google Scholar] [CrossRef] [PubMed]
- Milner, D.J.; Mavroidis, M.; Weisleder, N.; Capetanaki, Y. Desmin cytoskeleton linked to muscle mitochondrial distribution and respiratory function. J. Cell. Biol. 2000, 150, 1283–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, M.; Waele, L.D.; Hudson, J.; Eagle, M.; Sewry, C.; Marsh, J.; Charlton, R.; He, L.; Blakely, E.L.; Horrocks, I.; et al. Recessive desmin-null muscular dystrophy with central nuclei and mitochondrial abnormalities. Acta Neuropathol. (Berl.) 2013, 125, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Winter, L.; Wittig, I.; Peeva, V.; Eggers, B.; Heidler, J.; Chevessier, F.; Kley, R.A.; Barkovits, K.; Strecker, V.; Berwanger, C.; et al. Mutant desmin substantially perturbs mitochondrial morphology, function and maintenance in skeletal muscle tissue. Acta Neuropathol. 2016, 132, 453–473. [Google Scholar] [CrossRef] [Green Version]
- McCormick, E.M.; Kenyon, L.; Falk, M.J. Desmin common mutation is associated with multi-systemic disease manifestations and depletion of mitochondria and mitochondrial DNA. Front. Genet. 2015, 6, 199. [Google Scholar] [CrossRef] [Green Version]
- Hartmannova, H.; Kubanek, M.; Sramko, M.; Piherova, L.; Noskova, L.; Hodanova, K.; Stranecky, V.; Pristoupilova, A.; Sovova, J.; Marek, T.; et al. Isolated X-linked hypertrophic cardiomyopathy caused by a novel mutation of the Four-and-a-half LIM domain 1 gene. Circ. Cardiovasc. Genet. 2013, 6, 543–551. [Google Scholar] [CrossRef] [Green Version]
- Kalia, S.S.; Adelman, K.; Bale, S.J.; Chung, W.K.; Eng, C.; Evans, J.P.; Herman, G.E.; Hufnagel, S.B.; Klein, T.E.; Korf, B.R.; et al. Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): A policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 2017, 19, 249–255. [Google Scholar] [CrossRef] [Green Version]
- Brodehl, A.; Dieding, M.; Biere, N.; Unger, A.; Klauke, B.; Walhorn, V.; Gummert, J.; Schulz, U.; Linke, W.A.; Gerull, B.; et al. Functional characterization of the novel DES mutation p.L136P associated with dilated cardiomyopathy reveals a dominant filament assembly defect. J. Mol. Cell. Cardiol. 2016, 91, 207–214. [Google Scholar] [CrossRef]
- Brodehl, A.; Pour Hakimi, S.A.; Stanasiuk, C.; Ratnavadivel, S.; Hendig, D.; Gaertner, A.; Gerull, B.; Gummert, J.; Paluszkiewicz, L.; Milting, H. Restrictive Cardiomyopathy is Caused by a Novel Homozygous Desmin (DES) Mutation p.Y122H Leading to a Severe Filament Assembly Defect. Genes (Basel) 2019, 11, 918. [Google Scholar] [CrossRef] [Green Version]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, D.C.; Hrapchak, B.B. Theory and Practice of Histotechnology, 2nd ed.; Mosby: Columbus, OH, USA, 1987. [Google Scholar]
- Makinen, M.W.; Lee, C.P. Biochemical studies of skeletal muscle mitochondria. I. Microanalysis of cytochrome content, oxidative and phosphorylative activities of mammalian skeletal muscle mitochondria. Arch. Biochem. Biophys. 1968, 126, 75–82. [Google Scholar] [CrossRef]
- Melenovsky, M.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial Iron Content and Mitochondrial Function in Heart Failure: Direct Tissue Analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef]
- Diokmetzidou, A.; Soumaka, E.; Kloukina, I.; Tsikitis, M.; Makridakis, M.; Varela, A.; Davos, C.H.; Georgopoulos, S.; Anesti, A.; Vlachou, A.; et al. Desmin and αB-Crystallin Interplay in.Maintenance of Mitochondrial Homeostasis and Cardiomyocyte Survival. J. Cell Sci. 2016, 129, 3705–3720. [Google Scholar] [CrossRef] [Green Version]
- Bär, H.; Goudeau, B.; Wälde, S.; Casteras-Simon, M.; Mücke, N.; Shatunov, A.; Goldberg, Y.P.; Clarke, C.; Holton, J.L.; Eymard, B.; et al. Conspicuous involvement of desmin tail mutations in diverse cardiac and skeletal myopathies. Hum. Mutat. 2007, 28, 374–386. [Google Scholar] [CrossRef]
