
Short-Term Stability of Serum and Liver Extracts for Untargeted Metabolomics and Lipidomics
 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Sample Preparation
2.2.1. Serum
2.2.2. Liver
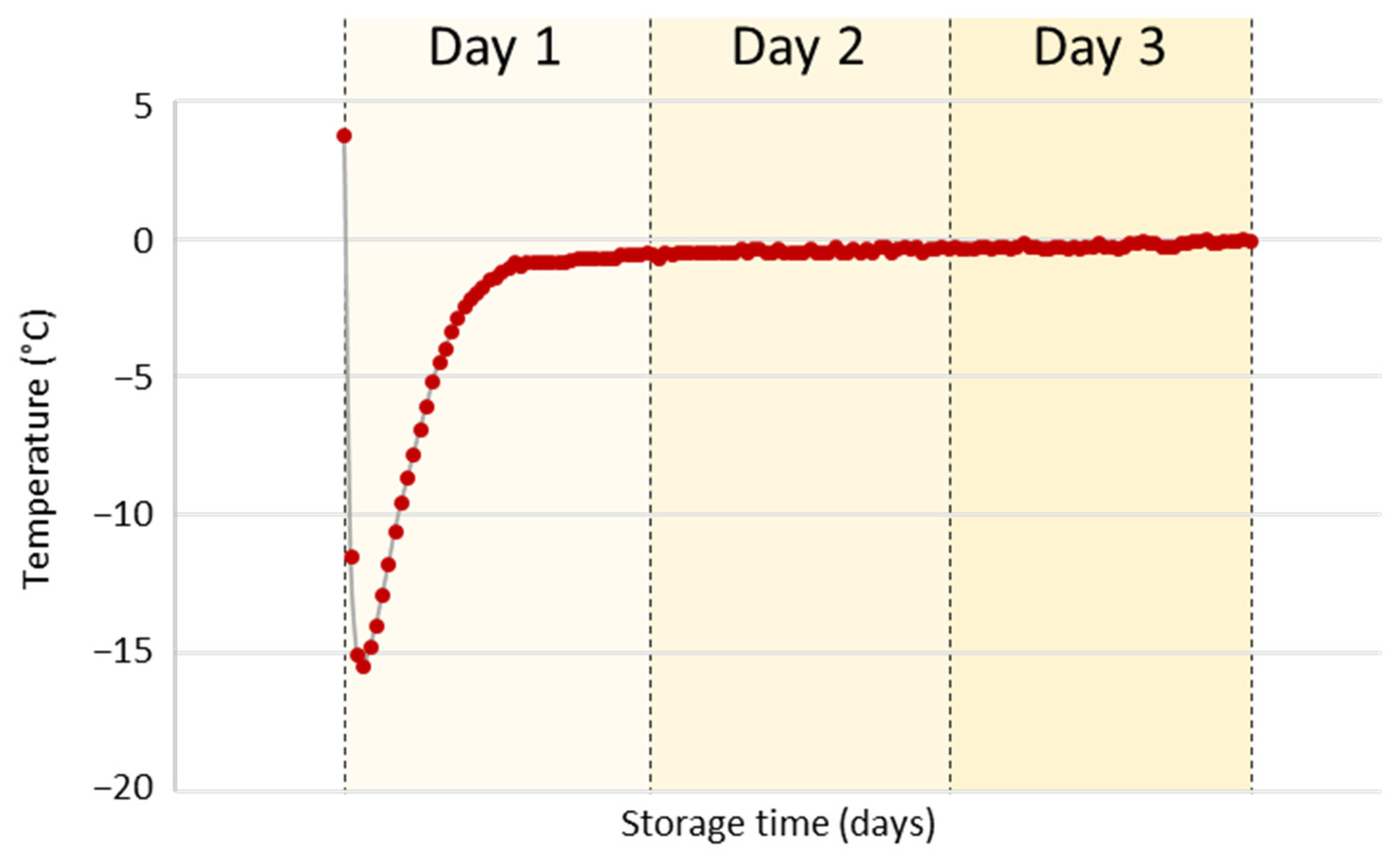
2.3. Storage Conditions
2.4. LC-MS Conditions
2.4.1. Untargeted Metabolomics
2.4.2. Untargeted Lipidomics
2.4.3. Iterative MS/MS Acquisition
2.5. Quality Control
2.6. Data Processing
2.7. Statistical Analysis
3. Results and Discussion
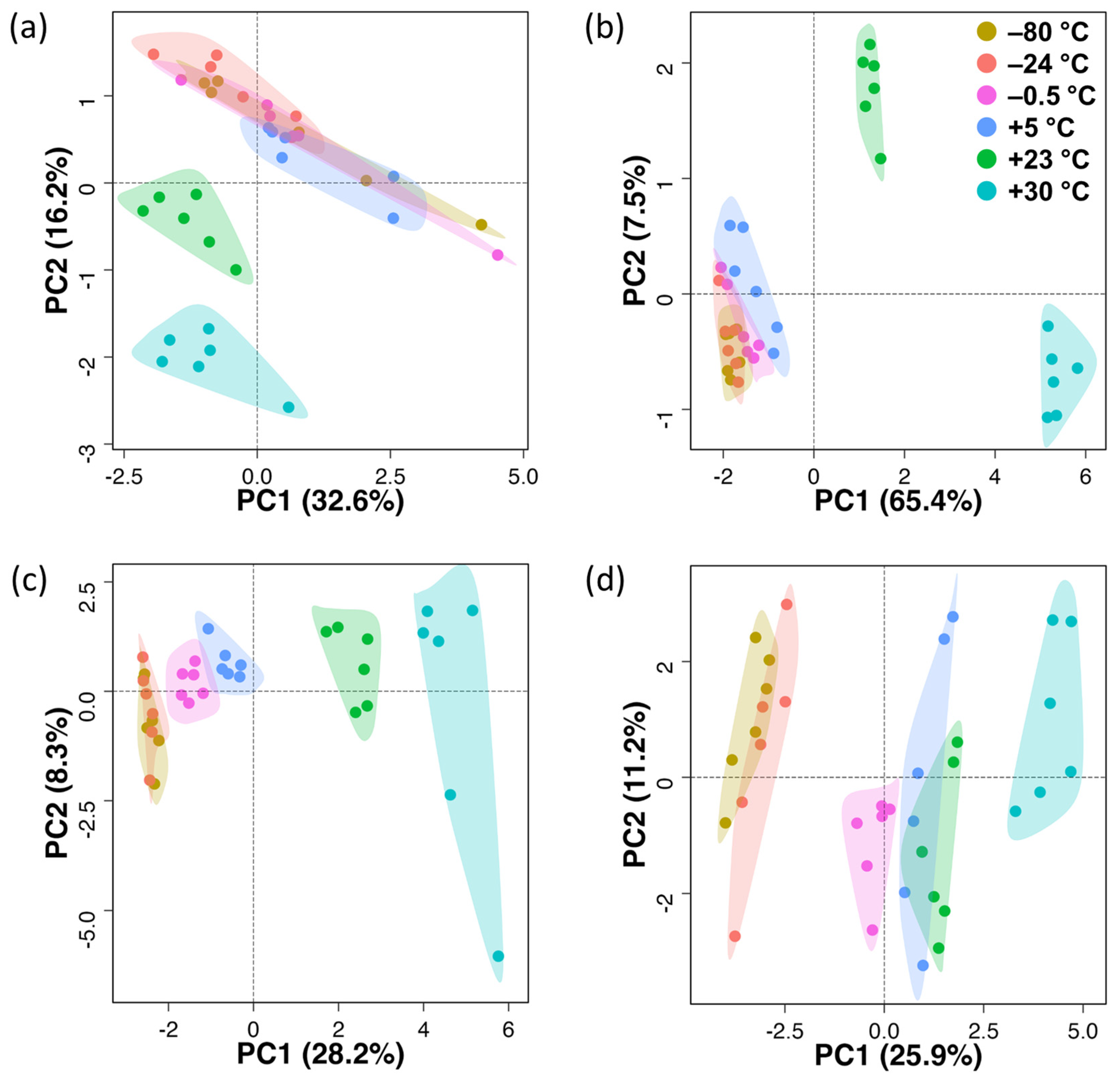
3.1. Untargeted Metabolomics
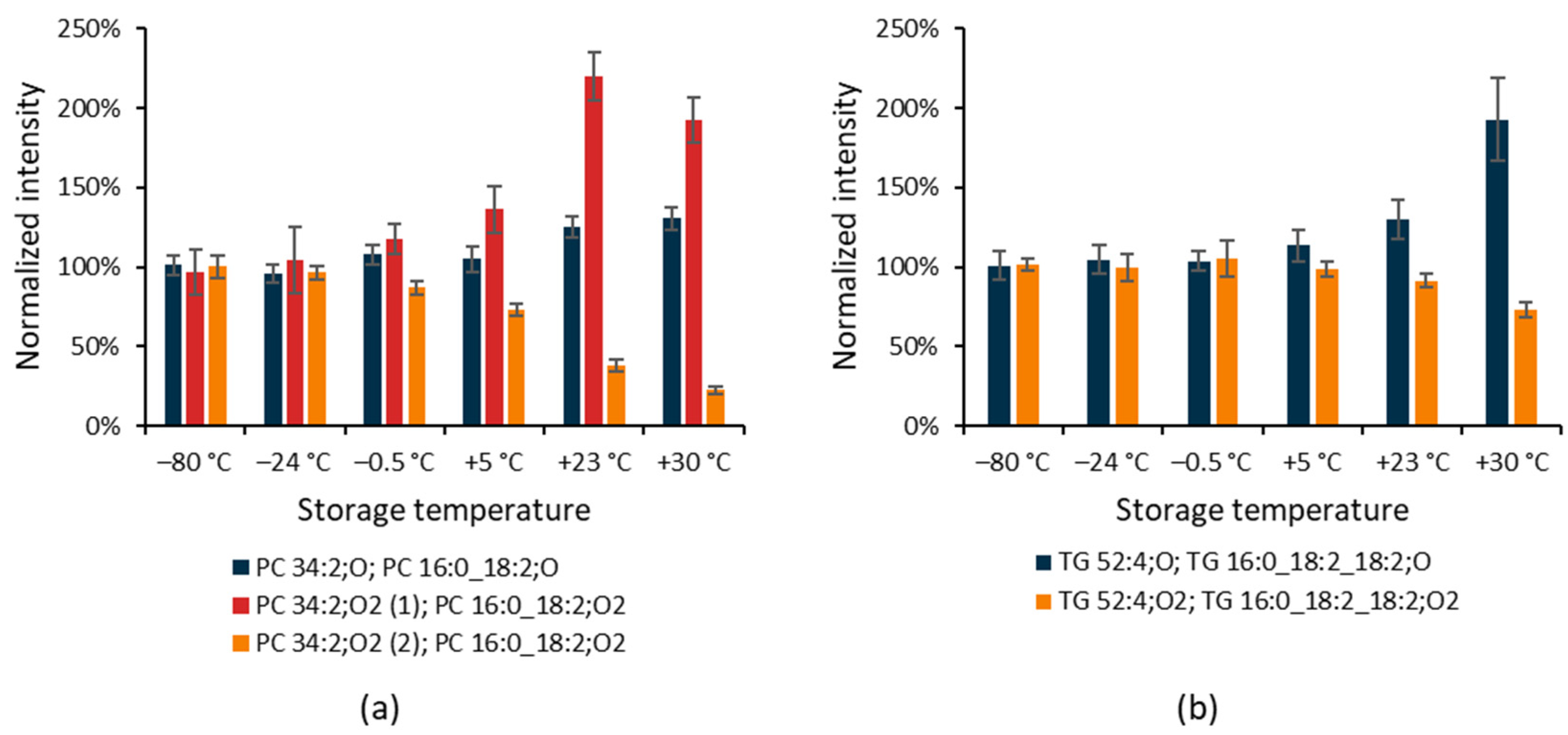
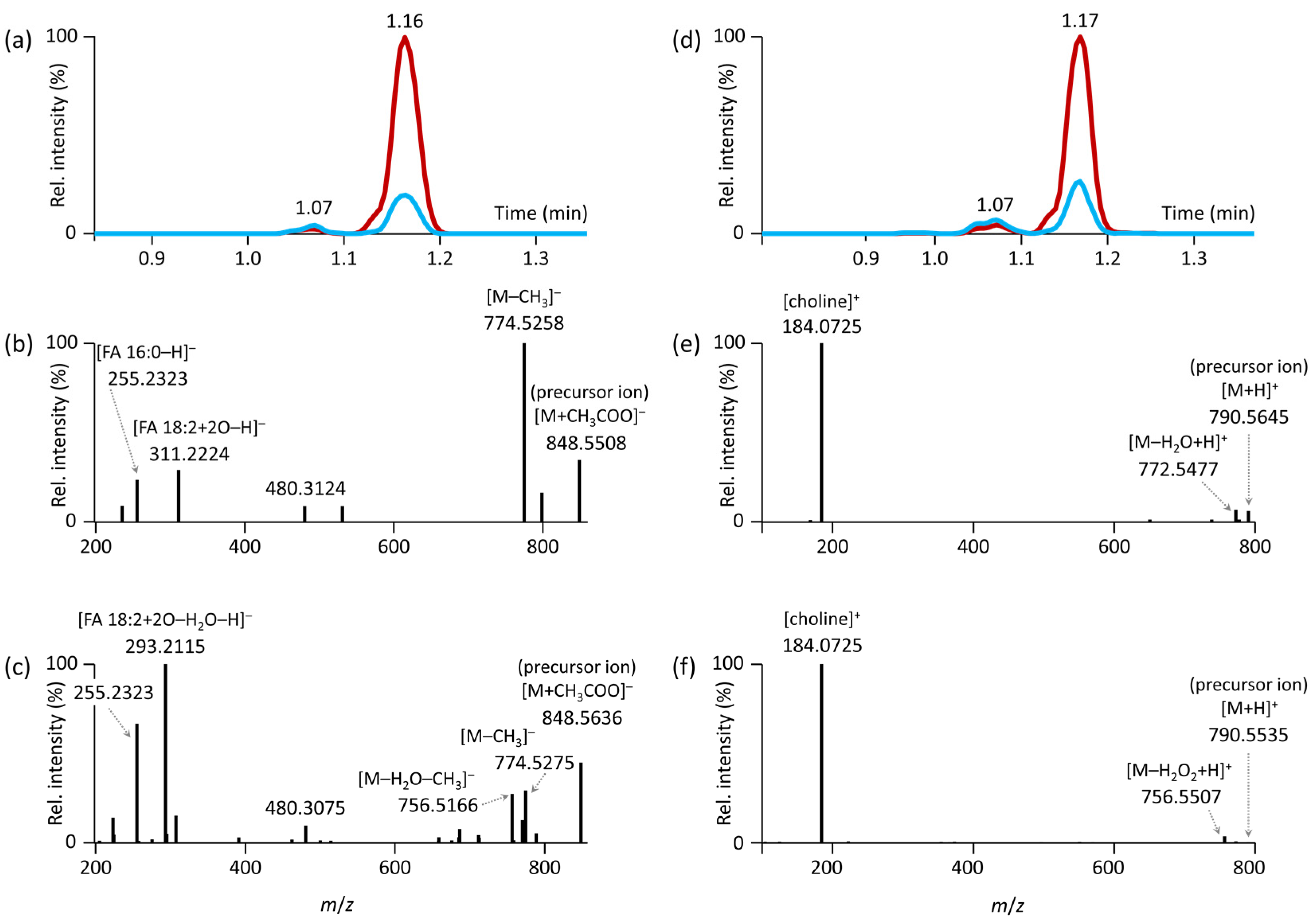
3.2. Untargeted Lipidomics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dettmer, K.; Aronov, P.A.; Hammock, B.D. Mass spectrometry-based metabolomics. Mass Spectrom. Rev. 2007, 26, 51–78. [Google Scholar] [CrossRef] [PubMed]
- Alseekh, S.; Aharoni, A.; Brotman, Y.; Contrepois, K.; D’auria, J.; Ewald, J.; Ewald, J.C.; Fraser, P.D.; Giavalisco, P.; Hall, R.D.; et al. Mass spectrometry-based metabolomics: A guide for annotation, quantification and best reporting practices. Nat. Methods 2021, 18, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Stevens, V.L.; Hoover, E.; Wang, Y.; Zanetti, K.A. Pre-analytical factors that affect metabolite stability in human urine, plasma, and serum: A review. Metabolites 2019, 9, 156. [Google Scholar] [CrossRef] [PubMed]
