
Aryl Hydrocarbon Receptor (AhR) Limits the Inflammatory Responses in Human Lung Adenocarcinoma A549 Cells via Interference with NF-κB Signaling

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Line
2.3. Generation of A549 Knock-Out Cells
2.4. Western Blotting
2.5. Luciferase Gene Reporter Assay
2.6. Real-Time Quantitative RT-PCR (RT-qPCR)
2.7. Liquid Chromatography-Tandem Mass Spectrometry (LC-MS/MS)
2.8. Enzyme-Linked Immunosorbent Assays (ELISA)
2.9. Statistical Analyses
3. Results
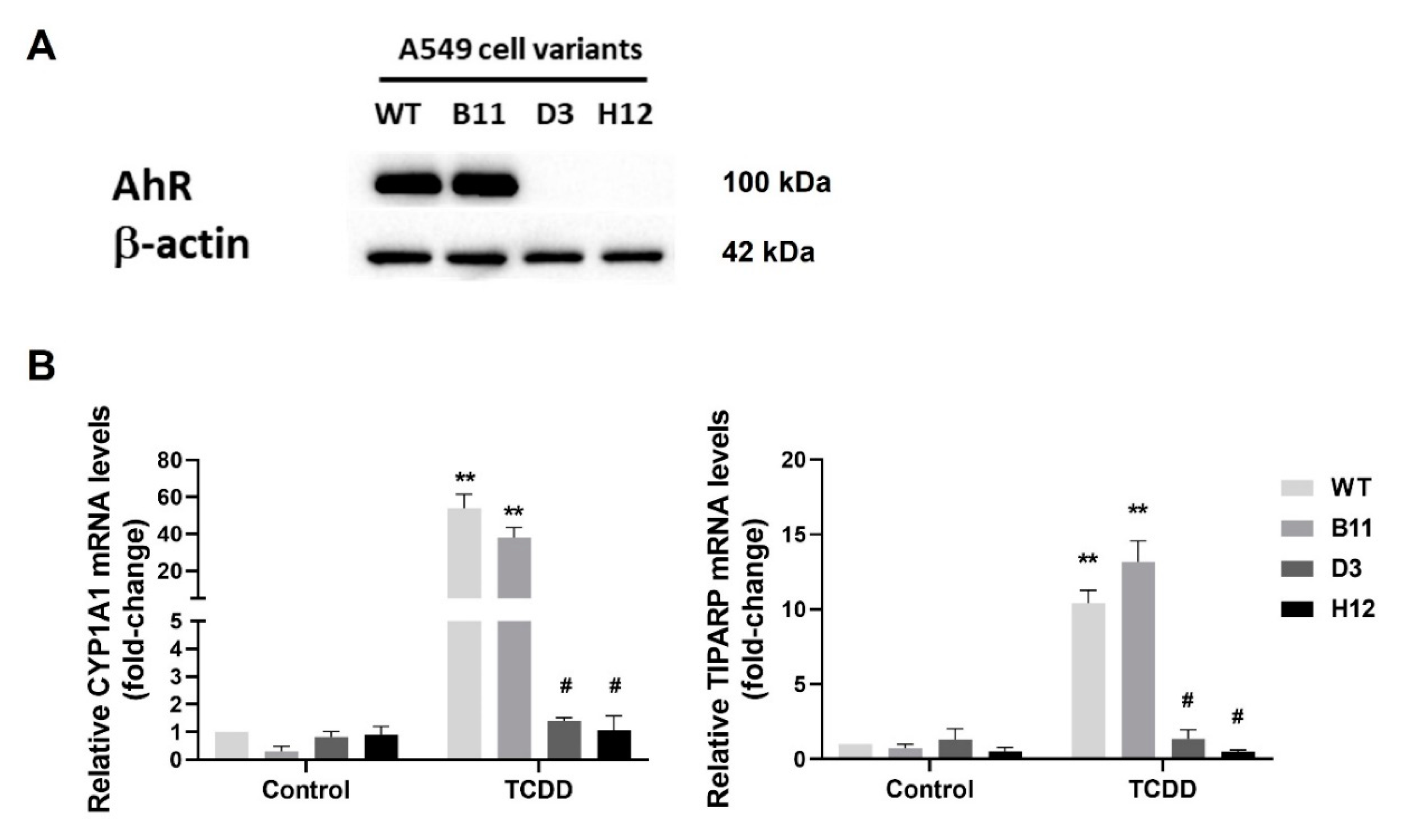
3.1. Generation of the AhR Knockout Cells by CRISPR/Cas9
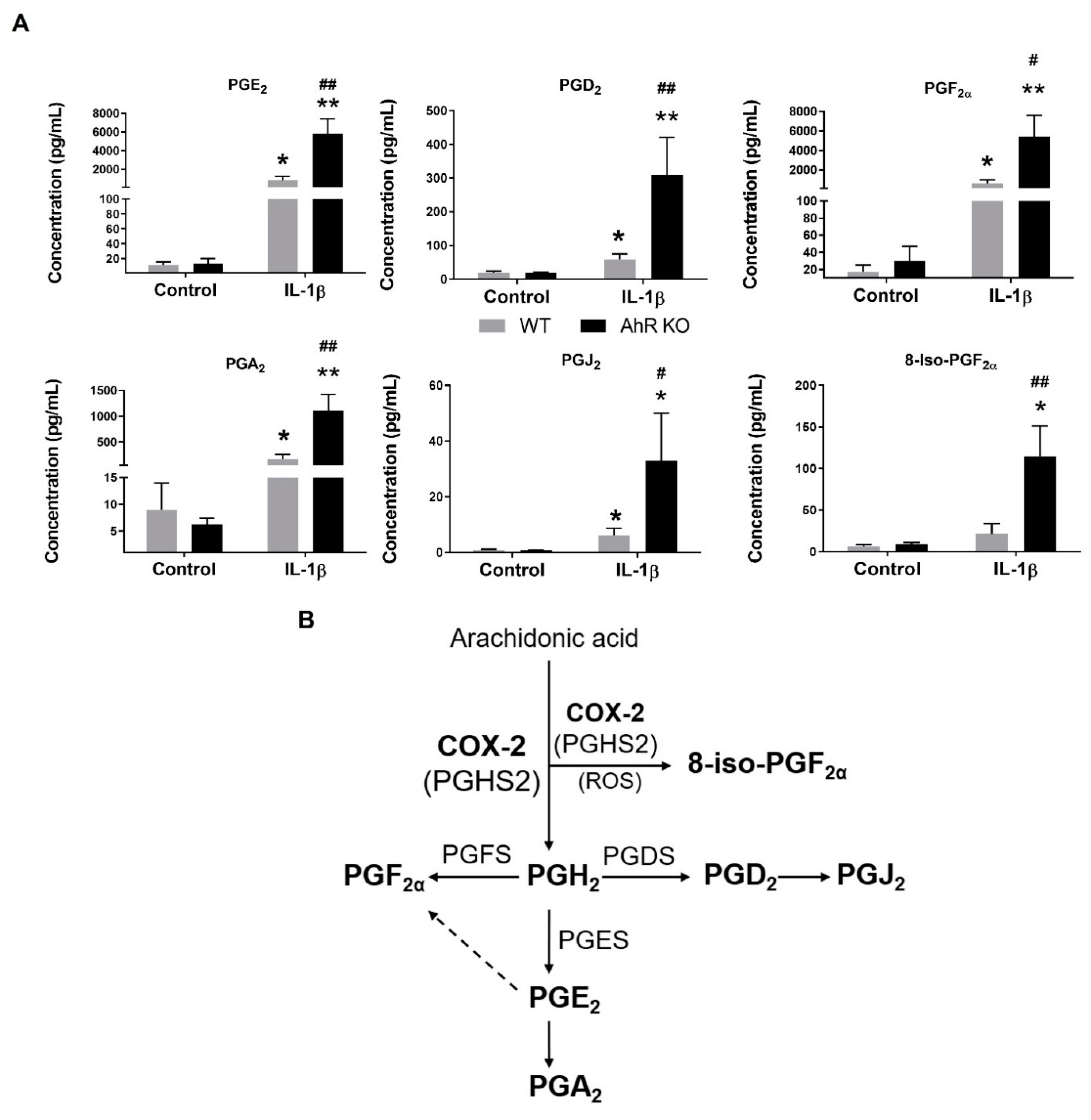
3.2. AhR Deficiency Increases Production of Prostaglandins in A549 Cells
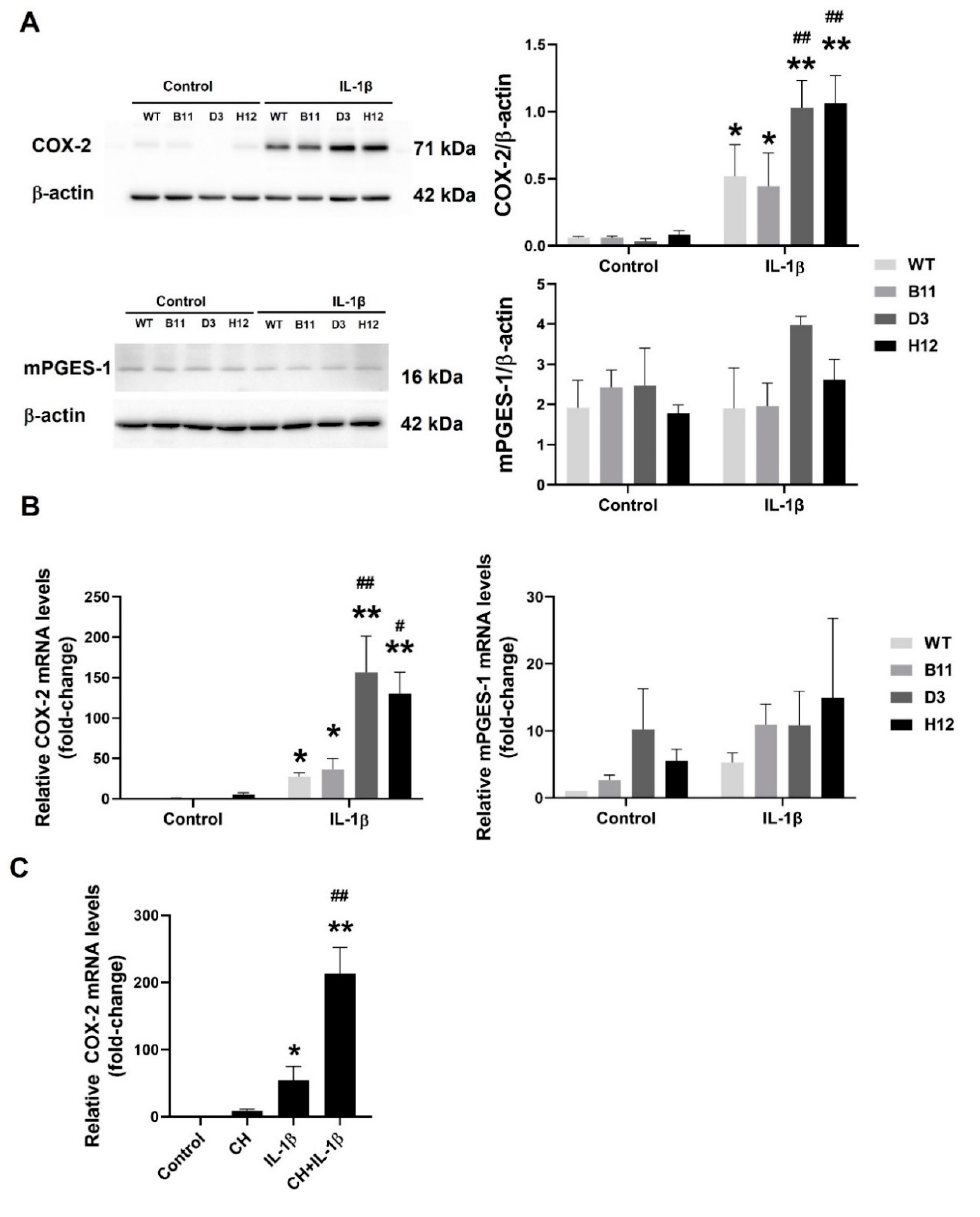
3.3. The AhR Disruption Increases Inducibility of COX-2 in A549 Cells
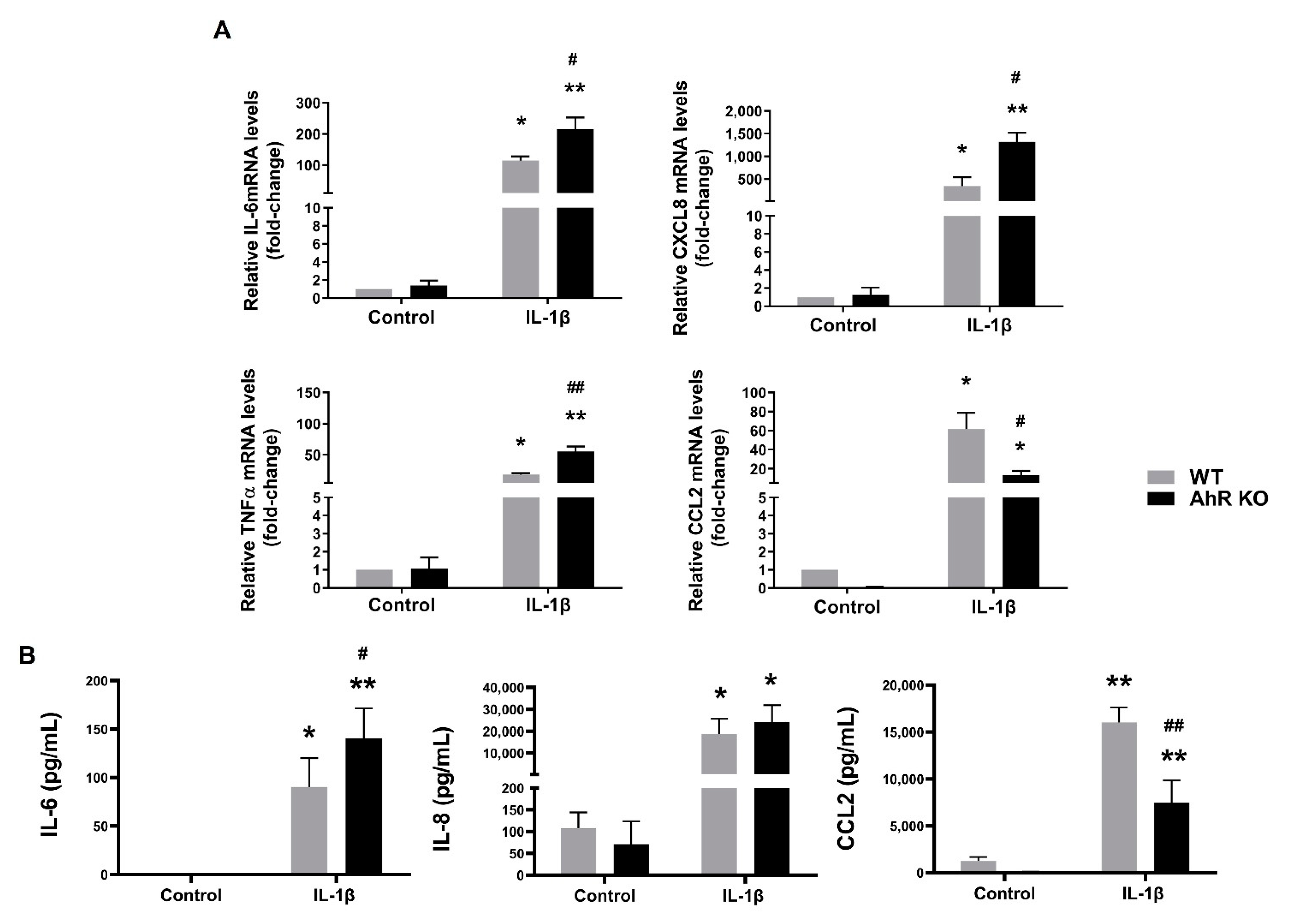
3.4. Disruption of the AhR Signaling Leads to an Increased Production of Some Pro-Inflammatory Cytokines in A549 Cells
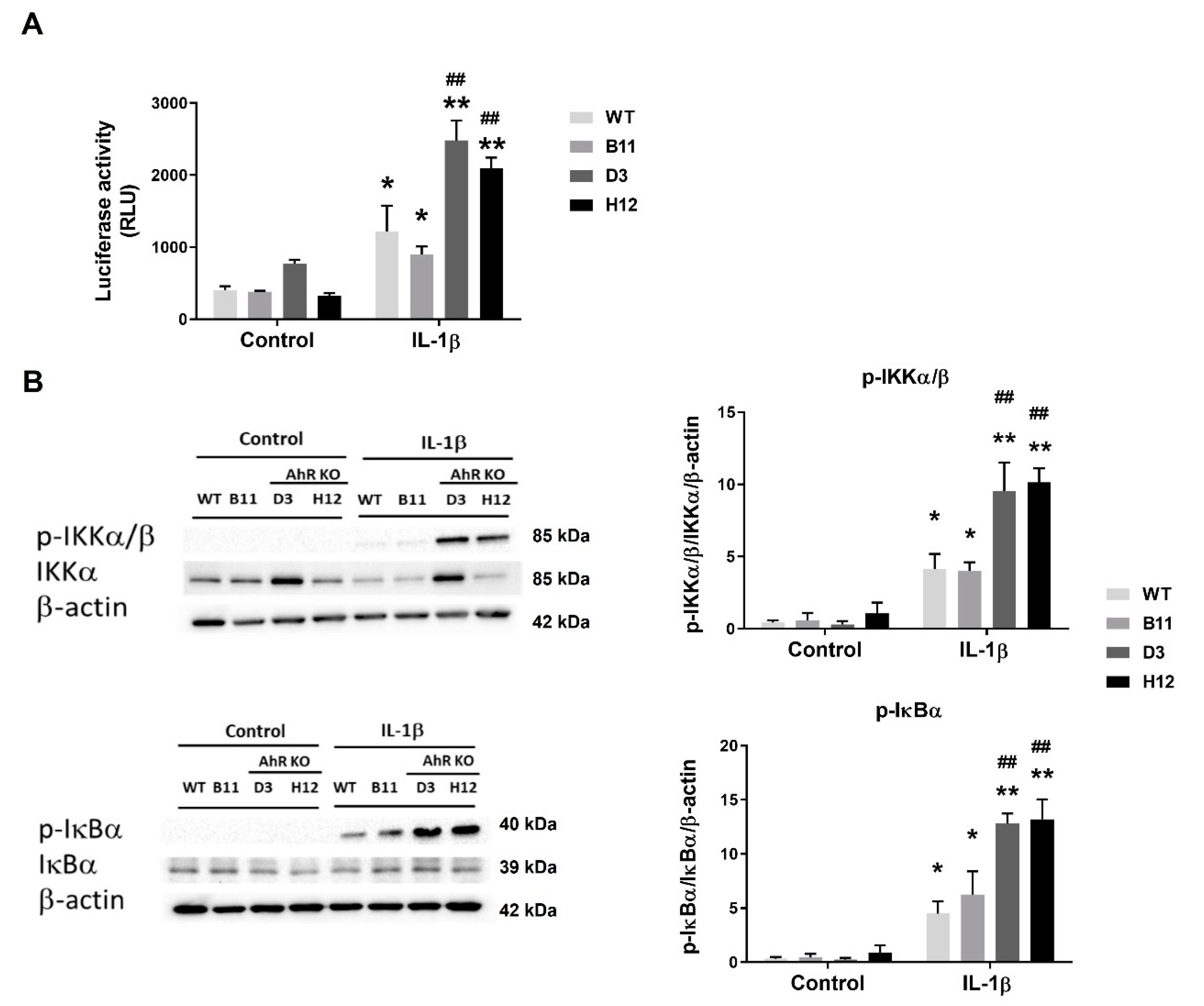
3.5. Disruption of the AhR Pathway Promotes NF-κB Activity in A549 Cells
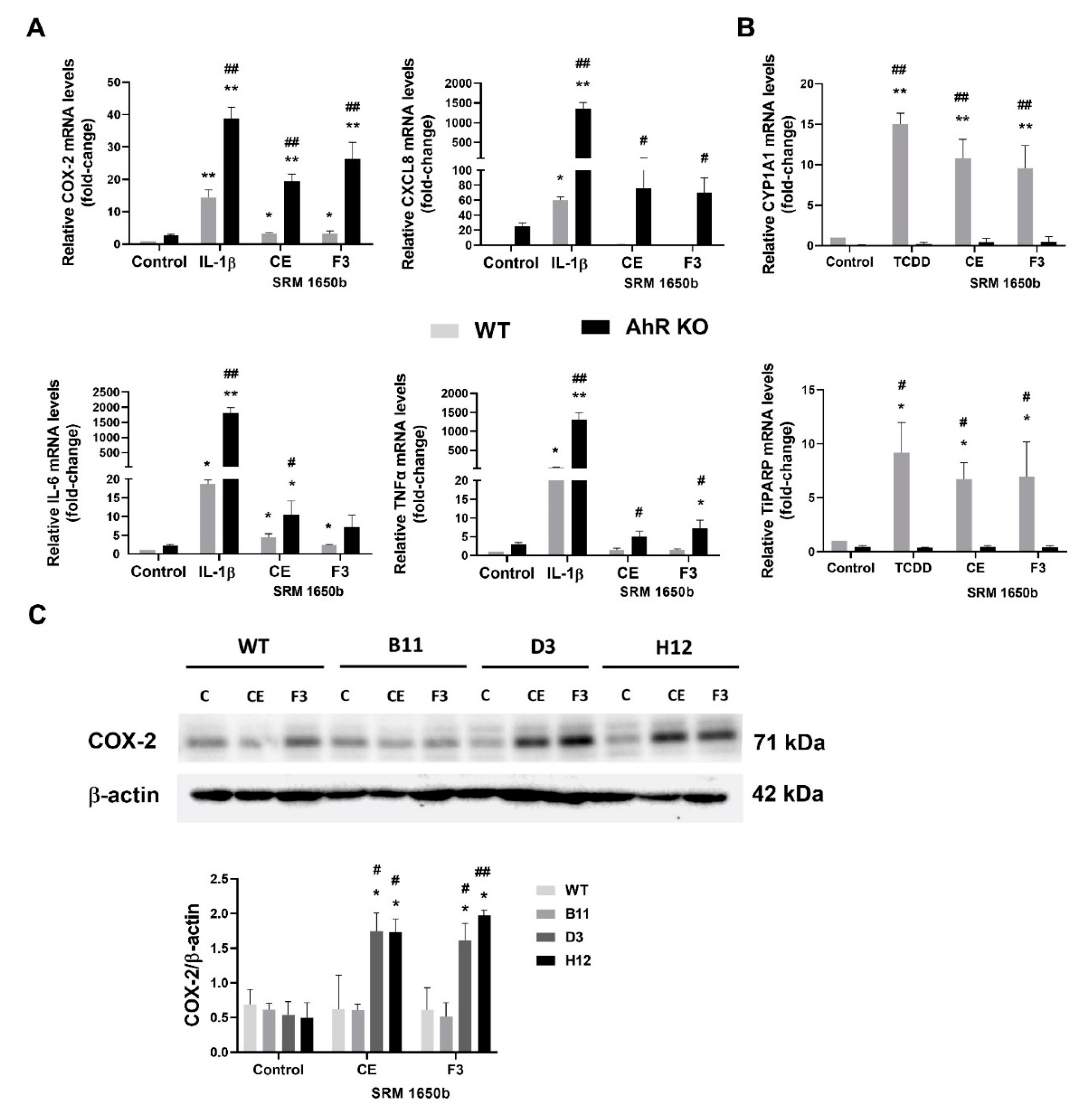
3.6. DEP Extract and Its Polar Fraction Induce Exacerbated Inflammatory Response in A549 AhR KO Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Traboulsi, H.; Guerrina, N.; Iu, M.; Maysinger, D.; Ariya, P.; Baglole, C.J. Inhaled pollutants: The molecular scene behind respiratory and systemic diseases associated with ultrafine particulate matter. Int. J. Mol. Sci. 2017, 18, 243. [Google Scholar] [CrossRef] [PubMed]
- Adler, K.B.; Fischer, B.M.; Wright, D.T.; Cohn, L.A.; Becker, S. Interactions between respiratory epithelial cells and cytokines: Relationships to lung inflammation. Ann. N. Y. Acad. Sci. 1994, 725, 128–145. [Google Scholar] [CrossRef] [PubMed]
- Nova, Z.; Skovierova, H.; Strnadel, J.; Halasova, E.; Calkovska, A. Short-term versus long-term culture of A549 cells for evaluating the effects of lipopolysaccharide on oxidative stress, surfactant proteins and cathelicidin LL-37. Int. J. Mol. Sci. 2020, 21, 1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehrenbach, H. Alveolar epithelial type II cell: Defender of the alveolus revisited. Respir. Res. 2001, 2, 33–46. [Google Scholar] [CrossRef]
- Guillot, L.; Nathan, N.; Tabary, O.; Thouvenin, G.; Le Rouzic, P.; Corvol, H.; Amselem, S.; Clement, A. Alveolar epithelial cells: Master regulators of lung homeostasis. Int. J. Biochem. Cell Biol. 2013, 45, 2568–2573. [Google Scholar] [CrossRef]
- Paine, R., 3rd; Rolfe, M.W.; Standiford, T.J.; Burdick, M.D.; Rollins, B.J.; Strieter, R.M. MCP-1 expression by rat type II alveolar epithelial cells in primary culture. J. Immunol. 1993, 150, 4561–4570. [Google Scholar]
