
C53 Interacting with UFM1-Protein Ligase 1 Regulates Microtubule Nucleation in Response to ER Stress

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Antibodies
2.3. Cell Cultures
2.4. DNA Constructs
2.5. Generation of UFL1 and C53 Deficient Cell Lines
2.6. Generation of Cell Lines Expressing Tagged Proteins
2.7. RNA Interference
2.8. Real-Time qRT-PCR
2.9. XBP1 mRNA Splicing Assay
2.10. Preparation of Cell Extracts
2.11. Centrosome Isolation
2.12. Gel Filtration Chromatography
2.13. Immunoprecipitation, GST Pull-Down Assay, Gel Electrophoresis, and Immunoblotting
2.14. Mass Spectrometry
2.15. Evaluation of Cell Growth and FACS Analysis
2.16. Microtubule Regrowth Experiments
2.17. Immunofluorescence Microscopy
2.18. Microtubule Nucleation Visualized by Time-Lapse Imaging
2.19. ER Area Quantification
2.20. Statistical Analysis
3. Results
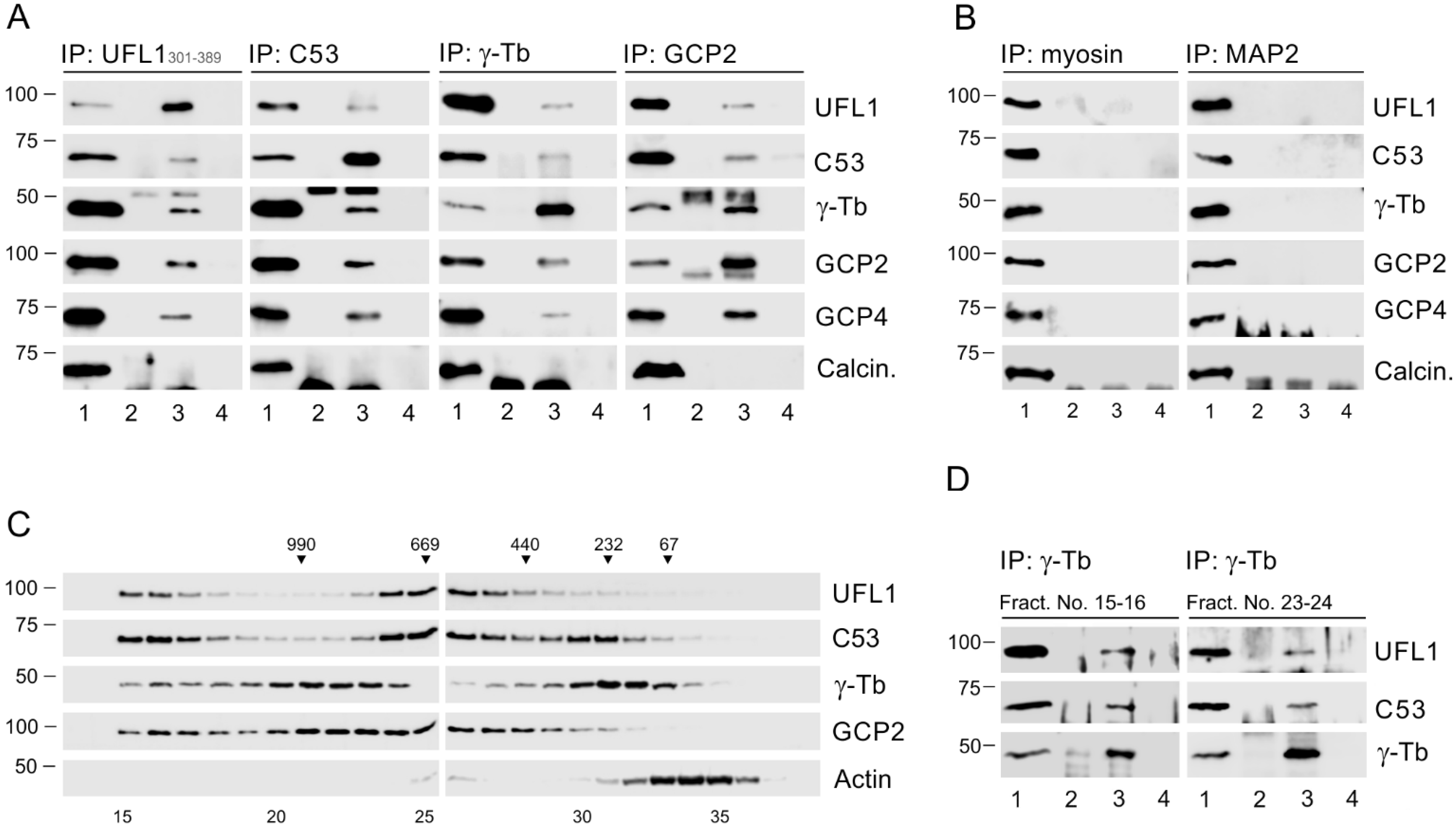
3.1. Identification of UFL1 as γ-Tubulin Interactor
3.2. UFL1 and C53 Associate with γTuRC Proteins
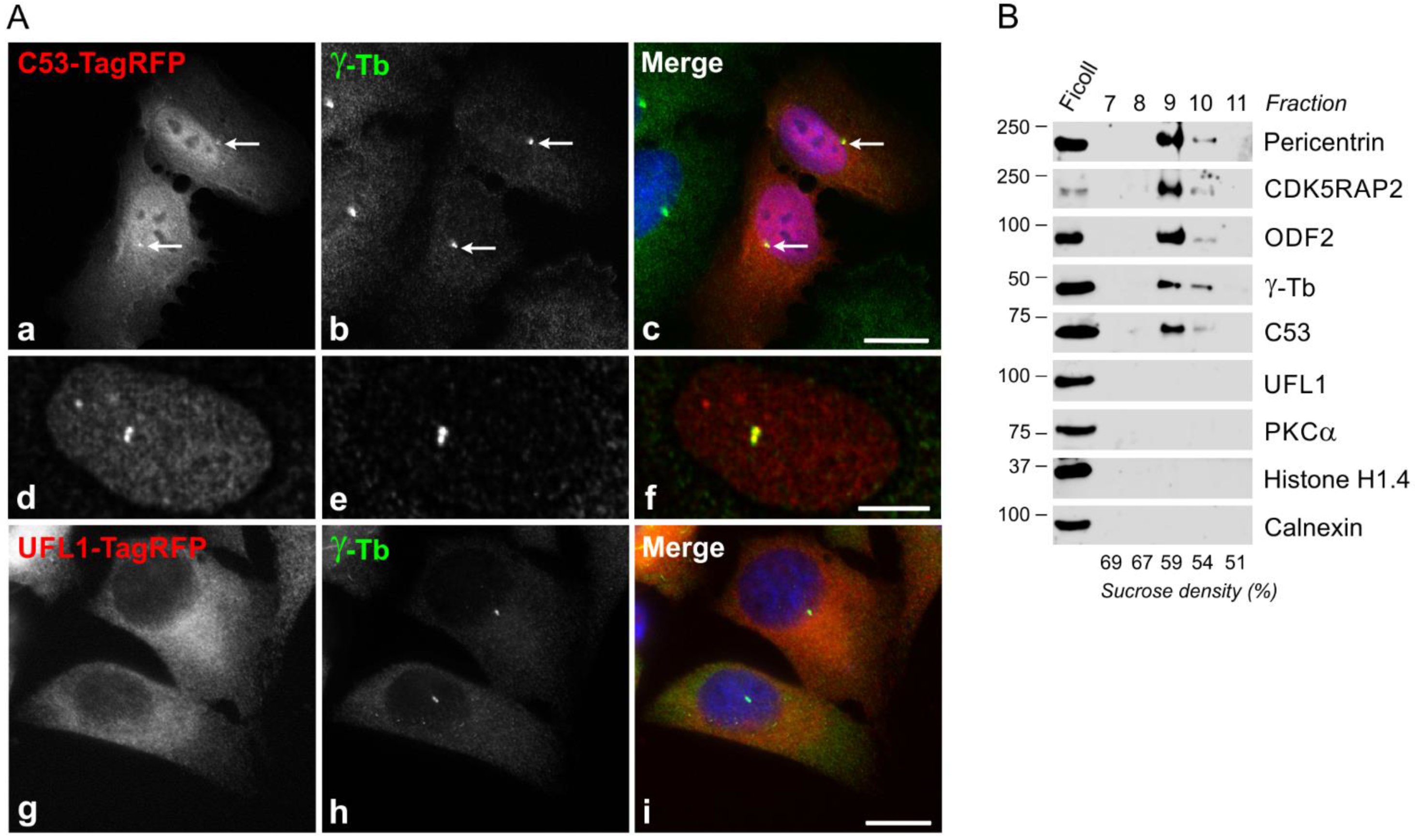
3.3. Association of Exogenous UFL1 and C53 with γTuRC Proteins and Centrosome
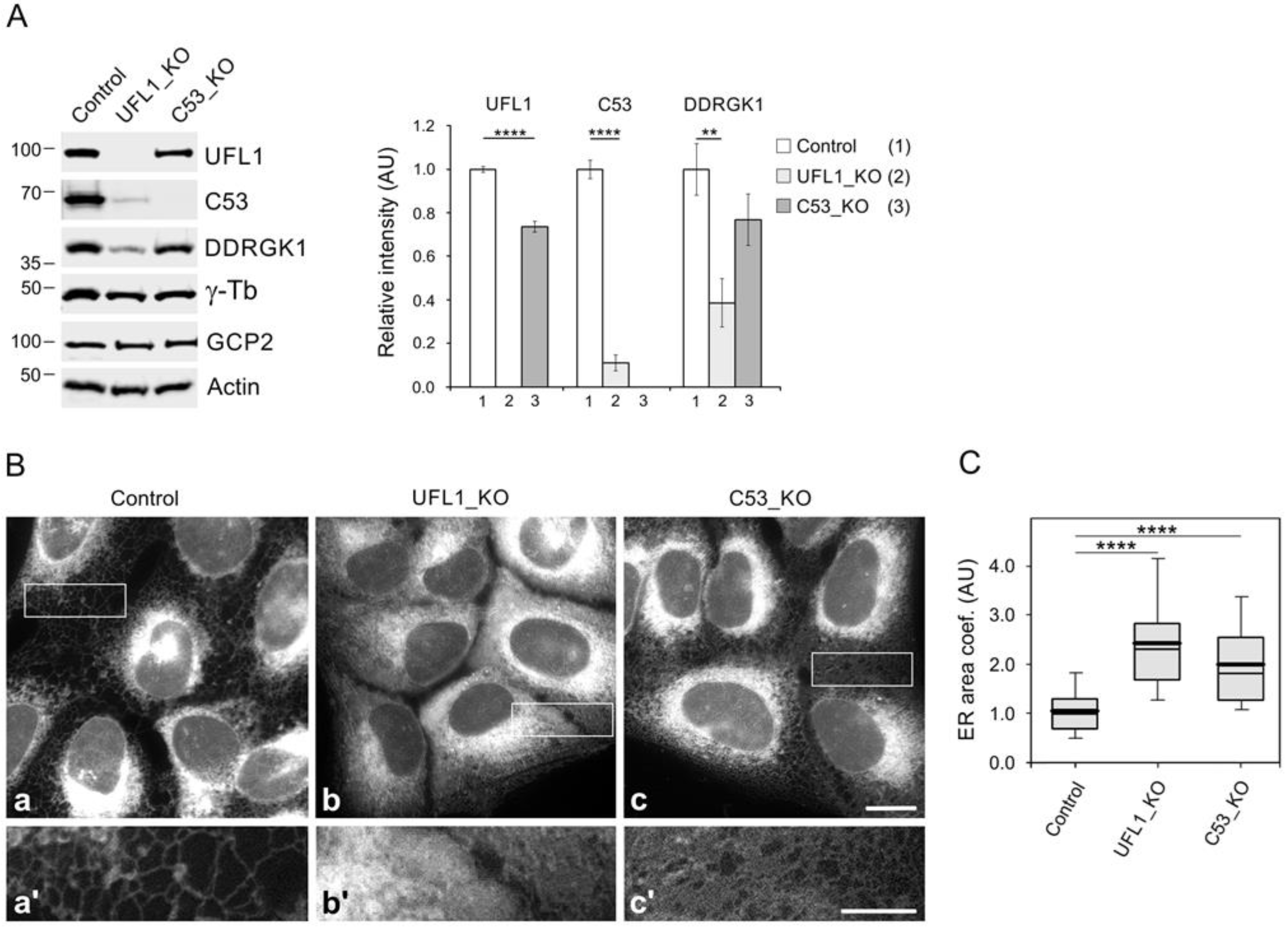
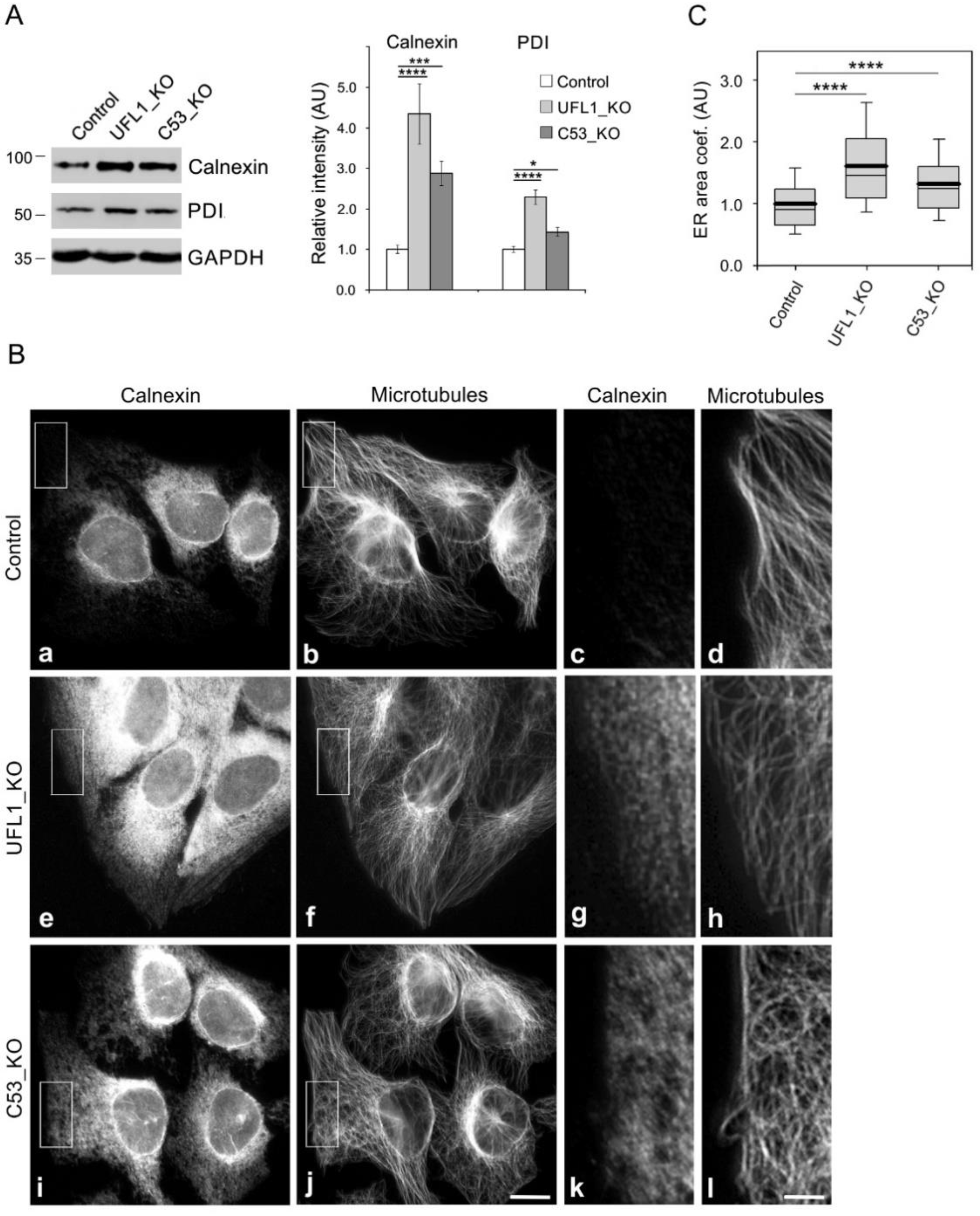
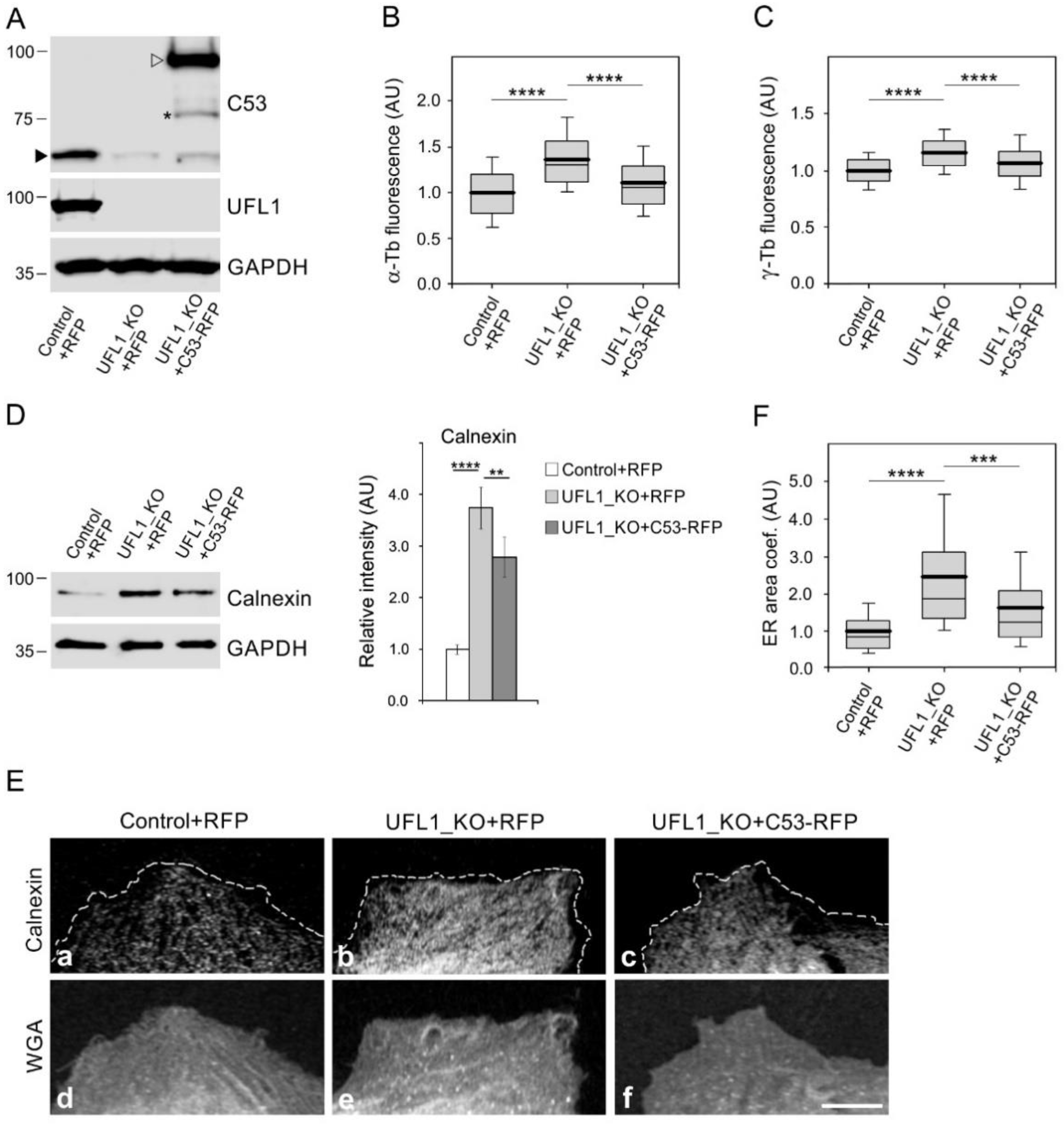
3.4. Preparation and Characterization of Cell Lines Lacking UFL1 or C53
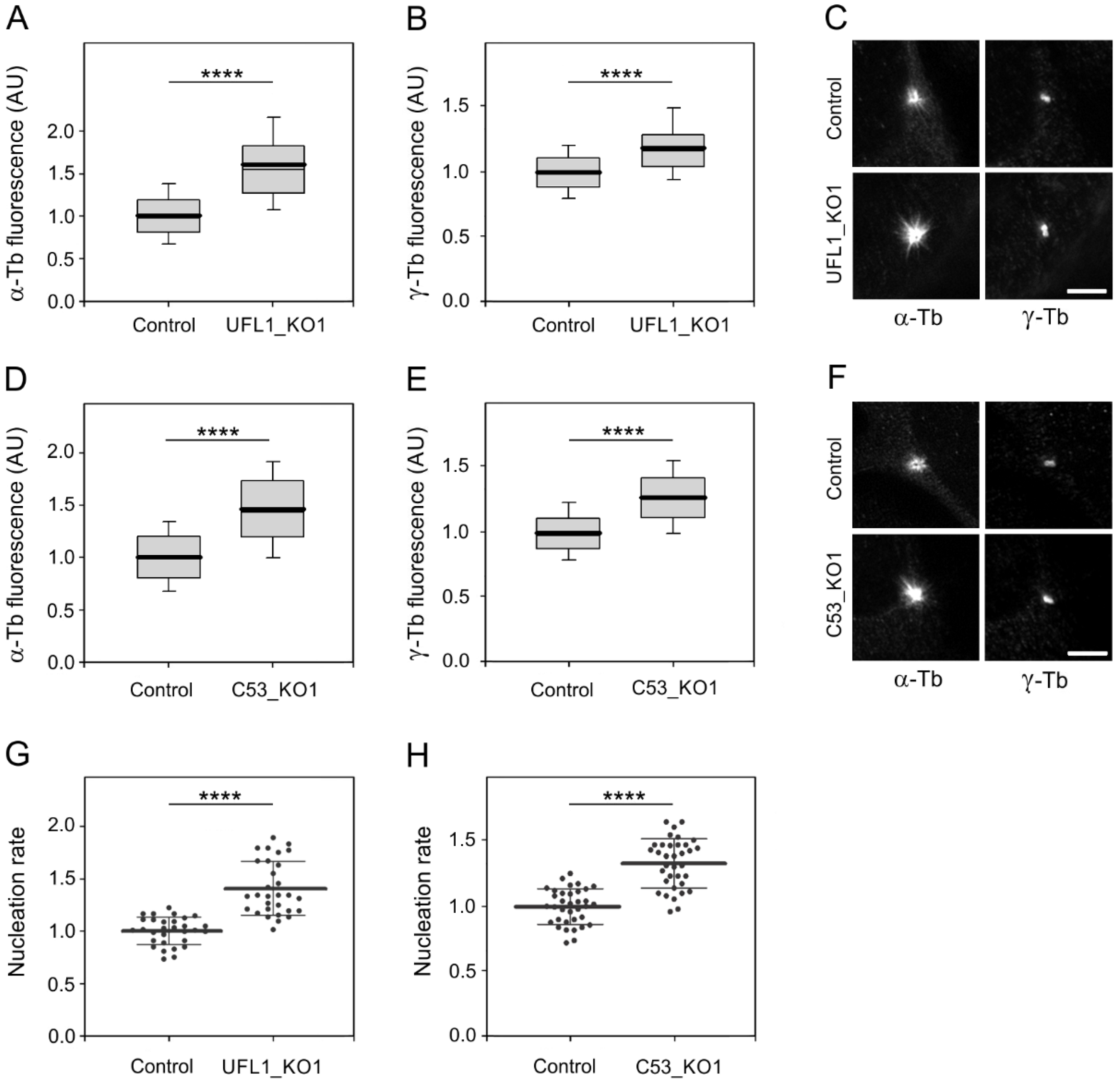
3.5. UFL1 or C53 Deficiency Increase Centrosomal Microtubule Nucleation
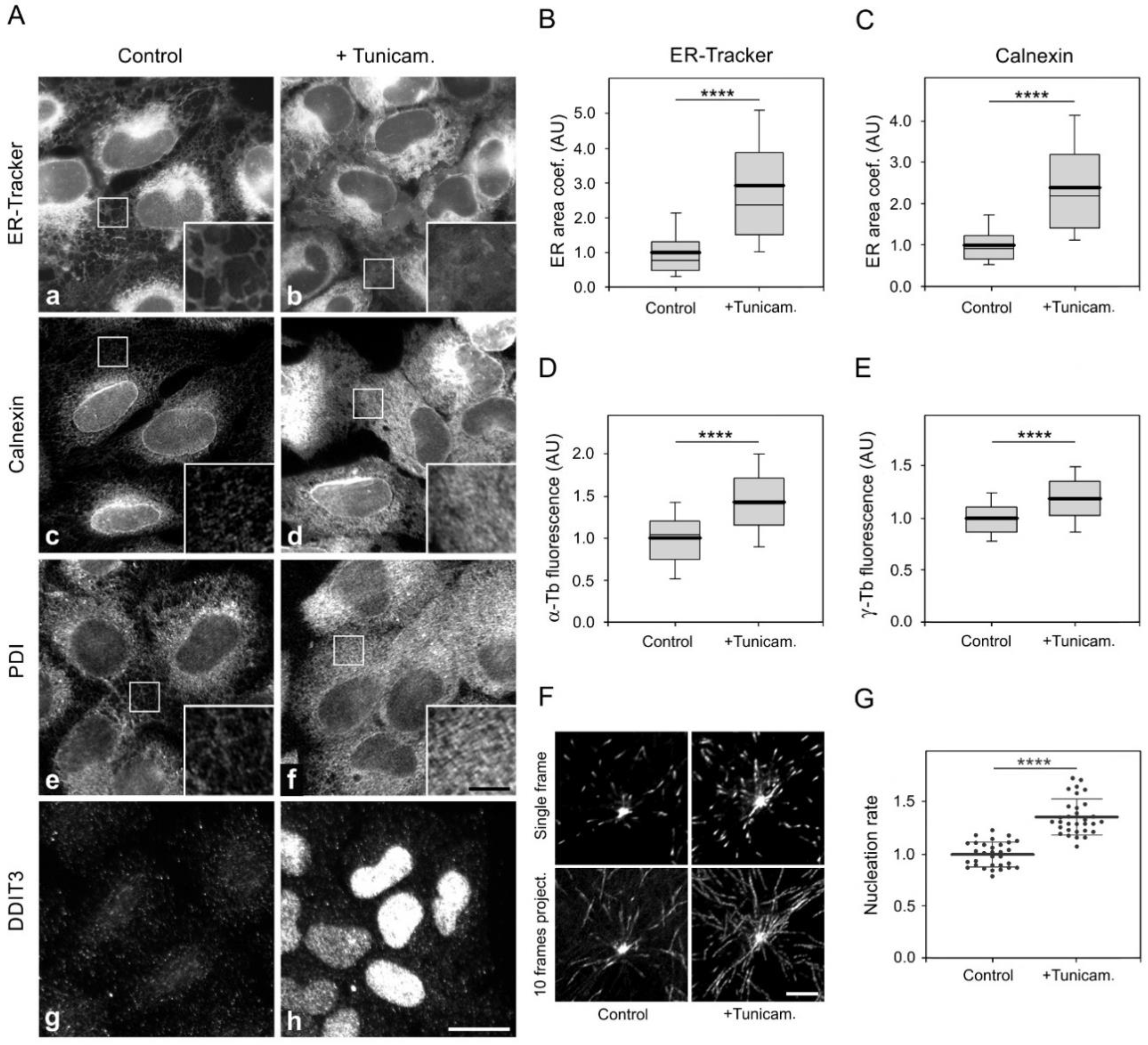
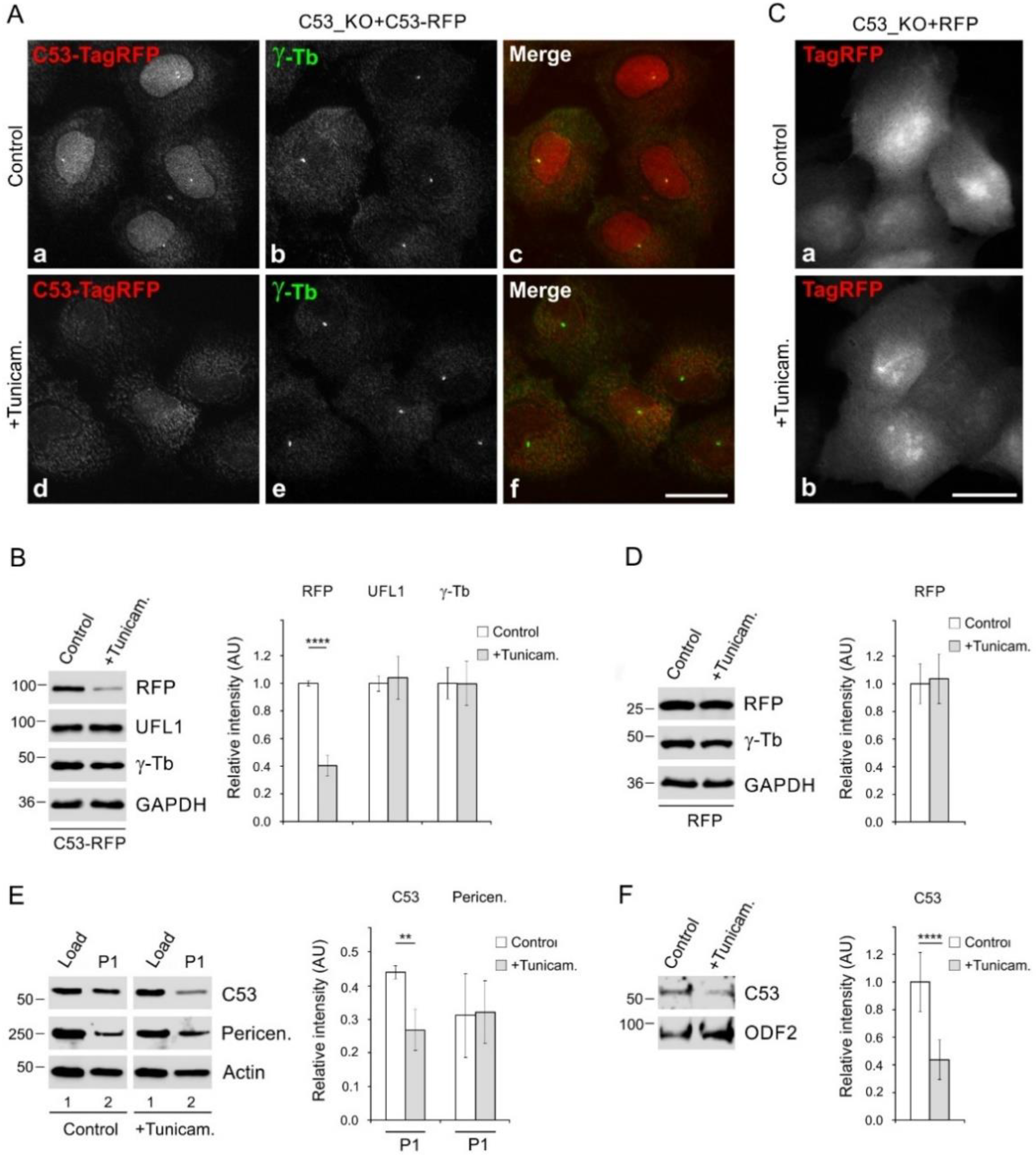
3.6. Enhancement of Microtubule Nucleation in Cells under ER Stress
4. Discussion
4.1. Interaction of UFL1 and C53 with γTuRC Proteins
4.2. Microtubule Nucleation in Cells Lacking UFL1 or C53
4.3. Regulatory Mechanisms by Which UFL1/C53 Can Control Microtubule Nucleation
4.4. Enhanced Microtubule Nucleation in Cells under ER Stress