- Ang, J.E.; Revell, V.; Mann, A.; Mäntele, S.; Otway, D.T.; Johnston, J.D.; Thumser, A.E.; Skene, D.J.; Raynaud, F. Identification of human plasma metabolites exhibiting time-of-day variation using an untargeted liquid chromatography–mass spectrometry metabolomic approach. Chronobiol. Int. 2012, 29, 868–881. [Google Scholar] [CrossRef]
- Bervoets, L.; Louis, E.; Reekmans, G.; Mesotten, L.; Thomeer, M.; Adriaensens, P.; Linsen, L. Influence of preanalytical sampling conditions on the 1H NMR metabolic profile of human blood plasma and introduction of the Standard PREanalytical Code used in biobanking. Metabolomics 2015, 11, 1197–1207. [Google Scholar] [CrossRef]
- Denery, J.R.; Nunes, A.A.K.; Dickerson, T.J. Characterization of differences between blood sample matrices in untargeted metabolomics. Anal. Chem. 2011, 83, 1040–1047. [Google Scholar] [CrossRef]
- Carayol, M.; Licaj, I.; Achaintre, D.; Sacerdote, C.; Vineis, P.; Key, T.J.; Moret, N.C.O.; Scalbert, A.; Rinaldi, S.; Ferrari, P. Reliability of serum metabolites over a two-year period: A targeted metabolomic approach in fasting and non-fasting samples from EPIC. PLoS ONE 2015, 10, e0135437. [Google Scholar] [CrossRef]
- Ammerlaan, W.; Trezzi, J.-P.; Lescuyer, P.; Mathay, C.; Hiller, K.; Betsou, F. Method validation for preparing serum and plasma samples from human blood for downstream proteomic, metabolomic, and circulating nucleic acid-based applications. Biopreserv. Biobank. 2014, 12, 269–280. [Google Scholar] [CrossRef]