- Standiford, T.J.; Kunkel, S.L.; Phan, S.H.; Rollins, B.J.; Strieter, R.M. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 1991, 266, 9912–9918. [Google Scholar] [CrossRef]
- Vanderbilt, J.N.; Mager, E.M.; Allen, L.; Sawa, T.; Wiener-Kronish, J.; Gonzalez, R.; Dobbs, L.G. CXC chemokines and their receptors are expressed in type II cells and upregulated following lung injury. Am. J. Respir. Cell Mol. Biol. 2003, 29, 661–668. [Google Scholar] [CrossRef]
- Esser, C.; Rannug, A. The aryl hydrocarbon receptor in barrier organ physiology, immunology, and toxicology. Pharmacol. Rev. 2015, 67, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Guarnieri, T.; Abruzzo, P.M.; Bolotta, A. More than a cell biosensor: Aryl hydrocarbon receptor at the intersection of physiology and inflammation. Am. J. Physiol. Cell Physiol. 2020, 318, C1078–C1082. [Google Scholar] [CrossRef] [PubMed]
- Baglole, C.J.; Maggirwar, S.B.; Gasiewicz, T.A.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J. Biol. Chem. 2008, 283, 28944–28957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico de Souza, A.; Zago, M.; Eidelman, D.H.; Hamid, Q.; Baglole, C.J. Aryl hydrocarbon receptor (AhR) attenuation of subchronic cigarette smoke-induced pulmonary neutrophilia is associated with retention of nuclear RelB and suppression of intercellular adhesion molecule-1 (ICAM-1). Toxicol. Sci. 2014, 140, 204–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Acosta, O.; Vega, L.; Estrada-Muniz, E.; Rodriguez, M.S.; Gonzalez, F.J.; Elizondo, G. Activation of aryl hydrocarbon receptor regulates the LPS/IFNgamma-induced inflammatory response by inducing ubiquitin-proteosomal and lysosomal degradation of RelA/p65. Biochem. Pharmacol. 2018, 155, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.H.; Chang, H.; Chang, J.T.; Lin, P. Aryl hydrocarbon receptor in association with RelA modulates IL-6 expression in non-smoking lung cancer. Oncogene 2012, 31, 2555–2565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martey, C.A.; Baglole, C.J.; Gasiewicz, T.A.; Sime, P.J.; Phipps, R.P. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 289, L391–L399. [Google Scholar] [CrossRef] [PubMed]
- Rogers, S.; de Souza, A.R.; Zago, M.; Iu, M.; Guerrina, N.; Gomez, A.; Matthews, J.; Baglole, C.J. Aryl hydrocarbon receptor (AhR)-dependent regulation of pulmonary miRNA by chronic cigarette smoke exposure. Sci. Rep. 2017, 7, 40539. [Google Scholar] [CrossRef] [Green Version]
