1. Introduction
Cardiovascular diseases belong to the most frequent causes of death or life quality impairment. Currently, autologous vessel replacements such as the saphenous vein represent the gold standard grafts for small-diameter vessels (below 6 mm), and in many factors, they outperform synthetic alternatives. Autologous grafts, however, are of limited availability, and an invasive approach for their harvesting is required. In many cases, they are unsuitable for use due to unfavorable anatomy (gracility, branching, thrombosis) or previous harvest [
1]. Allograft vessels from cadaveric donors provide an alternative source for bypass grafting. These grafts can be cold-stored or cryopreserved in tissue banks; nevertheless, they are not immediately available for emergent surgery, and also their long-term patency is limited [
2]. Synthetic grafts act in many cases as suitable replacements; however, they show a reduced clinical efficacy, especially in small-calibre applications [
3]. There is a great clinical demand for new biomaterials providing feasible solutions, including proper mechanical support, functional tissue regeneration, and a non-thrombogenic surface [
4].
Not only tubular grafts but also planar patches are required with similar properties as cardiovascular prostheses [
5]. These planar patches are indispensable in many vascular reconstruction procedures, i.e., endarterectomy [
6].
Moreover, with pliable, non-thrombogenic, non-immunogenic patches seeded with autologous cells, we are just one step ahead of new heart valves’ construction. Current heart valve bioprostheses, despite various types of advanced anti-calcification processing, are still subject to structural valve deterioration (SVD) in a long-term manner. The incidence of SVD is strongly dependent on age and the type of prosthesis, so it can vary widely between 3.3–20% at a 10-year follow up, even in the elderly population [
7]. SVD strongly limits the clinical use of bioprosthetic valves in younger patients, especially in children.
One possible solution to this problem is using xenogeneic grafts. However, animal tissues cannot be directly applied due to immune incapability, risk of infectious disease transfer, and other safety and regulatory issues. Therefore, a need for tissue processing and engineering is a crucial step toward practical application. One possible way is graft decellularization. This process removes the immunogenic cellular elements and produces a non-immunogenic substitute suitable as an implantable graft or scaffold for further modifications. Also, the mechanical strength is preserved similarly to the native blood vessel, which is suitable for recolonization with the recipient’s native cells in vivo [
8,
9].
The decellularization is typically accomplished by treating the tissues with a combination of detergents, solvents, enzymes, osmotic gradients, and chelating agents [
10,
11]. The process of decellularization affects the structural and functional features of the tissue, such as its swelling and stretching properties, suture retention, and the conductivity for cells [
12]. Decellularized matrices for cardiovascular use need further modification. The first major thing is to improve the antithrombogenicity. Second is the support of an overall cell recolonization and stimulate re-endothelization in vivo. These steps could involve chemical modifications employing growth factors such as VEGF (Vascular endothelial growth factor), bFGF (Basic fibroblast growth factor) [
13] to promote cell overgrowth, or use of heparin as an anti-thrombogenic agent, which also promotes cell grow [
14]. On the other hand, growth factor use can be problematic for fulfilling the regulatory requirements for tissue grafts intended for human use and also potentially can induce unpredictable cell growth of host cells.
The second approach to the modification of decellularized matrices is their recolonization with cells. Several studies have demonstrated the improved performance of these matrices in animal models. Cell-recolonized decellularized grafts are distinguished by better biocompatibility, growth potential, mechanical strength, and elasticity. They also reduce the risk of thrombus formation and calcification and induce the recruitment of progenitor cells and tissue regeneration in vivo in contrast to the artificial materials [
12]. These studies often used smooth muscle cells (SMCs) or endothelial cells (ECs) or their combinations [
15], or included the adipose tissue-derived stromal cells (ASCs) [
16]. One of the advantages of ASCs is their high yield and high differentiation potential in comparison with differentiated cells [
17]. Another advantage is the easiness of isolation, either from the liposuction-derived or excision-derived adipose tissue when compared to SMC or EC. These allow isolation of autologous cells for combined use with decellularized grafts. Dynamic cultivation of stem or stromal cells is an essential tool for promoting their proliferation, but also for inducing and establishing a contractile differentiated SMC-like phenotype in these cells. The differentiation of stem cells towards SMCs requires pulsatile stress and cyclic strain, which are components of the hemodynamic stress to which the blood vessels are exposed in vivo [
18].
Conventional preparation of recellularized grafts is time-consuming. Under the static conditions, it takes several weeks of cell cultivation to reach cell overgrowth and maturation toward differentiated cells/tissues [
15]. Dynamic bioreactors mimicking physiological conditions could speed-up recellularization and promote cell differentiation.
In this study, we demonstrate a novel cultivation system allowing the recellularization of planar decellularized tissues intended as cardiovascular patches. This system incorporates a combination of micro-perfusion with mechanical strain to stimulate the proliferation of ASC cells and their differentiation towards smooth muscle cells. As for substrate, we have used two types of decellularized tissues—porcine and ovine decellularized pericardium. With possible use in in vivo conditions, the porcine pericardium will be acting as allogenous decellularized tissue, and the ovine will be modelling xenogeneic decellularized tissue.
2. Materials and Methods
2.1. Porcine and Ovine Pericardium for Decellularization
The porcine pericardium was obtained from a local slaughterhouse (Steinhauser s.r.o, Skalice nad Svitavou, Czech Republic). The Large White breed of pigs with a weight of approximately 100 kg and age 5–6 months were used. The fresh tissues were extracted during the slaughter process. The tissue was then transported to our laboratory in phosphate-buffered saline (PBS) with 100 μg/mL Gentamicin and 10 μg/mL Fluconazole (both LEK PHARMACEUTICAL D.D., Ljubljana, Slovenia). In the laboratory, the pericardial tissue was mechanically cleaned from fat and other residues. Then the tissue was cut into smaller pieces (approx. 6 × 10 cm) to fit into the decellularization chamber described below, and frozen in a DMEM (Dulbecco’s Modified Eagle Medium) medium with 10% FBS (Fetal Bovine Serum), 1% ABAM (Antibiotic Antimycotic Solution, contains 100 units penicillin, 0.1 mg streptomycin, and 0.25 µg amphotericin B per ml of culture media) and 10% DMSO (Dimethyl sulfoxide). The freezing gradient was set to 1 °C/min up to −80 °C. All chemicals unless otherwise specified were from Sigma-Aldrich (Sigma-Aldrich, St. Louis, MO, USA).
The ovine pericardium was obtained from experimental sheep (Suffolk breed, animals were used for another experimental work; these tissues were harvested fulfilling 3R). After animals were euthanized, a median sternotomy was performed and was followed by thoracotomy. The pericardium was extracted and mechanically cleaned from surrounding tissue, mainly fat. The same protocol for handling and cryopreservation, as described above, was used. Due to a limited number of experimental sheep, optimization procedures were mainly done on the porcine pericardium.
2.2. Decellularization System and Pericardium Decellularization
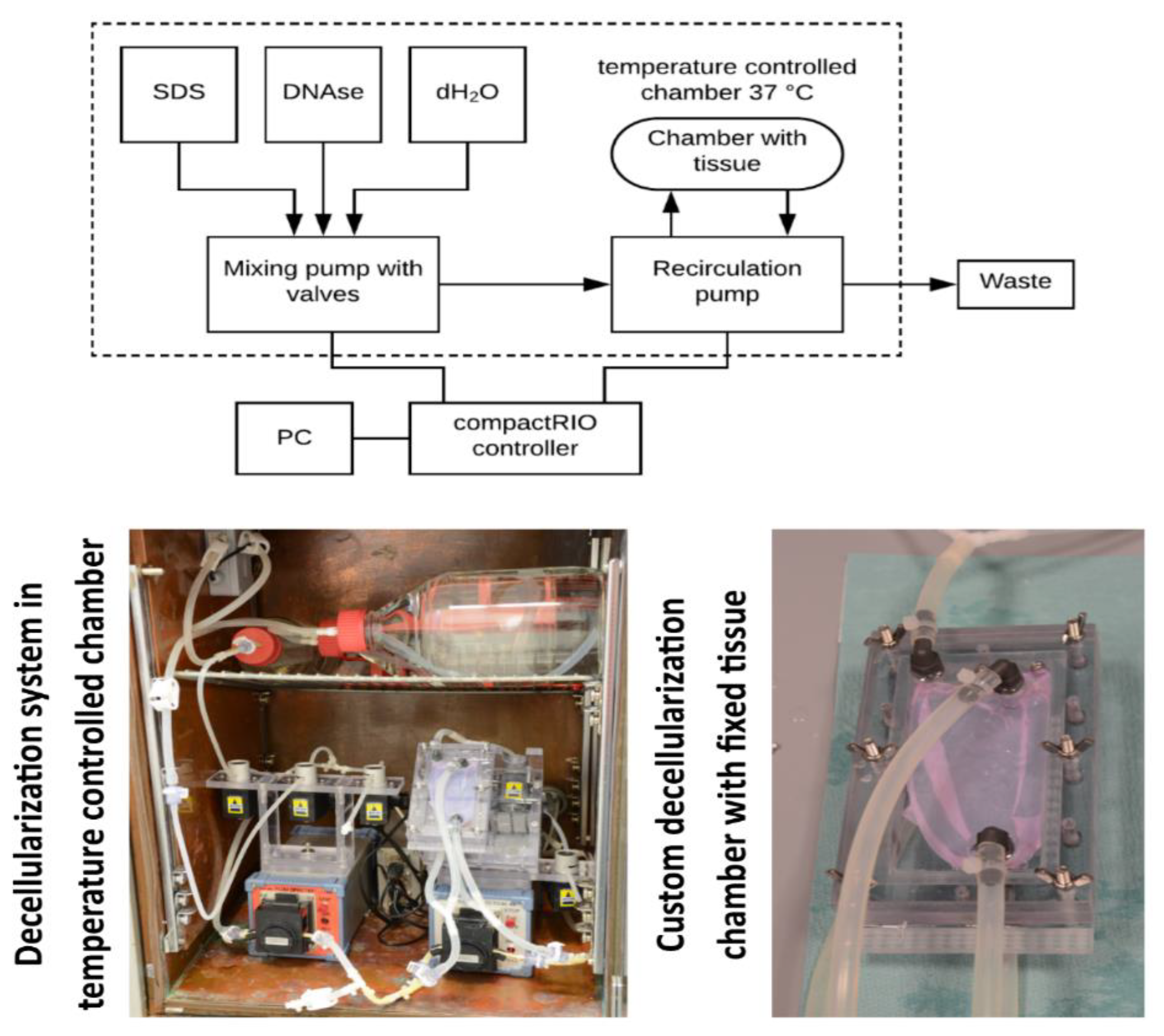
To automate the overall process of decellularization, a custom-made decellularization system was built. This solution consists of two peristaltic pumps with attached pinch valves. This system is controlled by an embedded controller compactRIO (National Instruments, Austin, TX, USA) with custom LabVIEW (also National Instruments) software to set appropriate parameters of the decellularization and to monitor the overall process. The first pump mixes and prepares decellularization agents, such as SDS (Sodium dodecyl sulfate) or DNAse, with dH2O (deionized water) and fills the decellularization chamber. The second pump maintains the recirculation of the agents in the decellularization chamber. This approach promotes the overall decellularization of large tissues.