- Breier, M.; Wahl, S.; Prehn, C.; Fugmann, M.; Ferrari, U.; Weise, M.; Banning, F.; Seissler, J.; Grallert, H.; Adamski, J.; et al. Targeted metabolomics identifies reliable and stable metabolites in human serum and plasma samples. PLoS ONE 2014, 9, e89728. [Google Scholar] [CrossRef]
- Anton, G.; Wilson, R.; Yu, Z.-H.; Prehn, C.; Zukunft, S.; Adamski, J.; Heier, M.; Meisinger, C.; Römisch-Margl, W.; Wang-Sattler, R.; et al. Pre-analytical sample quality: Metabolite ratios as an intrinsic marker for prolonged room temperature exposure of serum samples. PLoS ONE 2015, 10, e0121495. [Google Scholar] [CrossRef]
- Chetwynd, A.J.; Abdul-Sada, A.; Holt, S.G.; Hill, E.M. Use of a pre-analysis osmolality normalisation method to correct for variable urine concentrations and for improved metabolomic analyses. J. Chromatogr. A 2016, 1431, 103–110. [Google Scholar] [CrossRef]
- Bernini, P.; Bertini, I.; Luchinat, C.; Nincheri, P.; Staderini, S.; Turano, P. Standard operating procedures for pre-analytical handling of blood and urine for metabolomic studies and biobanks. J. Biomol. NMR 2011, 49, 231–243. [Google Scholar] [CrossRef]
- Rakusanova, S.; Fiehn, O.; Cajka, T. Toward building mass spectrometry-based metabolomics and lipidomics atlases for biological and clinical research. TrAC Trends Anal. Chem. 2023, 158, 116825. [Google Scholar] [CrossRef]