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chakrabarti, A.; Chen, A.W.; Varner, J.D. A review of the mammalian unfolded protein response. Biotechnol. Bioeng. 2011, 108, 2777–2793. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuck, S.; Prinz, W.A.; Thorn, K.S.; Voss, C.; Walter, P. Membrane expansion alleviates endoplasmic reticulum stress independently of the unfolded protein response. J. Cell Biol. 2009, 187, 525–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterman-Storer, C.M.; Salmon, E.D. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr. Biol. 1998, 8, 798–806. [Google Scholar] [CrossRef] [Green Version]
- Oakley, C.E.; Oakley, B.R. Identification of γ-tubulin, a new member of the tubulin superfamily encoded by mipA gene of Aspergillus nidulans. Nature 1989, 338, 662–664. [Google Scholar] [CrossRef]
- Kollman, J.M.; Merdes, A.; Mourey, L.; Agard, D.A. Microtubule nucleation by γ-tubulin complexes. Nat. Rev. Mol. Cell Biol. 2011, 12, 709–721. [Google Scholar] [CrossRef]
- Wieczorek, M.; Urnavicius, L.; Ti, S.C.; Molloy, K.R.; Chait, B.T.; Kapoor, T.M. Asymmetric molecular architecture of the human γ-tubulin ring complex. Cell 2020, 180, 165–175. [Google Scholar] [CrossRef]
- Consolati, T.; Locke, J.; Roostalu, J.; Chen, Z.A.; Gannon, J.; Asthana, J.; Lim, W.M.; Martino, F.; Cvetkovic, M.A.; Rappsilber, J.; et al. Microtubule nucleation properties of single human γTuRCs explained by their cryo-EM structure. Dev. Cell 2020, 53, 603–617. [Google Scholar] [CrossRef]
- Liu, P.; Zupa, E.; Neuner, A.; Böhler, A.; Loerke, J.; Flemming, D.; Ruppert, T.; Rudack, T.; Peter, C.; Spahn, C.; et al. Insights into the assembly and activation of the microtubule nucleator γ-TuRC. Nature 2020, 578, 467–471. [Google Scholar] [CrossRef]
- Sulimenko, V.; Hájková, Z.; Klebanovych, A.; Dráber, P. Regulation of microtubule nucleation mediated by γ-tubulin complexes. Protoplasma 2017, 254, 1187–1199. [Google Scholar] [CrossRef]
- Tovey, C.A.; Conduit, P.T. Microtubule nucleation by γ-tubulin complexes and beyond. Essays Biochem. 