- Thatcher, T.H.; Maggirwar, S.B.; Baglole, C.J.; Lakatos, H.F.; Gasiewicz, T.A.; Phipps, R.P.; Sime, P.J. Aryl hydrocarbon receptor-deficient mice develop heightened inflammatory responses to cigarette smoke and endotoxin associated with rapid loss of the nuclear factor-kappaB component RelB. Am. J. Pathol. 2007, 170, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.; Sciullo, E.; Matsumura, F. Activation of inflammatory mediators and potential role of ah-receptor ligands in foam cell formation. Cardiovasc. Toxicol. 2004, 4, 363–373. [Google Scholar] [CrossRef]
- Zago, M.; Sheridan, J.A.; Nair, P.; Rico de Souza, A.; Gallouzi, I.E.; Rousseau, S.; Di Marco, S.; Hamid, Q.; Eidelman, D.H.; Baglole, C.J. Aryl hydrocarbon receptor-dependent retention of nuclear HuR suppresses cigarette smoke-induced cyclooxygenase-2 expression independent of DNA-binding. PLoS ONE 2013, 8, e74953. [Google Scholar] [CrossRef] [Green Version]
- Zago, M.; Sheridan, J.A.; Traboulsi, H.; Hecht, E.; Zhang, Y.; Guerrina, N.; Matthews, J.; Nair, P.; Eidelman, D.H.; Hamid, Q.; et al. Low levels of the AhR in chronic obstructive pulmonary disease (COPD)-derived lung cells increases COX-2 protein by altering mRNA stability. PLoS ONE 2017, 12, e0180881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rico de Souza, A.; Traboulsi, H.; Wang, X.; Fritz, J.H.; Eidelman, D.H.; Baglole, C.J. The aryl hydrocarbon receptor attenuates acute cigarette smoke-induced airway neutrophilia independent of the dioxin response element. Front. Immunol. 2021, 12, 630427. [Google Scholar] [CrossRef] [PubMed]
- Brinchmann, B.C.; Skuland, T.; Rambol, M.H.; Szoke, K.; Brinchmann, J.E.; Gutleb, A.C.; Moschini, E.; Kubatova, A.; Kukowski, K.; Le Ferrec, E.; et al. Lipophilic components of diesel exhaust particles induce pro-inflammatory responses in human endothelial cells through AhR dependent pathway(s). Part. Fibre Toxicol. 2018, 15, 21. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, C.A.; Owens, L.A.; Gallo, M.E.; Hoffmann, E.J.; Afrazi, A.; Han, M.; Fechner, J.H.; Schauer, J.J.; Bradfield, C.A.; Mezrich, J.D. Differential effects of diesel exhaust particles on T cell differentiation and autoimmune disease. Part. Fibre Toxicol. 2018, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Schwarze, P.E.; Totlandsdal, A.I.; Lag, M.; Refsnes, M.; Holme, J.A.; Ovrevik, J. Inflammation-related effects of diesel engine exhaust particles: Studies on lung cells in vitro. BioMed Res. Int. 2013, 2013, 685142. [Google Scholar] [CrossRef] [Green Version]
- Standard Reference Material (SRM) 1650b; Diesel Particulate Matter. Certificate of Analysis. National Institute of Standards and Technology: Gaithersburg, MD, USA, 2006.