The process of decellularization works in cycles. First, the decellularization agent or water is filled into the chamber. Then, the agent/water is recirculated for a defined period. After that, the agent/water is flushed into waste. Finally, a new agent or water is filled again. These steps are repeated depending on the tissue properties or experimental setup. This multicycle approach maintains a high chemical gradient promoting the efficiency of agents and washing. The system is installed in a heated chamber that holds the temperature to 37 °C. Higher temperature promotes the activity of detergents. Also, a special chamber was designed for fixing the planar pericardial tissue. The flow in this chamber is optimized to be homogeneously spread over both sides of the tissue. This helps to decellularize the tissue homogenously and to avoid the persistence of non-decellularized areas. The tissue in this chamber is fixed on a silicone holder using surgical silk. The overall view of the decellularization system and the fixing chamber is in
Figure 1.
The frozen pericardium was thawed in a 37 °C water bath. Then, the tissue was installed in the decellularization chamber using a 4—0 suture. The 0.5% SDS solution was used as a decellularization agent. This solution was used in 6 recirculation cycles lasting 10 min each. Then the SDS was washed out using 5 recirculation cycles of dH
2O lasting 5 min each. After that, a 60 min-lasting DNAse cycle was performed. The DNAse was dissolved in a DNAse buffer to the concentration of 40 µg/mL. This treatment was crucial to remove the remaining residues of DNA spilt over the tissue as a result of the previous SDS treatment. Then, the overall process was finished with a thorough washing of the tissue, consisting of a hundred 10-min cycles of dH
2O. The agent concentration, the cycle repetitions, and the exposure times were optimized to neutralize the potentially toxic effects of the agents and to minimize the mechanical degradation of the tissue described earlier [
19]. The decellularized tissues were then sterilized in a 70% ethanol solution for 1 h, and then they were rinsed and stored in sterile PBS at 4 °C. To evaluate the effectiveness of the decellularization process, 10 µm-thin cryosections from randomly selected parts of the tissues were prepared. These sections were stained using DAPI to visualize the cell nuclei or the remaining DNA.
Cytotoxicity and sterility tests were also performed. The cultivation of cells in the leachate and directly on the decellularized tissue were done to verify the potential cytotoxic effects. The leachates were prepared in a laboratory. Decellularized and sterile tissue was incubated in DMEM culture media with 10% FBS for 72 h. Then, this culture media was added to cells, and proliferation was observed. The isolated porcine adipose tissue-derived stromal cells were used for these cytotoxic tests. The sterility tests were performed according to Ph. eur. 2.6.1 and ISO 11737-2 [
20] in thioglycollate broth and tryptic soy broth.
2.3. Fat Extraction and Isolation of Porcine Adipose-Derived Stromal Cells
Porcine adipose tissue-derived stromal cells (PrASCs) were isolated from fat that was surgically extracted from the neck area of experimental pigs. Prestice black-pied pigs weighing approximately 35–40 kg were used.
During the surgery under general anaesthesia, a small amount of subcutaneous fat (approx. 2 cm3) was excised. Harvesting subcutaneous fat from the neck area was an easy and rapid surgical procedure well tolerated by the animals. There were no complications regarding wound healing. The fat obtained from this procedure was stored in warm DMEM containing 10% FBS and 3% ABAM solution and was transferred to the laboratory for further cell isolation.
The protocol for cell isolation was set according to Estes et al. [
21] with some modifications developed in studies focused on adipose-derived stem or stromal cells [
22,
23]. These modifications included tissue homogenization and a red blood cell lysing step. First, the fat tissue was mechanically cleaned from other tissues and was rinsed several times in PBS with 3% ABAM solution. Then, the fat was mechanically minced and homogenized into small pieces, and put into a 0.1% collagenase I solution with 1% BSA in PBS. After 2 h of collagenase digestion, the cells were treated with a red blood cell lysis buffer, centrifuged, and seeded into cultivation flasks with DMEM with 15% FBS and 1% ABAM solution. In these primocultures, it took approximately 3–5 days to reach the cell confluence, which was taken as passage 0. After that, the cells were passaged into large 175 cm
2 culture flasks (1.2–1.5 million of cells per flask) and cultivated in a DMEM/F12 medium (ratio 1:1) supplemented with 10% FBS, 10 ng/mL FGF2 (FGF-basic (154aa), Genscript, Leiden, The Netherlands) and 1% ABAM solution. This medium was used to stimulate cell proliferation. After reaching confluence in passage 1, the cells were seeded on the decellularized pericardium. In addition, a flow cytometry analysis was performed to characterize the cells. The cells in the 2nd passage were also characterized by a flow cytometry analysis using NovoCyte Flow Cytometer (Acea Biosciences, Inc., CA, USA). Results of flow cytometry shown in
Table 1.
2.4. Cultivation Bioreactor
A custom-made dynamic culture system was used for recellularization of the decellularized tissue. This system consists of a specially designed cultivation chamber that allows fixing the pericardium, and of a pressure stimulation system generating pressure pulsation and culture media perfusion.