2018, 62, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Tatsumi, K.; Sou, Y.S.; Tada, N.; Nakamura, E.; Iemura, S.; Natsume, T.; Kang, S.H.; Chung, C.H.; Kasahara, M.; Kominami, E.; et al. A novel type of E3 ligase for the Ufm1 conjugation system. J. Biol. Chem. 2010, 285, 5417–5427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komatsu, M.; Chiba, T.; Tatsumi, K.; Iemura, S.; Tanida, I.; Okazaki, N.; Ueno, T.; Kominami, E.; Natsume, T.; Tanaka, K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004, 23, 1977–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerakis, Y.; Quintero, M.; Li, H.; Hetz, C. The UFMylation system in proteostasis and beyond. Trends Cell Biol. 2019, 29, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Cho, H.J.; Han, S.H.; No, J.G.; Kwon, J.Y.; Kim, H. A novel LZAP-binding protein, NLBP, inhibits cell invasion. J. Biol. Chem. 2010, 285, 12232–12240. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Lei, G.; Mei, M.; Tang, Y.; Li, H. A novel C53/LZAP-interacting protein regulates stability of C53/LZAP and DDRGK domain-containing Protein 1 (DDRGK1) and modulates NF-κB signaling. J. Biol. Chem. 2010, 285, 15126–15136. [Google Scholar] [CrossRef] [Green Version]
- Shiwaku, H.; Yoshimura, N.; Tamura, T.; Sone, M.; Ogishima, S.; Watase, K.; Tagawa, K.; Okazawa, H. Suppression of the novel ER protein Maxer by mutant ataxin-1 in Bergman glia contributes to non-cell-autonomous toxicity. EMBO J. 2010, 29, 2446–2460. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, K.; Moura, R.F.; Granvik, M.; Igoillo-Esteve, M.; Hohmeier, H.E.; Hendrickx, N.; Newgard, C.B.; Waelkens, E.; Cnop, M.; Schuit, F. Ubiquitin fold modifier 1 (UFM1) and its target UFBP1 protect pancreatic beta cells from ER stress-induced apoptosis. PLoS ONE 2011, 6, e18517. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, M.; Wu, J.; Lei, G.; Li, H. Transcriptional regulation of the Ufm1 conjugation system in response to disturbance of the endoplasmic reticulum homeostasis and inhibition of vesicle trafficking. PLoS ONE 2012, 7, e48587. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Bhatt, B.; Sivaprakasam, S.; Cai, Y.; Liu, S.; Kodeboyina, S.K.; Patel, N.; Savage, N.M.; Sharma, A.; Kaufman, R.J.; et al. Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat. Commun. 2019, 10, e1084. [Google Scholar] [CrossRef]