- Andrysík, Z.; Vondráček, J.; Marvanová, S.; Ciganek, M.; Neča, J.; Pěnčíková, K.; Mahadevan, B.; Topinka, J.; Baird, W.M.; Kozubík, A.; et al. Activation of the aryl hydrocarbon receptor is the major toxic mode of action of an organic extract of a reference urban dust particulate matter mixture: The role of polycyclic aromatic hydrocarbons. Mutat. Res. 2011, 714, 53–62. [Google Scholar] [CrossRef]
- Ciganek, M.; Neča, J.; Adamec, V.; Janošek, J.; Machala, M. A combined chemical and bioassay analysis of traffic-emitted polycyclic aromatic hydrocarbons. Sci. Total Environ. 2004, 334–335, 141–148. [Google Scholar] [CrossRef]
- Rynning, I.; Neča, J.; Vrbová, K.; Líbalová, H.; Rössner, P., Jr.; Holme, J.A.; Gutzkow, K.B.; Afanou, A.K.J.; Arnoldussen, Y.J.; Hruba, E.; et al. In vitro transformation of human bronchial epithelial cells by diesel exhaust particles: Gene expression profiling and early toxic responses. Toxicol. Sci. 2018, 166, 51–64. [Google Scholar] [CrossRef] [Green Version]
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef]
- van ‘t Erve, T.J.; Lih, F.B.; Kadiiska, M.B.; Deterding, L.J.; Eling, T.E.; Mason, R.P. Reinterpreting the best biomarker of oxidative stress: The 8-iso-PGF(2alpha)/PGF(2alpha) ratio distinguishes chemical from enzymatic lipid peroxidation. Free Radic. Biol. Med. 2015, 83, 245–251. [Google Scholar] [CrossRef] [Green Version]
- Moreno, J.J. New aspects of the role of hydroxyeicosatetraenoic acids in cell growth and cancer development. Biochem. Pharmacol. 2009, 77, 1–10. [Google Scholar] [CrossRef]
- Seo, M.J.; Oh, D.K. Prostaglandin synthases: Molecular characterization and involvement in prostaglandin biosynthesis. Prog. Lipid Res. 2017, 66, 50–68. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilera-Montilla, N.; Chamorro, S.; Nieto, C.; Sanchez-Cabo, F.; Dopazo, A.; Fernandez-Salguero, P.M.; Rodriguez-Fernandez, J.L.; Pello, O.M.; Andres, V.; Cuenda, A.; et al. Aryl hydrocarbon receptor contributes to the MEK/ERK-dependent maintenance of the immature state of human dendritic cells. Blood 2013, 121, e108–e117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, I.; Tatebe, J.; Namba, S.; Koizumi, M.; Yamazaki, J.; Morita, T. Activation of aryl hydrocarbon receptor mediates indoxyl sulfate-induced monocyte chemoattractant protein-1 expression in human umbilical vein endothelial cells. Circ. J. 2013, 77, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [Green Version]
- Dimitrakopoulos, F.D.; Kottorou, A.E.; Kalofonou, M.; Kalofonos, H.P. The fire within: NF-kappaB involvement in non-small cell lung cancer. Cancer Res. 2020, 80, 4025–4036. [Google Scholar] [CrossRef]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: Evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-κB activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Silverman, N.; Maniatis, T. NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev. 2001, 15, 2321–2342. [Google Scholar] [CrossRef] [Green Version]