- Saoi, M.; Britz-McKibbin, P. New advances in tissue metabolomics: A review. Metabolites 2021, 11, 672. [Google Scholar] [CrossRef]
- Fomenko, M.V.; Yanshole, L.V.; Tsentalovich, Y.P. Stability of metabolomic content during sample preparation: Blood and brain tissues. Metabolites 2022, 12, 811. [Google Scholar] [CrossRef]
- Lopes, M.; Brejchova, K.; Riecan, M.; Novakova, M.; Rossmeisl, M.; Cajka, T.; Kuda, O. Metabolomics atlas of oral 13C-glucose tolerance test in mice. Cell Rep. 2021, 37, 109833. [Google Scholar] [CrossRef]
- Sistilli, G.; Kalendova, V.; Cajka, T.; Irodenko, I.; Bardova, K.; Oseeva, M.; Zacek, P.; Kroupova, P.; Horakova, O.; Lackner, K.; et al. Krill oil supplementation reduces exacerbated hepatic steatosis induced by thermoneutral housing in mice with diet-induced obesity. Nutrients 2021, 13, 437. [Google Scholar] [CrossRef]
- Cajka, T.; Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O. Optimization of mobile phase modifiers for fast LC-MS-based untargeted metabolomics and lipidomics. Int. J. Mol. Sci. 2023, 24, 1987. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Koelmel, J.P.; Kroeger, N.M.; Gill, E.L.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Yost, R.A.; Garrett, T.J. Expanding lipidome coverage using LC-MS/MS data-dependent acquisition with automated exclusion list generation. J. Am. Soc. Mass Spectrom. 2017, 28, 908–917. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.