- Liu, D.; Wang, W.D.; Melville, D.B.; Cha, Y.I.; Yin, Z.; Issaeva, N.; Knapik, E.W.; Yarbrough, W.G. Tumor suppressor Lzap regulates cell cycle progression, doming, and zebrafish epiboly. Dev. Dyn. 2011, 240, 1613–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, H.; Wu, J.; He, C.; Yang, W.; Li, H. A tumor suppressor C53 protein antagonizes checkpoint kinases to promote cyclin-dependent kinase 1 activation. Cell Res. 2009, 19, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; An, H.; Mayo, M.W.; Baldwin, A.S.; Yarbrough, W.G. LZAP, a putative tumor suppressor, selectively inhibits NF-κB. Cancer Cell 2007, 12, 239–251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.J.; Pan, K.; Li, J.J.; Chen, Y.B.; Chen, J.G.; Lv, L.; Wang, D.D.; Pan, Q.Z.; Chen, M.S.; Xia, J.C. Identification of LZAP as a new candidate tumor suppressor in hepatocellular carcinoma. PLoS ONE 2011, 6, e26608. [Google Scholar] [CrossRef]
- Yang, R.; Wang, H.; Kang, B.; Chen, B.; Shi, Y.; Yang, S.; Sun, L.; Liu, Y.; Xiao, W.; Zhang, T.; et al. CDK5RAP3, a UFL1 substrate adaptor, is crucial for liver development. Development 2019, 146, dev169235. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Jiang, H.; Luo, S.; Zhang, M.; Zhang, Y.; Sun, F.; Huang, S.; Li, H. Caspase-mediated cleavage of C53/LZAP protein causes abnormal microtubule bundling and rupture of the nuclear envelope. Cell Res. 2013, 23, 691–704. [Google Scholar] [CrossRef] [Green Version]
- Hořejší, B.; Vinopal, S.; Sládková, V.; Dráberová, E.; Sulimenko, V.; Sulimenko, T.; Vosecká, V.; Philimonenko, A.; Hozák, P.; Katsetos, C.D.; et al. Nuclear γ-tubulin associates with nucleoli and interacts with tumor suppressor protein C53. J. Cell. Physiol. 2012, 227, 367–382. [Google Scholar] [CrossRef]
- Zheng, Y.; Jung, M.K.; Oakley, B.R. γ-Tubulin is present in Drosophila melanogaster and Homo sapiens and is associated with centrosome. Cell 1991, 65, 817–823. [Google Scholar] [CrossRef]
- Nováková, M.; Dráberová, E.; Schürmann, W.; Czihak, G.; Viklický, V.; Dráber, P. γ-Tubulin redistribution in taxol-treated mitotic cells probed by monoclonal antibodies. Cell Motil. Cytoskel. 1996, 33, 38–51. [Google Scholar] [CrossRef]