- Låg, M.; Øvrevik, J.; Refsnes, M.; Holme, J.A. Potential role of polycyclic aromatic hydrocarbons in air pollution-induced non-malignant respiratory diseases. Respir. Res. 2020, 21, 299. [Google Scholar] [CrossRef] [PubMed]
- Nemmar, A.; Holme, J.A.; Rosas, I.; Schwarze, P.E.; Alfaro-Moreno, E. Recent advances in particulate matter and nanoparticle toxicology: A review of the in vivo and in vitro studies. BioMed Res. Int. 2013, 2013, 279371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øvrevik, J.; Refsnes, M.; Låg, M.; Brinchmann, B.C.; Schwarze, P.E.; Holme, J.A. Triggering mechanisms and inflammatory effects of combustion exhaust particles with implication for carcinogenesis. Basic Clin. Pharmacol. Toxicol. 2017, 121 (Suppl. 3), 55–62. [Google Scholar] [CrossRef] [Green Version]
- Pěnčíková, K.; Ciganek, M.; Neča, J.; Illés, P.; Dvořák, Z.; Vondráček, J.; Machala, M. Modulation of endocrine nuclear receptor activities by polyaromatic compounds present in fractionated extracts of diesel exhaust particles. Sci. Total Environ. 2019, 677, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Vondráček, J.; Pěnčíková, K.; Neča, J.; Ciganek, M.; Grycová, A.; Dvořák, Z.; Machala, M. Assessment of the aryl hydrocarbon receptor-mediated activities of polycyclic aromatic hydrocarbons in a human cell-based reporter gene assay. Environ. Pollut. 2017, 220 Pt A, 307–316. [Google Scholar] [CrossRef]
- Li, N.; Xia, T.; Nel, A.E. The role of oxidative stress in ambient particulate matter-induced lung diseases and its implications in the toxicity of engineered nanoparticles. Free Radic. Biol. Med. 2008, 44, 1689–1699. [Google Scholar] [CrossRef] [Green Version]
- Longhin, E.; Capasso, L.; Battaglia, C.; Proverbio, M.C.; Cosentino, C.; Cifola, I.; Mangano, E.; Camatini, M.; Gualtieri, M. Integrative transcriptomic and protein analysis of human bronchial BEAS-2B exposed to seasonal urban particulate matter. Environ. Pollut. 2016, 209, 87–98. [Google Scholar] [CrossRef]
- Longhin, E.; Gualtieri, M.; Capasso, L.; Bengalli, R.; Mollerup, S.; Holme, J.A.; Ovrevik, J.; Casadei, S.; Di Benedetto, C.; Parenti, P.; et al. Physico-chemical properties and biological effects of diesel and biomass particles. Environ. Pollut. 2016, 215, 366–375. [Google Scholar] [CrossRef]
- Osgood, R.S.; Upham, B.L.; Bushel, P.R.; Velmurugan, K.; Xiong, K.N.; Bauer, A.K. Secondhand smoke-prevalent polycyclic aromatic hydrocarbon binary mixture-induced specific mitogenic and pro-inflammatory cell signaling events in lung epithelial cells. Toxicol. Sci. 2017, 157, 156–171. [Google Scholar] [CrossRef] [Green Version]