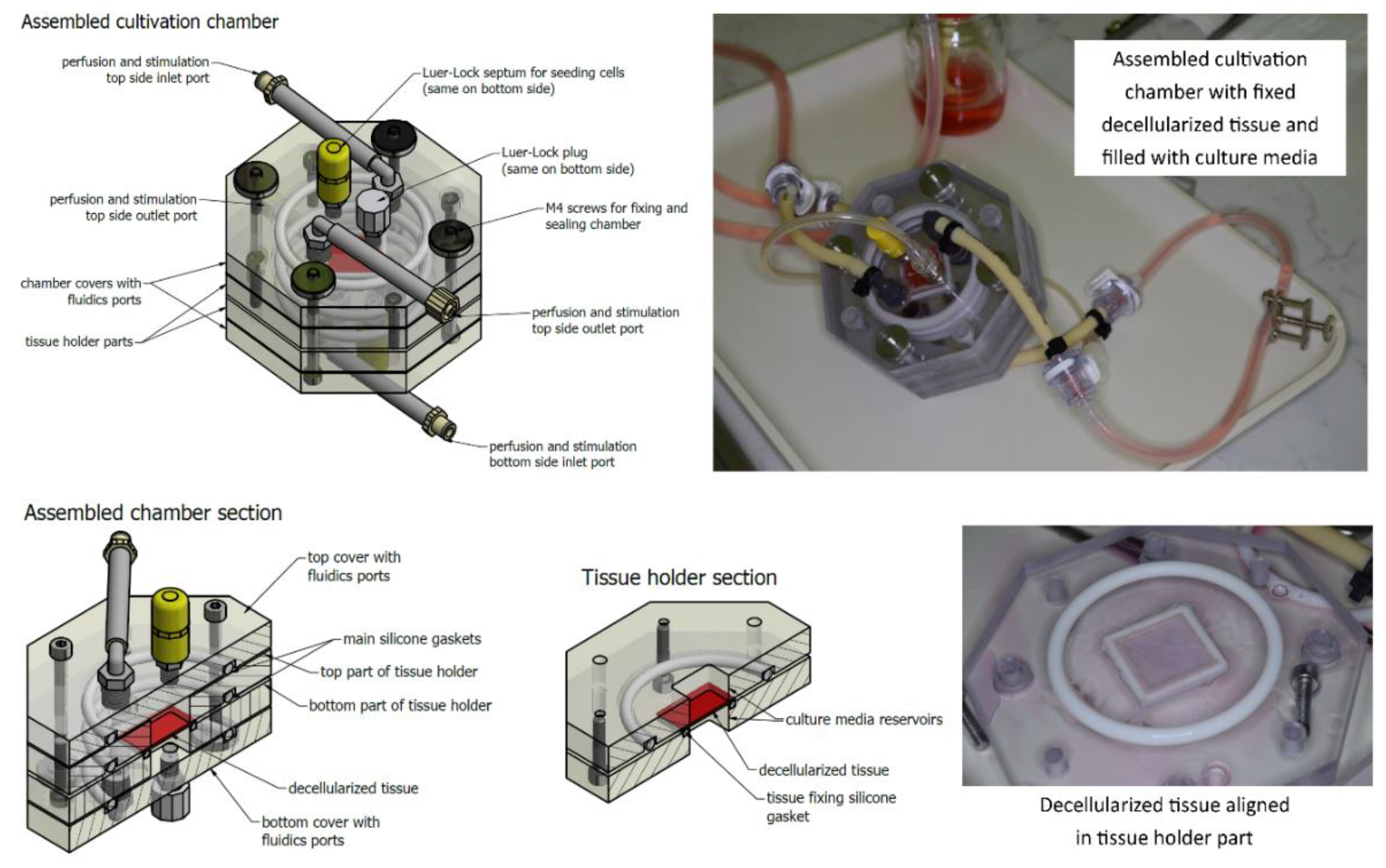
The cultivation chamber consists of four main parts. The middle two parts create a tissue holder. Both parts create reservoirs for culture media, and also, one part contains a small silicone gasket fixing the tissue. The decellularized tissue is cut to 35 × 35 mm2 squares. Then these squares are placed over the hole of the reservoir. The reservoir has a square shape and is limiting the area to 30 × 30 mm2 where cells are cultivated. The remaining 2.5 mm border of the prepared tissues is used for fixing them in place. A special inlet is put into this hole in order to prevent tissue bending. Then, the tissue is fixed with the second middle part of the chamber containing the silicone gasket that fixes the tissue in its position. A silicone O-ring gasket is also added to ensure the tightness of the chamber. Four stainless steel M4 screws tighten the holder. After this procedure, the helping inlet is removed.
Next, the chamber is closed by covers containing fluidics ports. Two more silicone O-ring gaskets are added to ensure the tightness. These covers are also tightened to the holder using stainless steel M4 screws with knurled nuts. To minimize the risk of any contamination or toxicity, these screws are not in contact with the tissue or the culture media. All other parts of the chamber are made of non-toxic materials including polycarbonate, polyamide, or silicone as mentioned above, and were previously tested for potential cytotoxicity. Fluidics ports include two elbow pieces with silicone hoses with Luer-Locks (LLs) or quick couplings for connecting the perfusion/stimulation system. There are also two female LLs located in the middle of the lid for connecting a sterile septum for cell seeding, auxiliary hoses for pressure sensing, etc. The overall view of the cultivation chamber and its sections are illustrated in
Figure 2. The technical solution is described in detail in our utility model [
24].
After assembling the chamber and ensuring its tightness, the chamber is connected to a perfusion circuit, a syringe with cultivation media and a working reservoir (described below). All the above-mentioned steps (i.e., tissue fixing, chamber assembling, connecting to perfusion circuit, etc.) are done in a biohazard box to ensure sterility. The cell seeding is then done using the sterile septum and a syringe with a needle. The chamber is vertically oriented during cultivation to minimize the risk of forming bubbles in the reservoirs. In order to provide multiple parallel sample cultivation, the chambers were daisy-chained.
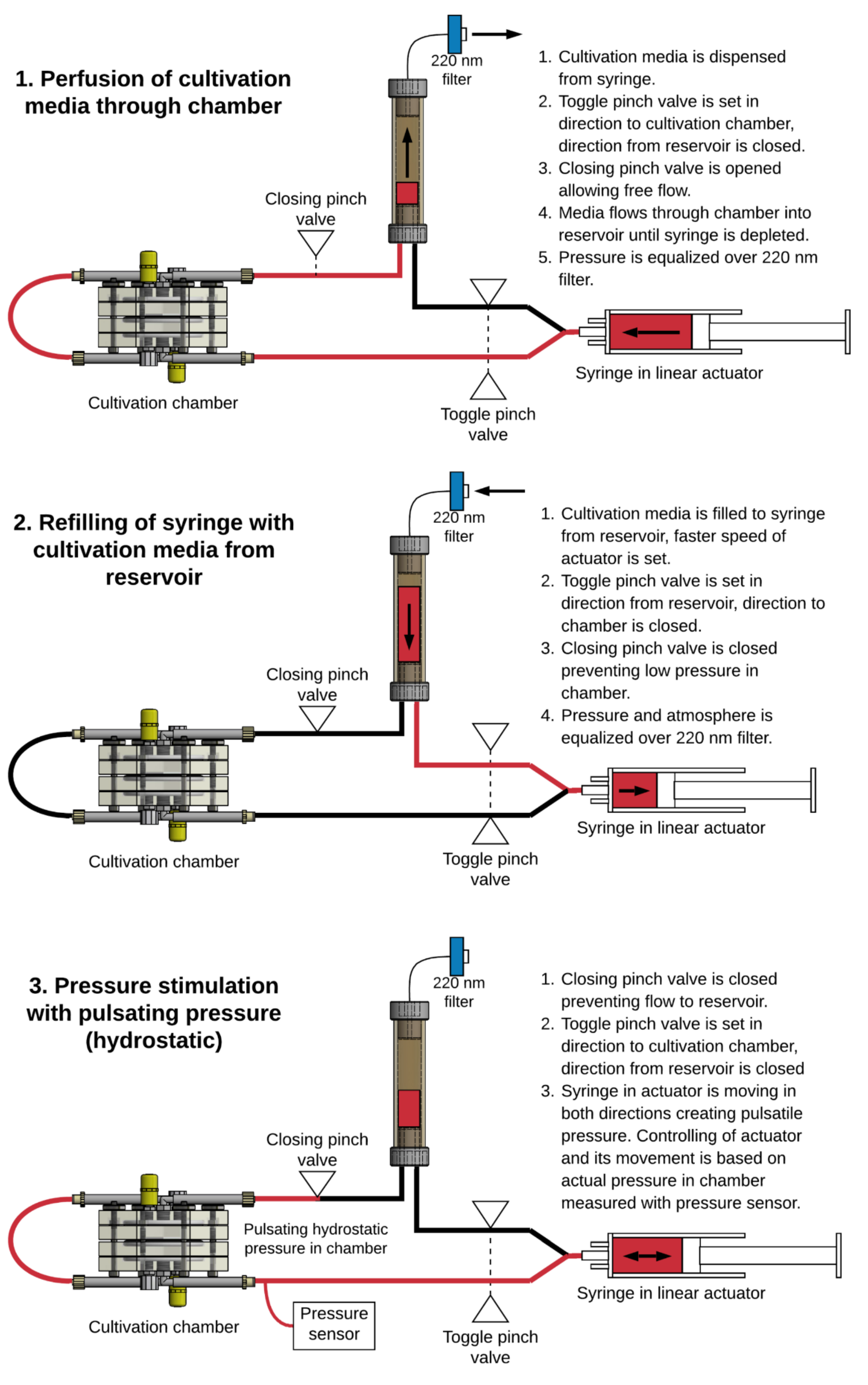
In order to maintain the perfusion of the tissue with cultivation media and to establish the stimulation of cells with pulsatile pressure waves, a unique system was also developed. This system is based on an electronically controlled linear actuator with the syringe and perfusion circuit connecting. The NI compactDAQ (National Instruments, Austin, TX, USA) platform was used with modules consisting of the digital input/output lines necessary for controlling the pump and valves (module cDAQ 9401), and with an analogue module for the pressure transducer (module cDAQ 9218). Custom LabVIEW (National Instruments, Austin, TX, USA) software was created to control the system. This software controls the movement of the linear actuator and implements a feedback control for the pressure stimulation. This linear actuator with electronics and the control software is depicted in
Figure 3. The design of the perfusion system is described in detail in our utility model [
25]. The perfusion circuit consists of a silicone tubing, a 20 mL syringe installed in the linear actuator, and a working reservoir for culture media. The perfusion/stimulation system in this study was optimized to operate in three modes. The modes of operation and the overall setup are illustrated in
Figure 4. In mode 1, perfusion of the cultivation media through the chamber, and in mode 2, refiling the syringe with cultivation media from the reservoir, the system moves at a constant speed until reaching the limit switch on the appropriate side of the movement. These modes were primarily used during the initial phase of experiments to equilibrate the pH and temperature of the cultivation media and to remove all bubbles from the cultivation chamber. Mode 3, pressure stimulation, uses feedback control. In the software, the frequency and two pressure thresholds are specified. Based on these settings, the actuator moves in both directions repeatedly until reaching high/low-pressure thresholds. The speed is adjusted to reach a specified frequency of pulses. The pressure measurement is done with a disposable pressure transducer used for measuring invasive blood pressure TruWave(Baxter, Irvine, CA, USA), which is connected to an auxiliary Luer-Lock in the cultivation chamber.