- Dráberová, E.; D’Agostino, L.; Caracciolo, V.; Sládková, V.; Sulimenko, T.; Sulimenko, V.; Sobol, M.; Maounis, N.F.; Tzelepis, E.; Mahera, E.; et al. Overexpression and nucleolar localization of γ-tubulin small complex proteins GCP2 and GCP3 in glioblastoma. J. Neuropathol. Exp. Neurol. 2015, 74, 723–742. [Google Scholar] [CrossRef] [Green Version]
- Viklický, V.; Dráber, P.; Hašek, J.; Bártek, J. Production and characterization of a monoclonal antitubulin antibody. Cell Biol. Int. Rep. 1982, 6, 725–731. [Google Scholar] [CrossRef]
- Zíková, M.; Dráberová, E.; Sulimenko, V.; Dráber, P. New monoclonal antibodies specific for microtubule-associated protein MAP2. Folia Biol. 2000, 46, 87–88. [Google Scholar]
- Bar-Peled, M.; Raikhel, N.V. A method for isolation and purification of specific antibodies to a protein fused to the GST. Anal. Biochem. 1996, 241, 140–142. [Google Scholar] [CrossRef] [PubMed]
- Kukharskyy, V.; Sulimenko, V.; Macurek, L.; Sulimenko, T.; Dráberová, E.; Dráber, P. Complexes of γ-tubulin with non-receptor protein tyrosine kinases Src and Fyn in differentiating P19 embryonal carcinoma cells. Exp. Cell Res. 2004, 298, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Vinopal, S.; Černohorská, M.; Sulimenko, V.; Sulimenko, T.; Vosecká, V.; Flemr, M.; Dráberová, E.; Dráber, P. γ-Tubulin 2 nucleates microtubules and is downregulated in mouse early embryogenesis. PLoS ONE 2012, 7, e29919. [Google Scholar] [CrossRef]
- Macurek, L.; Dráberová, E.; Richterová, V.; Sulimenko, V.; Sulimenko, T.; Dráberová, L.; Marková, V.; Dráber, P. Regulation of microtubule nucleation from membranes by complexes of membrane-bound γ-tubulin with Fyn kinase and phosphoinositide 3-kinase. Biochem. J. 2008, 416, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef]
- Flemr, M.; Buhler, M. Single-step generation of conditional knockout mouse embryonic stem cells. Cell Rep. 2015, 12, 709–716. [Google Scholar] [CrossRef] [Green Version]