- DiNatale, B.C.; Schroeder, J.C.; Francey, L.J.; Kusnadi, A.; Perdew, G.H. Mechanistic insights into the events that lead to synergistic induction of interleukin 6 transcription upon activation of the aryl hydrocarbon receptor and inflammatory signaling. J. Biol. Chem. 2010, 285, 24388–24397. [Google Scholar] [CrossRef] [Green Version]
- Hollingshead, B.D.; Beischlag, T.V.; Dinatale, B.C.; Ramadoss, P.; Perdew, G.H. Inflammatory signaling and aryl hydrocarbon receptor mediate synergistic induction of interleukin 6 in MCF-7 cells. Cancer Res. 2008, 68, 3609–3617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lahoti, T.S.; Boyer, J.A.; Kusnadi, A.; Muku, G.E.; Murray, I.A.; Perdew, G.H. Aryl hydrocarbon receptor activation synergistically induces lipopolysaccharide-mediated expression of proinflammatory chemokine (c-c motif) ligand 20. Toxicol. Sci. 2015, 148, 229–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Mei, J.; Gonzales, L.; Yang, G.; Dai, N.; Wang, P.; Zhang, P.; Favara, M.; Malcolm, K.C.; Guttentag, S.; et al. IL-17A and TNF-alpha exert synergistic effects on expression of CXCL5 by alveolar type II cells in vivo and in vitro. J. Immunol. 2011, 186, 3197–3205. [Google Scholar] [CrossRef] [Green Version]
- Thorley, A.J.; Ford, P.A.; Giembycz, M.A.; Goldstraw, P.; Young, A.; Tetley, T.D. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J. Immunol. 2007, 178, 463–473. [Google Scholar] [CrossRef]
- Ruan, D.; So, S.P. Prostaglandin E2 produced by inducible COX-2 and mPGES-1 promoting cancer cell proliferation in vitro and in vivo. Life Sci. 2014, 116, 43–50. [Google Scholar] [CrossRef]
- Cathcart, M.C.; O’Byrne, K.J.; Reynolds, J.V.; O’Sullivan, J.; Pidgeon, G.P. COX-derived prostanoid pathways in gastrointestinal cancer development and progression: Novel targets for prevention and intervention. Biochim. Biophys. Acta 2012, 1825, 49–63. [Google Scholar] [CrossRef]
- Liu, R.; Xu, K.P.; Tan, G.S. Cyclooxygenase-2 inhibitors in lung cancer treatment: Bench to bed. Eur. J. Pharmacol. 2015, 769, 127–133. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Z.; Wang, K. Combining sorafenib with celecoxib synergistically inhibits tumor growth of non-small cell lung cancer cells in vitro and in vivo. Oncol. Rep. 2014, 31, 1954–1960. [Google Scholar] [CrossRef]
- Nothdurft, S.; Thumser-Henner, C.; Breitenbucher, F.; Okimoto, R.A.; Dorsch, M.; Opitz, C.A.; Sadik, A.; Esser, C.; Holzel, M.; Asthana, S.; et al. Functional screening identifies aryl hydrocarbon receptor as suppressor of lung cancer metastasis. Oncogenesis 2020, 9, 102. [Google Scholar] [CrossRef]