2.5. Cell Seeding and Dynamic Cultivation
Before the cells were seeded, the pH and the temperature in the cultivation system and in the chamber were equilibrated. In this phase, the culture medium was perfused through the cultivation chamber. All bubbles were also removed. This phase took about 2 h.
Then the cell suspension was prepared. The cells were seeded at a density of 90,000 cells/cm2. This density was experimentally adjusted to form a homogeneous, confluent layer on the top of the decellularized tissue. As was mentioned above, 35 × 35 mm2 squares of the tissue were fixed in the chamber. The total area for cell growth was 30 × 30 mm2; the borders were used for fixing the tissue in the holder. Thus, there were 810,000 cells on one side of the tissue, which were suspended in 500 µl of the culture medium. This cell suspension was then injected through a sterile septum into the appropriate side of the cultivation chamber. Then, the chamber was aligned horizontally, allowing the cells to adhere. During this phase, there was no perfusion and no dynamic stimulation. The cell adhesion phase took 60 min. After that, mild perfusion (flow 2 mL/min) with the culture medium was started. The perfusion lasted for 2 h. After that, the stimulation was set to 15.9/10.6 kPa (120/80 mmHg) high/low pressure with a frequency of 1 Hz (60 pulses per minute). Every 2 h, the stimulation was changed to the perfusion mode for 10 min in order to refill the reservoirs of the chamber with a fresh culture medium. This cultivation was performed for 5 days and for 10 days.
The first goal of this study was to test how well the bioreactor promotes the ingrowth of the cells through the decellularized tissue, so only one side of the tissue was seeded with cells and analyzed. The second goal was to prepare fully recellularized cardiovascular patches. When these patches were being prepared, the cells were seeded from both sides. This procedure involved the described seeding on one side. Then, the cells were left for 60 min, allowing their adhesion. After this phase, the chamber was turned and then the same seeding procedure, but from the second side, was performed, also followed by 60 min of cell adhesion.
The cultivation medium consists of DMEM/F12 with a 1:1 mixture, supplemented by 10% of fetal bovine serum, 50 μg/mL of ascorbic acid, 2.5 ng/mL of TGF-
β (Transformation growth factor β), 2.5 ng/mL of BMP4 (Bone morphological protein 4), and 1% of ABAM antibiotics. This composition was set on the basis of our previous study [
17], in order to promote both the growth and the differentiation of the ASCs towards smooth muscle cells. This culture medium was replaced every two days. During replacement, the cultivation chamber and the perfusion circuit were still fully flooded, and only the remaining medium was refilled into a syringe from the working reservoir. Then, the syringe was replaced by a new syringe containing fresh culture medium. These syringe replacements were carried out in a biohazard box to ensure sterility.
A comparative control under static cultivation conditions was performed. In order to ensure the homogeneity of the experimental setup, the second cultivation system with cultivation chambers was used. The preparation included the same preparation steps as above, until the beginning of the 2 mL/min perfusion. Instead of perfusion followed by mechanical stimulation, the system with chambers was disassembled, and the tissues were inserted into standard six-well plates. The same culture medium was used and was replaced every two days.
2.6. Evaluation of Cell Ingrowing
After observed cultivation intervals (5 and 10 days), the chambers were disconnected from the perfusion/stimulation. The chambers were then disassembled, and the tissues were removed. After that, the tissues were fixed using 4% paraformaldehyde (PFA) and were washed with PBS. Then, 20 µm thick cryosections were made on SuperFrost Pluss glass (ThermoFisher, MA, USA). The cell membranes were then permeabilized using 0.1% Triton X-100 in PBS for 15 min at room temperature (RT). The cell nuclei were counterstained with DAPI and mounted with Fluoroshield mounting medium (Sigma-Aldrich, MO, USA).
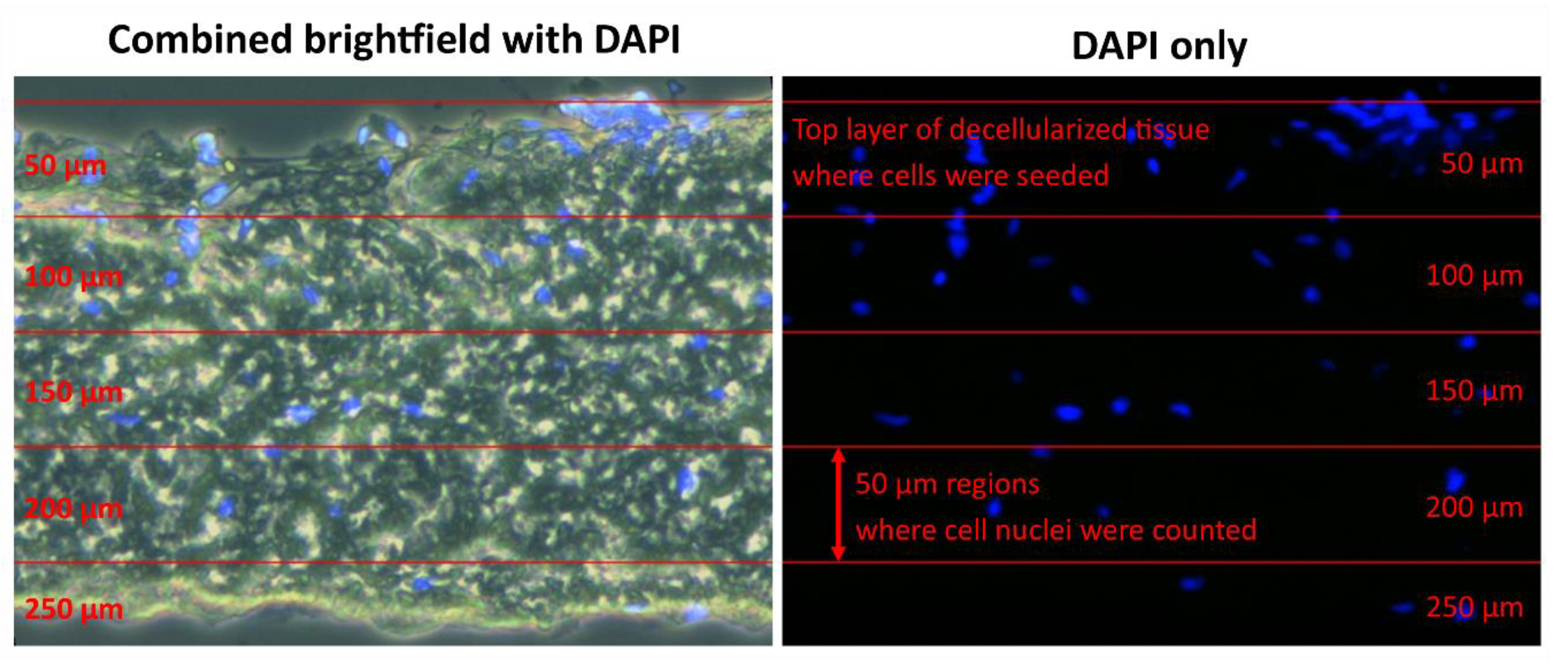
To quantify the overall cell ingrowth into the decellularized tissue, the images were sliced into 50 µm thick layers. The borders of the first layer were set from the surface of the decellularized tissue, where the cells were seeded. Then, based on the scale of the image, 50 µm layers were formed. In these layers, the cell nuclei were counted to quantify the cell ingrowth. This approach is illustrated for analysis in
Figure 5. Image processing and analysis were done in ImageJ (National Institutes of Health, Bethesda, Md, USA) and MATLAB (MathWorks, Natic, MA, USA)
To obtain relevant results, five random sections for each type of cultivation were analyzed in five randomly selected microscopic fields used for cell counting. The statistical significance was evaluated using nonparametric Kruskal-Wallis One Way Analysis of Variance on Ranks, Dunn’s Method, statistical significance (p ≤ 0.05) in MATLAB.
Immunofluorescence staining of h1-calponin, considered in various studies as an early marker or as an intermediate marker of SMC differentiation [
18], was used for evaluating the differentiation of ASCs towards SMCs. The calponin-h1 Monoclonal Antibody (CALP, MA5-11620, concentration 2 µg/mL, ThermoFisher, MA, USA), was applied as the primary antibody. The primary antibody was applied at RT for 120 min to samples in humidified chambers.