- Dráberová, E.; Sulimenko, V.; Vinopal, S.; Sulimenko, T.; Sládková, V.; D’Agostino, L.; Sobol, M.; Hozák, P.; Křen, L.; Katsetos, C.D.; et al. Differential expression of human γ-tubulin isotypes during neuronal development and oxidative stress points to a γ-tubulin-2 prosurvival function. FASEB J. 2017, 31, 1828–1846. [Google Scholar] [CrossRef] [Green Version]
- Chu, Q.; Martinez, T.F.; Novak, S.W.; Donaldson, C.J.; Tan, D.; Vaughan, J.M.; Chang, T.; Diedrich, J.K.; Andrade, L.; Kim, A.; et al. Regulation of the ER stress response by a mitochondrial microprotein. Nat. Commun. 2019, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mitchison, T.J.; Kirschner, M.W. Isolation of mammalian centrosomes. Methods Enzymol. 1986, 134, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Dráberová, E.; Dráber, P. A microtubule-interacting protein involved in coalignment of vimentin intermediate filaments with microtubules. J. Cell Sci. 1993, 106, 1263–1273. [Google Scholar] [CrossRef] [PubMed]
- Klebanovych, A.; Sládková, V.; Sulimenko, T.; Vosecká, V.; Rubíková, Z.; Čapek, M.; Dráberová, E.; Dráber, P.; Sulimenko, V. Regulation of microtubule nucleation in mouse bone marrow-derived mast cells by protein tyrosine kinase SHP-1. Cells 2019, 8, 345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yue, G.; Ma, W.; Zhang, A.; Zou, J.; Cai, Y.; Tang, X.; Wang, J.; Liu, J.; Li, H.; et al. Ufm1-specific ligase Ufl1 regulates endoplasmic reticulum homeostasis and protects against heart failure. Circ. Heart Fail. 2018, 11, e004917. [Google Scholar] [CrossRef]
- Zhang, M.; Zhu, X.; Zhang, Y.; Cai, Y.; Chen, J.; Sivaprakasam, S.; Gurav, A.; Pi, W.; Makala, L.; Wu, J.; et al. RCAD/Ufl1, a Ufm1 E3 ligase, is essential for hematopoietic stem cell function and murine hematopoiesis. Cell Death Differ. 2015, 22, 1922–1934. [Google Scholar] [CrossRef]
- Quintero, M.; Liu, S.; Xia, Y.; Huang, Y.; Zou, Y.; Li, G.; Hu, L.; Singh, N.; Blumberg, R.; Cai, Y.; et al. Cdk5rap3 is essential for intestinal Paneth cell development and maintenance. Cell Death Dis. 2021, 12, e131. [Google Scholar] [CrossRef]