- Tsay, J.J.; Tchou-Wong, K.M.; Greenberg, A.K.; Pass, H.; Rom, W.N. Aryl hydrocarbon receptor and lung cancer. Anticancer Res. 2013, 33, 1247–1256. [Google Scholar]
- Vancheri, C.; Mastruzzo, C.; Sortino, M.A.; Crimi, N. The lung as a privileged site for the beneficial actions of PGE2. Trends Immunol. 2004, 25, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Maher, S.A.; Dekkak, B.; Jones, V.; Wong, S.; Brook, P.; Belvisi, M.G. Anti-inflammatory effects of PGE2 in the lung: Role of the EP4 receptor subtype. Thorax 2015, 70, 740–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bärnthaler, T.; Maric, J.; Platzer, W.; Konya, V.; Theiler, A.; Hasenöhrl, C.; Gottschalk, B.; Trautmann, S.; Schreiber, Y.; Graier, W.F.; et al. The Role of PGE2 in Alveolar Epithelial and Lung Microvascular Endothelial Crosstalk. Sci. Rep. 2017, 7, 7923. [Google Scholar] [CrossRef] [PubMed]
- Øvrevik, J.; Låg, M.; Lecureur, V.; Gilot, D.; Lagadic-Gossmann, D.; Refsnes, M.; Schwarze, P.E.; Skuland, T.; Becher, R.; Holme, J.A. AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun. Signal. 2014, 12, 48. [Google Scholar] [CrossRef] [Green Version]
- Kurita, H.; Schnekenburger, M.; Ovesen, J.L.; Xia, Y.; Puga, A. The Ah receptor recruits IKKalpha to its target binding motifs to phosphorylate serine-10 in histone H3 required for transcriptional activation. Toxicol. Sci. 2014, 139, 121–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.; Liu, S.; Shi, Y.; Liu, N.; Chen, L.; Wang, X.; Xiao, D.; Liu, X.; Mao, C.; Jiang, Y.; et al. Activation of AhR with nuclear IKKalpha regulates cancer stem-like properties in the occurrence of radioresistance. Cell Death Dis. 2018, 9, 490. [Google Scholar] [CrossRef] [Green Version]
- Vineis, P.; Husgafvel-Pursiainen, K. Air pollution and cancer: Biomarker studies in human populations. Carcinogenesis 2005, 26, 1846–1855. [Google Scholar] [CrossRef] [Green Version]
- De Kok, T.M.; Driece, H.A.; Hogervorst, J.G.; Briede, J.J. Toxicological assessment of ambient and traffic-related particulate matter: A review of recent studies. Mutat. Res. 2006, 613, 103–122. [Google Scholar] [CrossRef]
- Lewtas, J. Air pollution combustion emissions: Characterization of causative agents and mechanisms associated with cancer, reproductive, and cardiovascular effects. Mutat. Res. 2007, 636, 95–133. [Google Scholar] [CrossRef]
- Ji, J.; Upadhyay, S.; Xiong, X.; Malmlof, M.; Sandstrom, T.; Gerde, P.; Palmberg, L. Multi-cellular human bronchial models exposed to diesel exhaust particles: Assessment of inflammation, oxidative stress and macrophage polarization. Part. Fibre Toxicol. 2018, 15, 19. [Google Scholar] [CrossRef] [Green Version]