-É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Samino, S.; Saez, I.; Duran, J.; Guinovart, J.J.; Yanes, O. A guideline to univariate statistical analysis for LC/MS-based untargeted metabolomics-derived data. Metabolites 2012, 2, 775–795. [Google Scholar] [CrossRef]
- Haid, M.; Muschet, C.; Wahl, S.; Römisch-Margl, W.; Prehn, C.; Möller, G.; Adamski, J. Long-term stability of human plasma metabolites during storage at −80 °C. J. Proteome Res. 2018, 17, 203–211. [Google Scholar] [CrossRef] [PubMed]
- Polson, C.; Sarkar, P.; Incledon, B.; Raguvaran, V.; Grant, R. Optimization of protein precipitation based upon effectiveness of protein removal and ionization effect in liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2003, 785, 263–275. [Google Scholar] [CrossRef]
- Wright, H.T.; Urry, D.W. Nonenzymatic deamidation of asparaginyl and glutaminyl residues in protein. Crit. Rev. Biochem. Mol. Biol. 1991, 26, 1–52. [Google Scholar] [CrossRef]
- Savino, R.J.; Kempisty, B.; Mozdziak, P. The potential of a protein model synthesized absent of methionine. Molecules 2022, 27, 3679. [Google Scholar] [CrossRef]
- Wyrzykowski, D.; Hebanowska, E.; Nowak-Wiczk, G.; Makowski, M.; Chmurzyński, L. Thermal behaviour of citric acid and isomeric aconitic acids. J. Therm. Anal. Calorim. 2011, 104, 731–735. [Google Scholar] [CrossRef]