Alexa Fluor 488®-conjugated F(ab‘) fragment of goat anti-mouse IgG (H1L, Cat. No. A11017, Sigma-Aldrich; diluted in PBS + 1% BSA, concentration 1 µg/mL), together with Phalloidin conjugated with TRITC (P1951, Sigma-Aldrich; concentration 2 µg/mL), was applied as the secondary antibody, after the mouse monoclonal primary antibody. The cell nuclei were counterstained with DAPI (Thermo-Fisher, MA, USA; D1306, 300 nM concentration), which was added together with the secondary antibody. The samples were incubated with the secondary antibody at RT for 60 min in humidified chambers. Microscopic images were taken using Carl Zeiss LSM 880 NLO confocal microscope and Leica DMi-8 fluorescence microscope.
4. Discussion
The operating protocol involving the decellularization agents, such as SDS, DNAse, and deionized water, was adopted and modified according to the studies by Crapo et al. and by Gilbert et al. [
10,
11]. Many of the methods described require repeated steps involving changing decellularization agents or water after a certain period to maintain a chemical concentration gradient. Also, to improve the method and ensure homogenous decellularization, agitation in a shaker is used. These steps can be time-consuming. To ease these steps and ensure uninterrupted automation of the overall process, a novel system was built. This system implements a cyclic-based process of changing agents and washing water based on the desired setting. This approach alters the manual changing of them. Also, a special cultivation chamber was used where the tissue is fixed, and the chamber allows the flowing of used liquids to flow over the tissue, improving homogeneity.
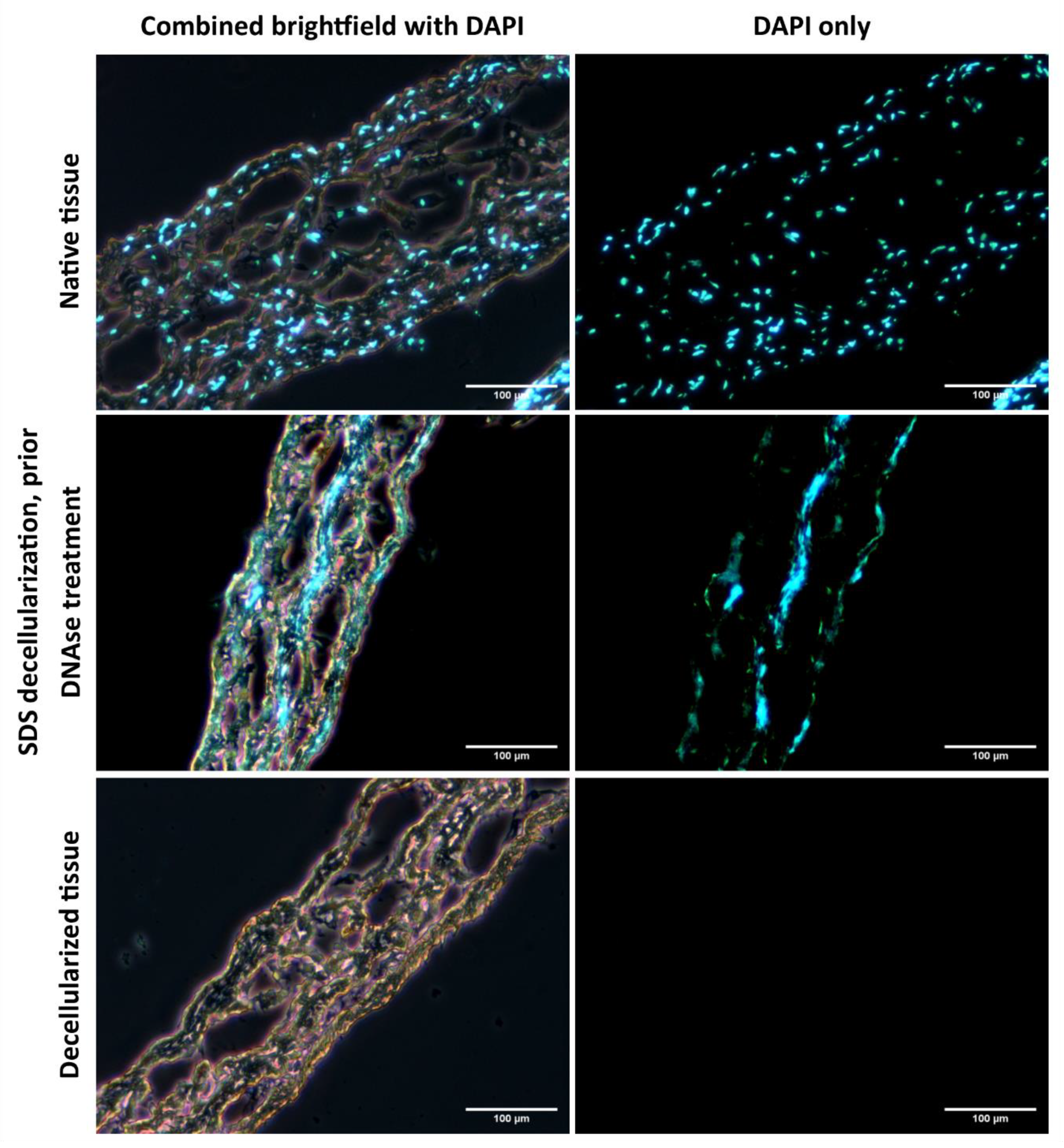
As demonstrated in
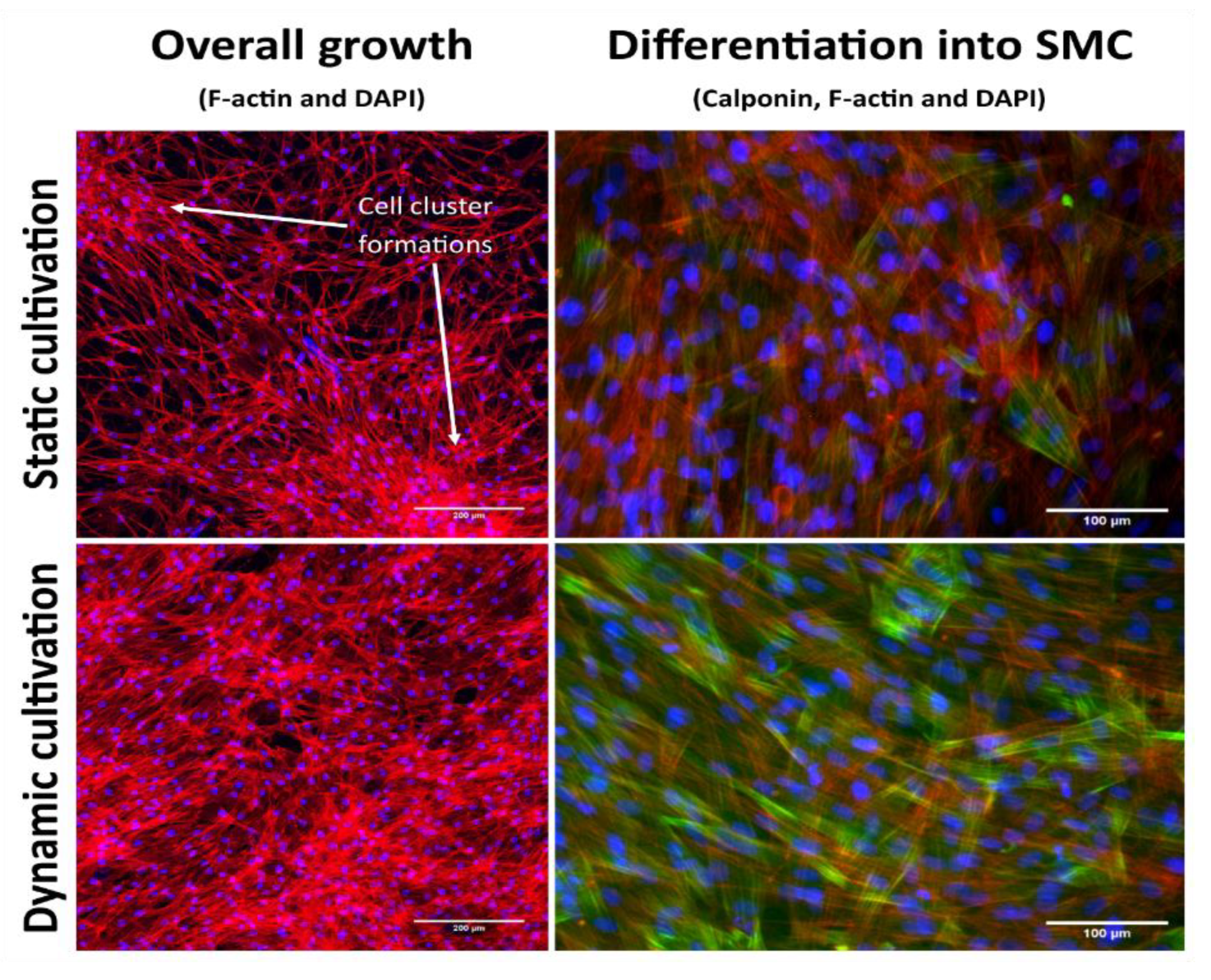
Figure 6, the native tissue has visible cell nuclei, whereas in the fully decellularized tissues there were no visible cell nuclei or DNA residues. The cell cultivation, both in the dynamic cultivation system and in static conditions, also demonstrated that the tissue decellularized by the method described here is suitable for recolonization with cells. The cells adhering to the surface of the decellularized tissue were able to penetrate the tissue. However, under static conditions, the cells formed clusters instead of a homogeneous spread on the surface of the tissue when observed under dynamic conditions, as illustrated in
Figure 9.