- Capetanaki, Y.; Papathanasiou, S.; Diokmetzidou, A.; Vatsellas, G.; Tsikitis, M. Desmin related disease: A matter of cell survival failure. Curr. Opin. Cell Biol. 2015, 32, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Kostera-Pruszczyk, A.; Pruszczyk, P.; Kamińska, A.; Lee, H.S.; Goldfarb, L.G. Diversity of cardiomyopathy phenotypes caused by mutations in desmin. Int. J. Cardiol. 2008, 131, 146–147. [Google Scholar] [CrossRef]
- Clemen, C.S.; Herrmann, H.; Strelkov, S.V.; Schröder, R. Desminopathies: Pathology and mechanisms. Acta Neuropathol. 2013, 125, 47–75. [Google Scholar] [CrossRef] [Green Version]
- Bermúdez-Jiménez, F.J.; Carriel, V.; Brodehl, A.; Alaminos, M.; Campos, A.; Schirmer, I.; Milting, H.; Abril, B.Á.; Álvarez, M.; López-Fernández, S.; et al. The Novel Desmin Mutation p.Glu401Asp Impairs Filament Formation, Disrupts Cell Membrane Integrity and Causes Severe Arrhythmogenic Left Ventricular Cardiomyopathy/Dysplasia. Circulation 2018, 137, 1595–1610. [Google Scholar] [CrossRef] [PubMed]
- Cetin, N.; Balci-Hayta, B.; Gundesli, H.; Korkusuz, P.; Purali, N.; Talim, B.; Tan, E.; Selcen, D.; Erdem-Ozdamar, S.; Dincer, P. A novel desmin mutation leading to autosomal recessive limb-girdle muscular dystrophy: Distinct histopathological outcomes compared with desminopathies. J. Med. Genet. 2013, 50, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Arbustini, E.; Favalli, V.; Narula, N.; Serio, A.; Grasso, M. Left Ventricular Noncompaction: A Distinct Genetic Cardiomyopathy? J. Am. Coll. Cardiol. 2016, 68, 949–966. [Google Scholar] [CrossRef] [PubMed]
- van Waning, J.I.; Caliskan, K.; Hoedemaekers, Y.M.; van Spaendonck-Zwarts, K.Y.; Baas, A.F.; Boekholdt, S.M.; van Melle, J.P.; Teske, A.J.; Asselbergs, F.W.; Backx, A.P.C.M.; et al. Genetics, Clinical Features, and Long-Term Outcome of Noncompaction Cardiomyopathy. J. Am. Coll. Cardiol. 2018, 71, 711–722. [Google Scholar] [CrossRef]
- Marakhonov, A.V.; Brodehl, A.; Myasnikov, R.P.; Sparber, P.A.; Kiseleva, A.V.; Kulikova, O.V.; Meshkov, A.N.; Zharikova, A.A.; Koretsky, S.N.; Kharlap, M.S.; et al. Noncompaction cardiomyopathy is caused by a novel in-frame desmin (DES) deletion mutation within the 1A coiled-coil rod segment leading to a severe filament assembly defect. Hum. Mutat. 2019, 40, 734–741. [Google Scholar] [CrossRef]
- Miszalski-Jamka, K.; Jefferies, J.L.; Mazur, W.; Głowacki, J.; Hu, J.; Lazar, M.; Gibbs, R.A.; Liczko, J.; Kłyś, J.; Venner, E.; et al. Novel Genetic Triggers and Genotype-Phenotype Correlations in Patients with Left Ventricular. Noncompact. Circ. Cardiovasc. Genet. 2017, 10, e001763. [Google Scholar] [CrossRef] [Green Version]
- Schröder, R.; Goudeau, B.; Simon, M.C.; Fischer, D.; Eggermann, T.; Clemen, C.S.; Li, Z.; Reimann, J.; Xue, Z.; Rudnik-Schöneborn, S.; et al. On noxious desmin: Functional effects of a novel heterozygous desmin insertion mutation on the extrasarcomeric desmin cytoskeleton and mitochondria. Hum. Mol. Genet. 2003, 12, 657–669. [Google Scholar] [CrossRef]
- Vincent, A.E.; Grady, J.P.; Rocha, M.C.; Alston, C.L.; Rygiel, K.A.; Barresi, R.; Taylor, R.W.; Turnbull, D.M. Mitochondrial dysfunction in myofibrillar myopathy. Neuromuscul. Disord. 2016, 26, 691–701. [Google Scholar] [CrossRef] [Green Version]
- Galata, Z.; Kloukina, I.; Kostavasili, I.; Varela, A.; Davos, C.H.; Makridakis, M.; Bonne, G.; Capetanaki, Y. Amelioration of desmin network defects by αB-crystallin overexpression confers cardioprotection in a mouse model of dilated cardiomyopathy caused by LMNA gene mutation. J. Mol. Cell. Cardiol. 2018, 125, 73–86. [Google Scholar] [CrossRef]