- Morana, A.; Stiuso, P.; Colonna, G.; Lamberti, M.; Cartenì, M.; De Rosa, M. Stabilization of S-adenosyl-l-methionine promoted by trehalose. BBA-Gen. Subj. 2002, 1573, 105–108. [Google Scholar] [CrossRef]
- Reis, G.B.; Rees, J.C.; Ivanova, A.A.; Kuklenyik, Z.; Drew, N.M.; Pirkle, J.L.; Barr, J.R. Stability of lipids in plasma and serum: Effects of temperature-related storage conditions on the human lipidome. J. Mass Spectrom. Adv. Clin. Lab 2021, 22, 34–42. [Google Scholar] [CrossRef]
- Liebisch, G.; Fahy, E.; Aoki, J.; Dennis, E.A.; Durand, T.; Ejsing, C.S.; Fedorova, M.; Feussner, I.; Griffiths, W.J.; Köfeler, H.; et al. Update on LIPID MAPS classification, nomenclature, and shorthand notation for MS-derived lipid structures. J. Lipid Res. 2020, 61, 1539–1555. [Google Scholar] [CrossRef]
- Ni, Z.; Angelidou, G.; Hoffmann, R.; Fedorova, M. LPPtiger software for lipidome-specific prediction and identification of oxidized phospholipids from LC-MS datasets. Sci. Rep. 2017, 7, 15138. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Takahashi, M.; Sugiura, Y.; Izumi, Y.; Nishiyama, K.; Nishida, M.; Suematsu, M.; Bamba, T.; Yamada, K.-I. Structural library and visualization of endogenously oxidized phosphatidylcholines using mass spectrometry-based techniques. Nat. Commun. 2021, 12, 6339. [Google Scholar] [CrossRef]
- Ikeda, K.; Oike, Y.; Shimizu, T.; Taguchi, R. Global analysis of triacylglycerols including oxidized molecular species by reverse-phase high resolution LC/ESI-QTOF MS/MS. J. Chromatogr. B 2009, 877, 2639–2647. [Google Scholar] [CrossRef]