It can, therefore, be summarized that this protocol, implemented into our decellularization system, provided a decellularized porcine pericardium matrix that is non-toxic, sterile, and ready for further recellularization and use as an experimental cardiovascular patch. The whole process can be completed within 24 h. The decellularization chamber designed for fixing the planar tissue can prepare tissues up to 12 × 8 cm
2 in size in a single run. As was mentioned above, the porcine pericardium was harvested from a local slaughterhouse and was a cheap and easily obtainable source of tissue for validating the method. The ovine pericardium was also decellularized and was further tested to form a xenogeneic substrate with autologous cells (PrASCs) for further implantation into a porcine in vivo model to verify low immunogenicity of decellularized matrices reported in the literature [
8,
9,
26].
Cell seeding is an important component of vascular tissue engineering that decreases graft thrombogenicity and promotes graft patency, regardless of the cell type used. A possible mechanism is the recruitment of autologous cells in a paracrine manner and accelerated regeneration of the seeded matrices into a neovessel. A method of static seeding passively introduces cells onto a scaffold. The cell maturation and penetration into the scaffold is low.
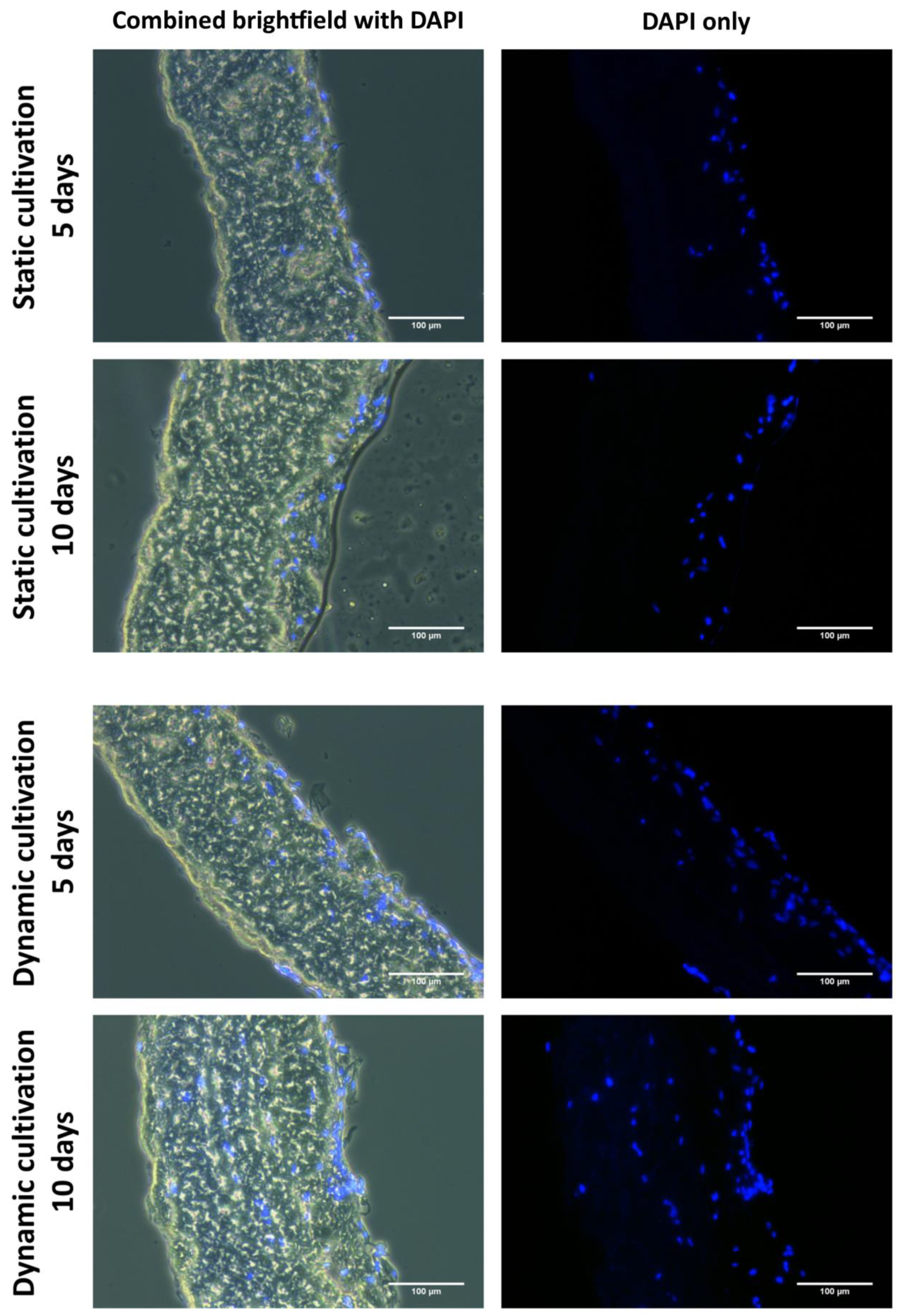
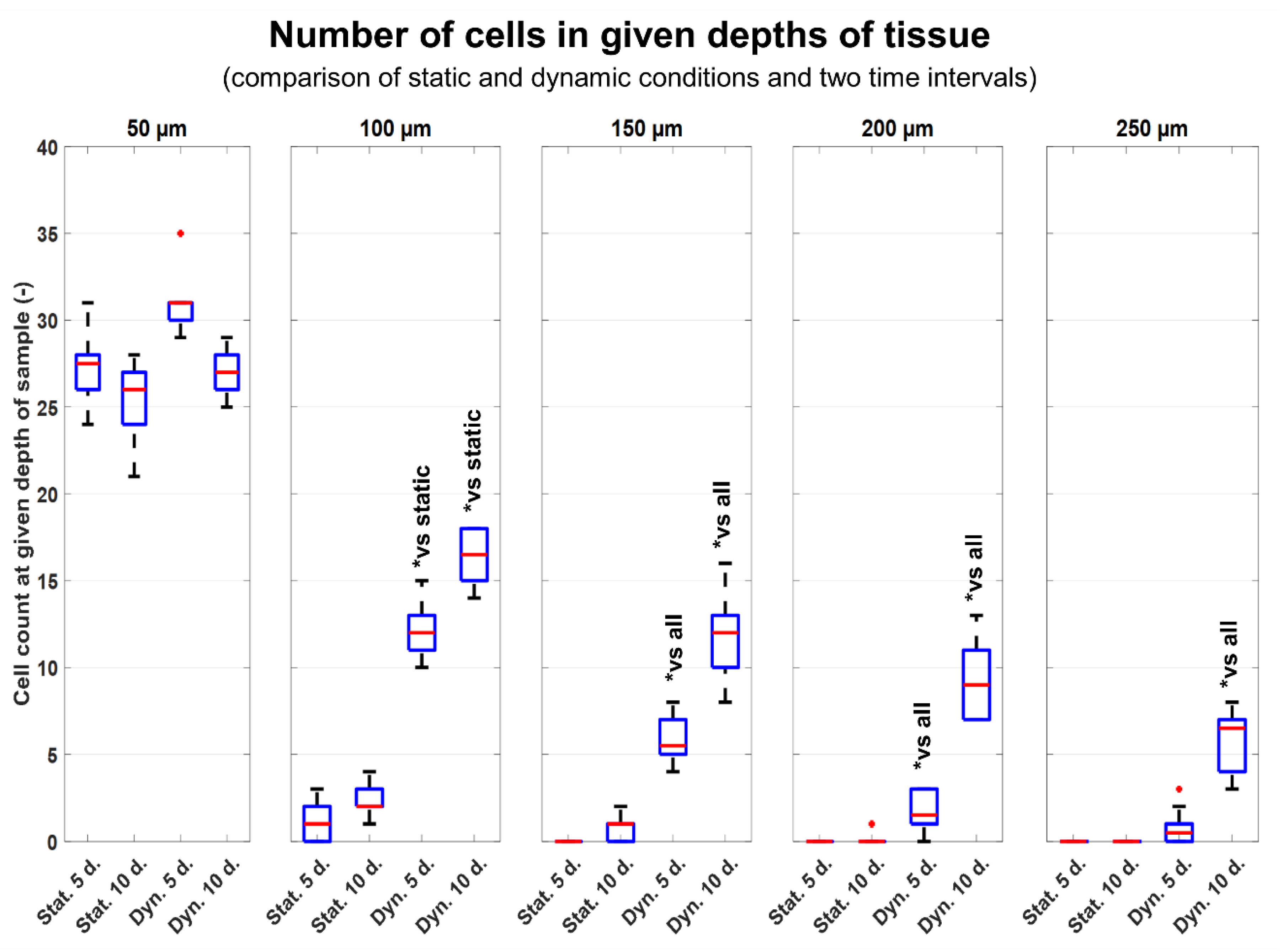
As has been described above, the process of recellularization in static conditions does not give satisfactory results. As is illustrated in
Figure 7 and
Figure 8, only 1/5 (50 µm) of the decellularized tissue was penetrated with cells after 10 days of static cultivation. Similar results have also been obtained in other studies, in which several weeks of cultivation were needed to overgrow various matrices with cells and to achieve cell maturation [
15]. The speed of the overall recellularization process is increased when perfusion bioreactors are used to provide mechanical stimulation of the cells according to studies by Melchiorri et al. [
27]. Also, many studies have investigated the correlation between pressure strain, cell proliferation, and stem cell differentiation towards SMCs. Cyclic physiological pressure (CP) of 120/80 torr altered the cellular morphology and increased the proliferation rates in stem cells over a 7-day period as described in the study by Maul et al. [
28]. Moreover, dynamic seeding using a bioreactor enables preconditioning of the cells under more physiological conditions, as described in the study by Villalona et al. [
15].