- Sriburi, R.; Jackowski, S.; Mori, K.; Brewer, J.W. XBP1: A link between the unfolded protein response, lipid biosynthesis, and biogenesis of the endoplasmic reticulum. J. Cell Biol. 2004, 167, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Černohorská, M.; Sulimenko, V.; Hájková, Z.; Sulimenko, T.; Sládková, V.; Vinopal, S.; Dráberová, E.; Dráber, P. GIT1/βPIX signaling proteins and PAK1 kinase regulate microtubule nucleation. BBA Mol. Cell. Res. 2016, 1863, 1282–1297. [Google Scholar] [CrossRef]
- Colello, D.; Reverte, C.G.; Ward, R.; Jones, C.W.; Magidson, V.; Khodjakov, A.; LaFlamme, S.E. Androgen and Src signaling regulate centrosome activity. J. Cell Sci. 2010, 123, 2094–2102. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.X.; Ni, H.M.; Gao, W.; Hou, Y.F.; Melan, M.A.; Chen, X.; Stolz, D.B.; Shao, Z.M.; Yin, X.M. Differential effects of endoplasmic reticulum stress-induced autophagy on cell survival. J. Biol. Chem. 2007, 282, 4702–4710. [Google Scholar] [CrossRef] [Green Version]
- Liao, W.; Chan, L. Tunicamycin induces ubiquitination and degradation of apolipoprotein B in HepG2 cells. Biochem. J. 2001, 353, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; de Heus, C.; Liu, Q.; Bouchet, B.P.; Noordstra, I.; Jiang, K.; Hua, S.; Martin, M.; Yang, C.; Grigoriev, I.; et al. Molecular pathway of microtubule organization at the Golgi apparatus. Dev. Cell 2016, 39, 44–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hehnly, H.; Doxsey, S. Rab11 endosomes contribute to mitotic spindle organization and orientation. Dev. Cell 2014, 28, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teixidó-Travesa, N.; Roig, J.; Lüders, J. The where, when and how of microtubule nucleation—One ring to rule them all. J. Cell Sci. 2012, 125, 4445–4456. [Google Scholar] [CrossRef] [Green Version]
- Stephani, M.; Picchianti, L.; Gajic, A.; Beveridge, R.; Skarwan, E.; de Hernandez, V.S.M.; Mohseni, A.; Clavel, M.; Zeng, Y.; Naumann, C.; et al. A cross-kingdom conserved ER-phagy receptor maintains endoplasmic reticulum homeostasis during stress. Elife 2020, 9, e58396. [Google Scholar] [CrossRef] [PubMed]