- Fabritius, M.; Yang, B. Direct infusion and ultra-high-performance liquid chromatography/electrospray ionization tandem mass spectrometry analysis of phospholipid regioisomers. Rapid Commun. Mass Spectrom. 2021, 35, e9151. [Google Scholar] [CrossRef]
- Gladine, C.; Ostermann, A.I.; Newman, J.W.; Schebb, N.H. MS-based targeted metabolomics of eicosanoids and other oxylipins: Analytical and inter-individual variabilities. Free Radic. Biol. Med. 2019, 144, 72–89. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
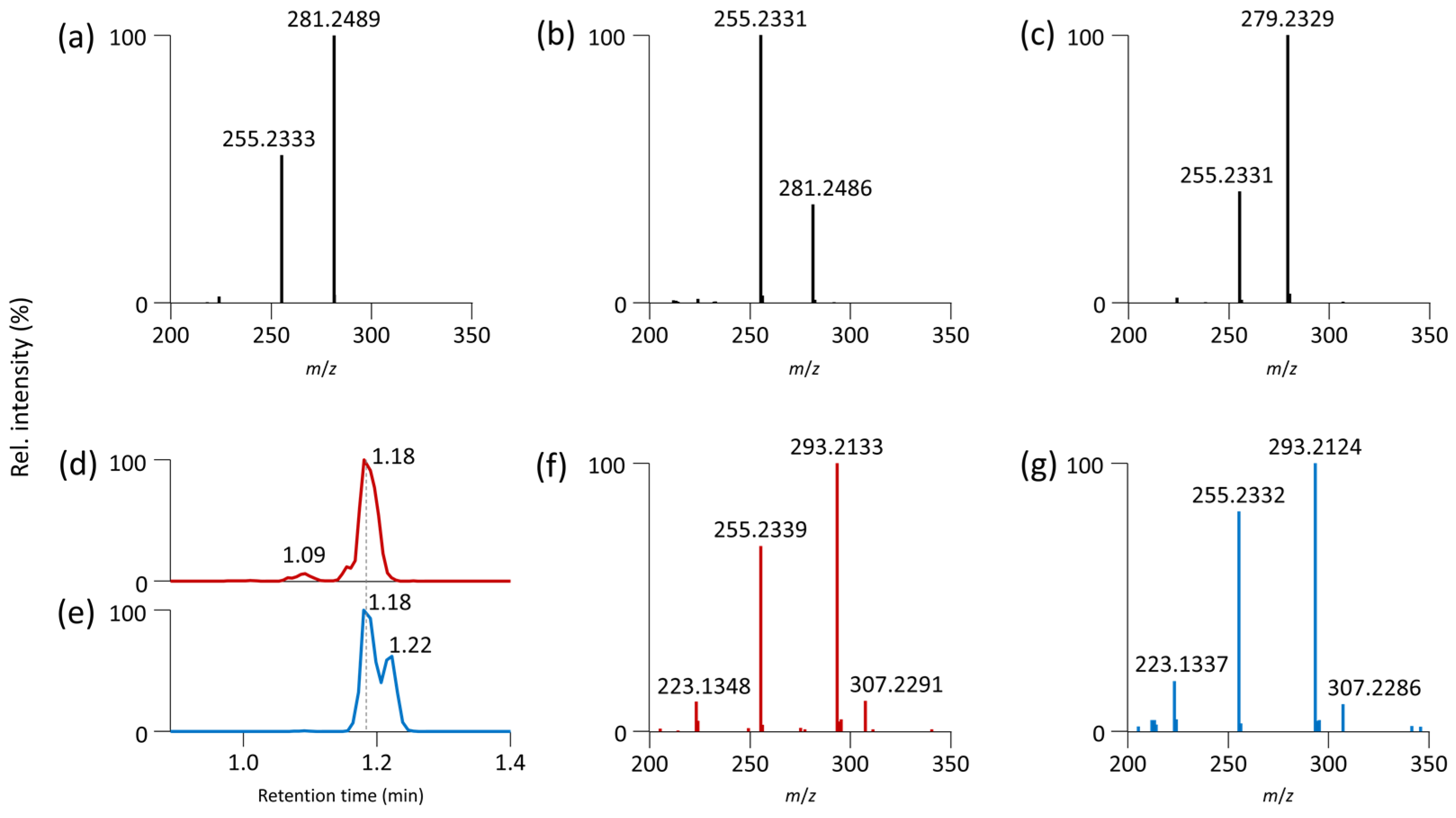{kind=link}
| Temperature | Storage | Serum | Liver |
|---|---|---|---|
| −24 °C | freezer | 0.0% | 0.0% |
| −0.5 °C | box with ice packs | 0.0% | 0.6% |
| +5 °C | refrigerator | 0.0% | 2.8% |
| +23 °C | laboratory | 2.6% | 22.2% |
| +30 °C | thermostat | 3.3% | 50.0% |
| Temperature | Storage | Serum | Liver |
|---|---|---|---|
| −24 °C | freezer | 0.0% | 0.0% |
| −0.5 °C | box with ice packs | 0.9% | 2.6% |
| +5 °C | refrigerator | 2.4% | 2.6% |
| +23 °C | laboratory | 8.8% | 4.2% |
| +30 °C | thermostat | 13.9% | 5.9% |
| Trend | Dry Serum Extracts | Dry Liver Extracts |
|---|---|---|