- Vogel, C.F.A.; Van Winkle, L.S.; Esser, C.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor as a target of environmental stressors—Implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020, 34, 101530. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Fan, L.; Feng, J.; Lv, S.; Wu, M.; Li, B.; Zang, Y.S. Genotoxic and inflammatory effects of organic extracts from traffic-related particulate matter in human lung epithelial A549 cells: The role of quinones. Toxicol. In Vitro 2013, 27, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.W.; Chang, Y.C.; Ho, C.C.; Tsai, M.H.; Lin, P. Increase of carcinogenic risk via enhancement of cyclooxygenase-2 expression and hydroxyestradiol accumulation in human lung cells as a result of interaction between BaP and 17-beta estradiol. Carcinogenesis 2007, 28, 1606–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Øvrevik, J.; Refsnes, M.; Holme, J.A.; Schwarze, P.E.; Låg, M. Mechanisms of chemokine responses by polycyclic aromatic hydrocarbons in bronchial epithelial cells: Sensitization through toll-like receptor-3 priming. Toxicol. Lett. 2013, 219, 125–132. [Google Scholar] [CrossRef]
- Guerrina, N.; Traboulsi, H.; Eidelman, D.H.; Baglole, C.J. The aryl hydrocarbon receptor and the maintenance of lung health. Int. J. Mol. Sci. 2018, 19, 3882. [Google Scholar] [CrossRef] [Green Version]
- Beamer, C.A.; Shepherd, D.M. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin. Immunopathol. 2013, 35, 693–704. [Google Scholar] [CrossRef] [Green Version]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vázquez-Gómez, G.; Karasová, M.; Tylichová, Z.; Kabátková, M.; Hampl, A.; Matthews, J.; Neča, J.; Ciganek, M.; Machala, M.; Vondráček, J. Aryl Hydrocarbon Receptor (AhR) Limits the Inflammatory Responses in Human Lung Adenocarcinoma A549 Cells via Interference with NF-κB Signaling. Cells 2022, 11, 707. https://doi.org/10.3390/cells11040707
Vázquez-Gómez G, Karasová M, Tylichová Z, Kabátková M, Hampl A, Matthews J, Neča J, Ciganek M, Machala M, Vondráček J. Aryl Hydrocarbon Receptor (AhR) Limits the Inflammatory Responses in Human Lung Adenocarcinoma A549 Cells via Interference with NF-κB Signaling. Cells. 2022; 11(4):707. https://doi.org/10.3390/cells11040707
Chicago/Turabian StyleVázquez-Gómez, Gerardo, Martina Karasová, Zuzana Tylichová, Markéta Kabátková, Aleš Hampl, Jason Matthews, Jiří Neča, Miroslav Ciganek, Miroslav Machala, and Jan Vondráček. 2022. "Aryl Hydrocarbon Receptor (AhR) Limits the Inflammatory Responses in Human Lung Adenocarcinoma A549 Cells via Interference with NF-κB Signaling" Cells 11, no. 4: 707. https://doi.org/10.3390/cells11040707