- Tsikitis, M.; Galata, Z.; Mavroidis, M.; Psarras, S.; Capetanaki, Y. Intermediate filaments in cardiomyopathy. Bioph. Rev. 2018, 10, 1007–1031. [Google Scholar] [CrossRef]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme Activity of Muscle Homogenates (nmol/min/mg protein) | P4 | P5 | P6 | Controls n = 30 |
|---|---|---|---|---|
| Complex IV | 130.1 | 38.1 | 81.8 | 68–213 |
| Citrate synthase (CS) | 109.5 | 41.4 | 97.8 | 48–128 |
| Complex IV/CS | 1.19 | 0.92 | 0.84 | 080–160 |
| Coenzyme Q10 content (pmol/mg) | 282.9 | 140.5 | 112.5 | 180–460 |
| Respiratory Activity of Permeabilized Muscle Fibers (pmol O2/s/mg protein) | P6 | Controls n = 9 |
|---|---|---|
| ADP-stimulated oxidation of NADH-dependent substrates | 7.4 | 16–26 |
| ADP-stimulated oxidation of succinate | 10.7 | 9–18 |
| Cytochrome c oxidase respiration | 63 | 43–83 |
| Enzyme Activity of Isolated Mitochondria (nmol/min/mg protein) | P4 | P5 | P6 | Controls n = 30 |
|---|---|---|---|---|
| Complex I | 328.5 | 230.8 | 131.2 | 110–290 |
| Complex I+III | 94.1 | 18.7 | 53.2 | 126–316 |
| Complex II | 69.7 | 50.5 | 49.5 | 21–93 |
| Complex II+III | 174.2 | 92.9 | 146.7 | 82–251 |
| Complex III | 303.0 | 342.7 | 535.0 | 200–600 |
| Complex IV | 578.4 | 311.6 | 236.6 | 658–1552 |
| Citrate synthase | 372.5 | 240.4 | 384.2 | 435–1234 |
| Complex I/CS | 0.88 | 0.96 | 0.34 | 0.17–0.41 |
| Complex I+III/CS | 0.25 | 0.07 | 0.13 | 0.07–0.27 |
| Complex II/CS | 0.19 | 0.21 | 0.13 | 0.04–0.12 |
| Complex II+III/CS | 0.47 | 0.39 | 0.38 | 0.35–0.36 |
| Complex III/CS | 0.81 | 1.43 | 1.39 | 0.56–1.46 |
| Complex IV/CS | 1.55 | 1.30 | 0.62 | 0.82–1.88 |
| Respiratory/Enzyme Activity | P2 Left Ventricle | P2 Right Ventricle | P6 Left Ventricle | P6 Right Ventricle | Controls n = 38 |
|---|---|---|---|---|---|
| (pmol O2/s/mg) NADH respiration | 245 | 203 | 448 | 229 | 235–2356 |
| Succinate respiration | 295 | 242 | 254 | 319 | 365–1529 |
| Cytochrome c oxidase respiration | 766 | 991 | 1003 | 1047 | 561–4120 |
| (nmol/min/mg) Complex I+III | 26.1 | 30.3 | 145.1 | 83.9 | 44–386 |
| Complex II+III | 88.0 | 71.3 | 59.5 | 56.7 | 27–195 |
| Complex IV | 262.6 | 432.4 | 430.5 | 276.8 | 389–1989 |
| Citrate synthase (CS) | 915.9 | 975.2 | 562.2 | 483.6 | 446–1207 |
| (activity ratio) | |||||
| Complex I+III/CS | 0.03 | 0.03 | 0.26 | 0.17 | 0.09–0.63 |
| Complex II+III/CS | 0.10 | 0.07 | 0.11 | 0.12 | 0.04–0.37 |
| Complex IV/CS | 0.29 | 0.44 | 0.77 | 0.57 | 0.54–2.60 |
| mtDNA content | |||||
| (2−ΔCt) | |||||
| D-loop/GAPDH | 4980 | 5499 | 2863 | 3592 | 2052–10519 |
| 16S RNA/GAPDH | 11629 | 10914 | 8017 | 7299 | 3715–15843 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kubánek, M.; Schimerová, T.; Piherová, L.; Brodehl, A.; Krebsová, A.; Ratnavadivel, S.; Stanasiuk, C.; Hansíková, H.; Zeman, J.; Paleček, T.; et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. J. Clin. Med. 2020, 9, 937. https://doi.org/10.3390/jcm9040937
Kubánek M, Schimerová T, Piherová L, Brodehl A, Krebsová A, Ratnavadivel S, Stanasiuk C, Hansíková H, Zeman J, Paleček T, et al. Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction. Journal of Clinical Medicine. 2020; 9(4):937. https://doi.org/10.3390/jcm9040937
Chicago/Turabian StyleKubánek, Miloš, Tereza Schimerová, Lenka Piherová, Andreas Brodehl, Alice Krebsová, Sandra Ratnavadivel, Caroline Stanasiuk, Hana Hansíková, Jiří Zeman, Tomáš Paleček, and et al. 2020. "Desminopathy: Novel Desmin Variants, a New Cardiac Phenotype, and Further Evidence for Secondary Mitochondrial Dysfunction" Journal of Clinical Medicine 9, no. 4: 937. https://doi.org/10.3390/jcm9040937