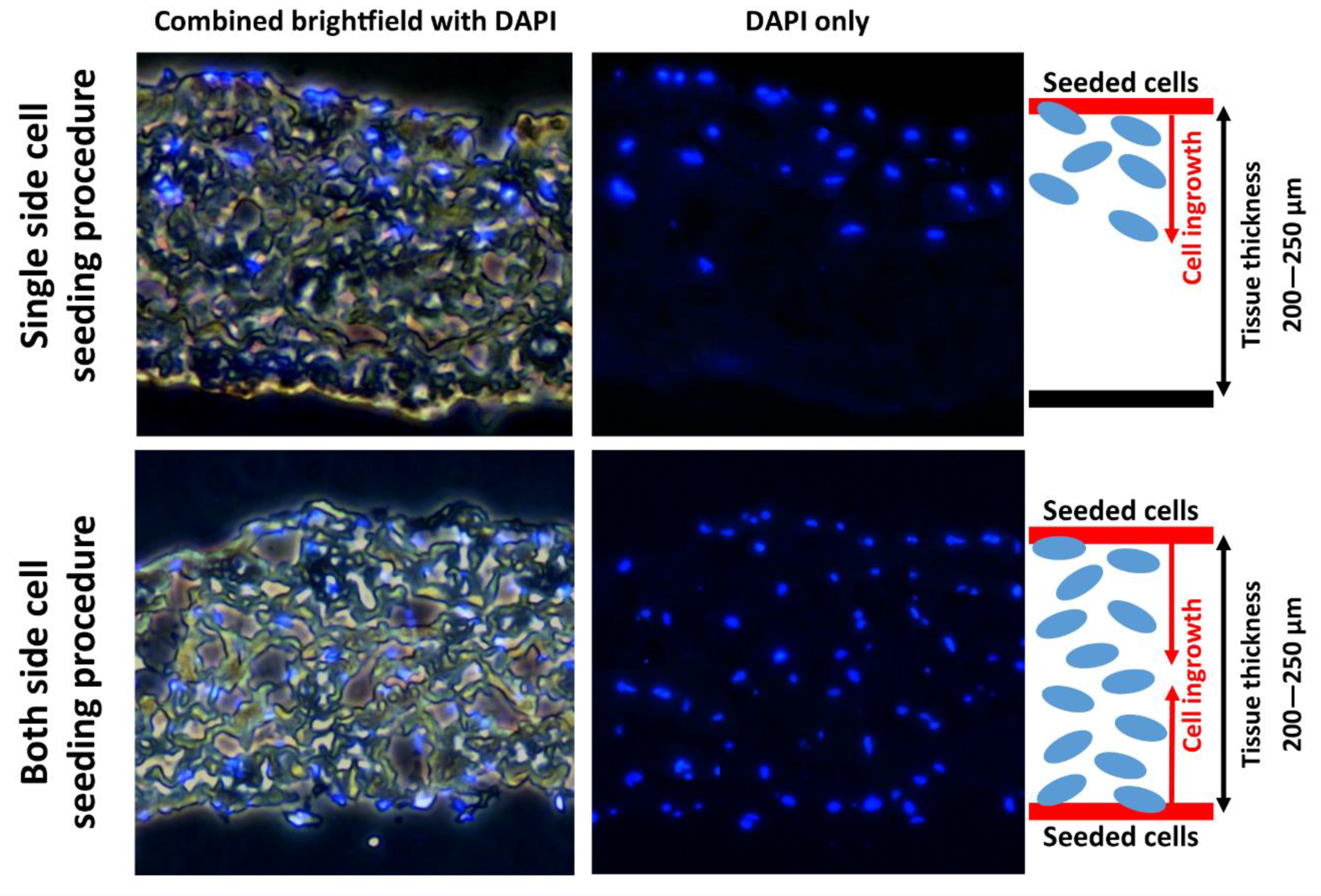
A custom dynamic cultivation system built in our laboratory was used in this study. This system generates a microperfusion flow and pulsatile hydrostatic pressure. As mentioned above, in static conditions, the cells penetrated only in 50 µm depth. The tissues cultivated in dynamic conditions proved significantly better penetration rates of cells. Significant results are observable from 100 µm depths. At depths >150 µm and more, there is nearly no evidence of cells ingrowing in static conditions. As is illustrated in
Figure 7 and
Figure 8 after 10 days of dynamic cultivation, most of the tissue has been newly recellularized with cells. As was mentioned, we tried to demonstrate the efficiency of a dynamic cultivation bioreactor in stimulating cell ingrowth inside the decellularized tissue. So, the technique of seeding utilized only one side of the decellularized tissue. Thus, the number of cells decreased with the penetration depth. To promote more homogenous ingrowing of cells, the cultivation chamber was optimized for dual side seeding of cells. In this approach, both sides of decellularized tissues were seeded with the same density of cells prior to cultivation. As is illustrated in
Figure 11 and
Figure 12, the cells are homogeneously distributed inside the recellularized tissue. Also, the time period used to achieve this was reduced to 5 days.
Also, the differentiation of ASCs towards the SMC phenotype can be achieved via biochemical and mechanical stimulation. A positive effect on the differentiation of stem cells towards SMCs has been demonstrated in studies using a combination of TGF-ß and BMP4 growth factors. This led to an increase in the production of specific cytoskeletal protein markers of differentiation, such as α-actin and h1-calponin, which are present only in differentiated SMCs [
29], after 7 and more days of static cultivation. Cell differentiation towards the SMC phenotype can be further enhanced by appropriate mechanical stimulation in a dynamic cell culture system. The positive effect of mechanical stimulation on differentiation towards SMCs has been described for cells cultured either with or without the presence of TGF-ß and BMP4 [
17,
30]. In addition, mechanical stimulation helps to keep the cells in a differentiated state and prevents de-differentiation [
31].
As for mechanical stimulation, we applied physiological hemodynamic force, which is stimulation with cyclic hydrostatic pressure (CP) 120/80 mmHg. The CP induced differentiation of the seeded adipose-derived stromal cells towards the SMC phenotype. This was proven by the expression of an SMC marker calponin. Greater cell population density and greater upregulation of early differentiation markers α-actin and calponin were achieved in a pressure-stimulated culture than in static conditions [
18,
32]. In this context, two mechanosensitive signaling pathways associated with SMC differentiation have been described: the RhoA-associated pathway, and the FAK kinase-associated pathway. Activation of these signaling pathways leads to increased synthesis of SMC differentiation markers in the cells [
33].
An analysis of the growth and differentiation of cells on the surface of the decellularized matrix showed that there is a major difference between static cultivation and dynamic cultivation after 10 days of cultivation. As shown in
Figure 9, under static conditions, the cells form clusters instead of spreading homogeneously over the entire surface. The signal from h1-calponin is also weaker and is present in a smaller number of cells. Under dynamic conditions, however, a homogeneous cell layer is formed, and h1-calponin is present in most of the cells, including cells with well-developed calponin fibers. In this case, increased cell growth and overall improved growth of cells on the surface of decellularized tissue can also be observed under dynamic culture conditions.
Analogous to our study, Kobayashi et al. found upregulated expression of SMC markers, alpha smooth muscle actin (aSMA), and smooth muscle myosin heavy chain (SMMHC) in rat bone marrow stromal cells after exposure to pressure-dominant forces or combined flow and pressure forces for 3 days [
34]. The authors attribute this differentiation to longer pre-incubation periods (7–21 days). The effects of different types of mechanical stimulation in isolation cyclic strain and cyclic pressure, including cyclic pressure alone, were previously studied in detail in rat bone marrow mesenchymal stem cells plated on collagen type-I coated culture slips [
28,
35]. In contrast to our study, they found no SMC markers (including no calponin) via immunohistochemistry after 5 days of CP of different magnitudes. Although the cells were increasing in number under CP in the study by Maul et al. [
35], in our study, we initially seeded the cells in a much higher density (90,000 cells/cm
2 vs. 200 cells/cm
2) to achieve confluence on a 3D biological material—decellularized pericardium. Our setting most probably resulted in simultaneous signaling from more sources: cell-cell contacts, cell-material contacts (natural matrix environment), soluble factors (TGF-β1 and BMP-4), and the mechanical load. These stimuli create synergy in conditioning the cells [
36]. This may explain the fact that the cells differentiated towards the intended SMC-phenotype in our system. Additionally, the cells penetrated through the entire wall of the matrix, thus resembling a newly formed vascular tunica media. Cyclic pressure is a known factor that induces the proliferation of several cell types (EC, SMC and MSC) [
28,
35,
37]. We believe that these are desired events with respect to vascular tissue engineering and future implantation of this matrix as a cell-seeded cardiovascular patch.