| Increasing | oxFA (O; O2; O3), LPI, oxPC (O; O2), oxPI (O), oxTG (O), FA, LPC, 5′-S-methyl-5′-thioadenosine, α-ketoglutaric acid | oxFA (O), LPE, oxPC (O; O2), oxPE (O2), oxPI (O; O2), 5′-S-methyl-5′-thioadenosine, arginine, creatinine, pyridoxamine, N-acetylphenylalanine, N-ε-dimethyllysine, citric acid, inosine 5′-monophosphate, methioninesulfoxide, N6,N6,N6-trimethyllysine |
| Decreasing | LPE, oxPC (O2), PE, etherPE, oxTG (O2), glucose, taurine, uric acid | ether-TG, 1-/3-methylhistidine, acetylcholine, cis-aconitic acid, adenosine, dipeptides, citrulline, histidine, hypotaurine, pentose phosphates, hexose phosphates, TMAO, pyridoxal 5′-phosphate, ornithine, N-acetylhistidine, N-α-acetyllysine, cystathionine, glutamic acid, glutamine, methionine |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hricko, J.; Rudl Kulhava, L.; Paucova, M.; Novakova, M.; Kuda, O.; Fiehn, O.; Cajka, T. Short-Term Stability of Serum and Liver Extracts for Untargeted Metabolomics and Lipidomics. Antioxidants 2023, 12, 986. https://doi.org/10.3390/antiox12050986
Hricko J, Rudl Kulhava L, Paucova M, Novakova M, Kuda O, Fiehn O, Cajka T. Short-Term Stability of Serum and Liver Extracts for Untargeted Metabolomics and Lipidomics. Antioxidants. 2023; 12(5):986. https://doi.org/10.3390/antiox12050986
Chicago/Turabian StyleHricko, Jiri, Lucie Rudl Kulhava, Michaela Paucova, Michaela Novakova, Ondrej Kuda, Oliver Fiehn, and Tomas Cajka. 2023. "Short-Term Stability of Serum and Liver Extracts for Untargeted Metabolomics and Lipidomics" Antioxidants 12, no. 5: 986. https://doi.org/10.3390/antiox12050986