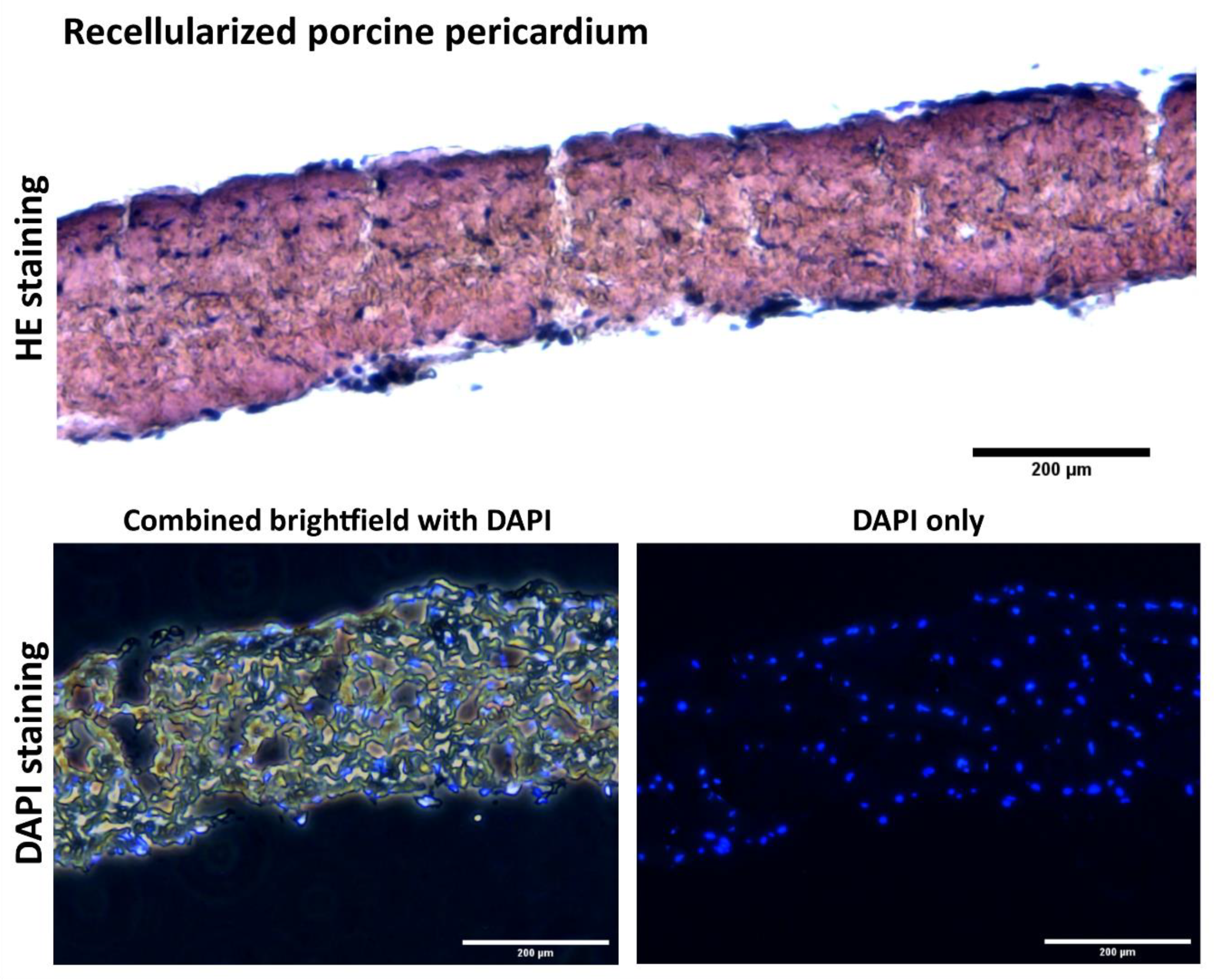
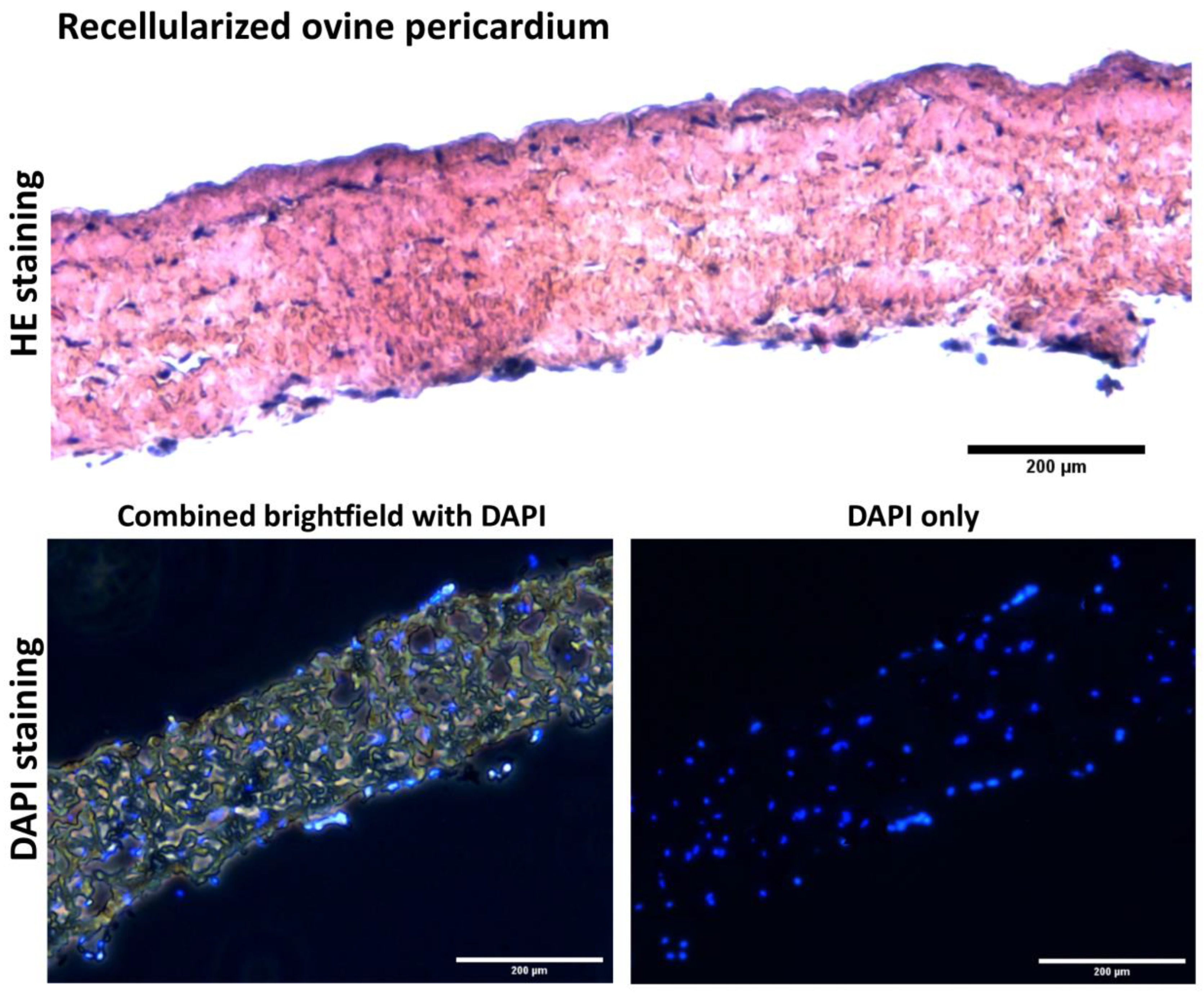
In a future study, we plan to prepare autologous cell-seeded cardiovascular patches based on the protocol described here. Cultivation substrate will involve both autologous (porcine)
Figure 11, and xenogenous (ovine)
Figure 12 decellularized tissues as a substrate. In this planned study, the animals (Prestice pigs) will undergo two surgeries—the first surgery for fat tissue extraction and ASC isolation, and the second surgery in which a patch will be implanted onto an experimental carotid artery defect. Based on the protocol presented here, the time needed for preparing the recellularized autologous patch will be approximately 8–10 days for preparing the autologous ASCs and then 5 days of cultivation in the dynamic bioreactor to prepare the recellularized autologous seeded grafts.

 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}