- Wamsley, J.J.; Issaeva, N.; An, H.; Lu, X.; Donehower, L.A.; Yarbrough, W.G. LZAP is a novel Wip1 binding partner and positive regulator of its phosphatase activity in vitro. Cell Cycle 2017, 16, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Borek, W.E.; Groocock, L.M.; Samejima, I.; Zou, J.; de Lima Alves, F.; Rappsilber, J.; Sawin, K.E. Mto2 multisite phosphorylation inactivates non-spindle microtubule nucleation complexes during mitosis. Nat. Commun. 2015, 6, 7929. [Google Scholar] [CrossRef]
- Yu, L.; Li, G.; Deng, J.; Jiang, X.; Xue, J.; Zhu, Y.; Huang, W.; Tang, B.; Duan, R. The UFM1 cascade times mitosis entry associated with microcephaly. FASEB J. 2020, 34, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Chen, Q.; Feng, J.; Hou, J.; Yang, F.; Liu, J.; Jiang, Q.; Zhang, C. Sequential phosphorylation of Nedd1 by Cdk1 and Plk1 is required for targeting of the γTuRC to the centrosome. J. Cell Sci. 2009, 122, 2240–2251. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Li, D.; Zhang, S.; Yang, Y.; Liu, J.J.; Wang, X.; Liu, C.; Milkie, D.E.; Moore, R.P.; Tulu, U.S.; et al. Visualizing intracellular organelle and cytoskeletal interactions at nanoscale resolution on millisecond timescales. Cell 2018, 175, 1430–1442. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.R.; Lingeman, E.; Luong, T.; Ahmed, S.; Muhar, M.; Nguyen, T.; Olzmann, J.A.; Corn, J.E. A genome-wide ER-phagy screen highlights key roles of mitochondrial metabolism and ER-resident UFMylation. Cell 2020, 180, 1160–1177.e20. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klebanovych, A.; Vinopal, S.; Dráberová, E.; Sládková, V.; Sulimenko, T.; Sulimenko, V.; Vosecká, V.; Macůrek, L.; Legido, A.; Dráber, P. C53 Interacting with UFM1-Protein Ligase 1 Regulates Microtubule Nucleation in Response to ER Stress. Cells 2022, 11, 555. https://doi.org/10.3390/cells11030555
Klebanovych A, Vinopal S, Dráberová E, Sládková V, Sulimenko T, Sulimenko V, Vosecká V, Macůrek L, Legido A, Dráber P. C53 Interacting with UFM1-Protein Ligase 1 Regulates Microtubule Nucleation in Response to ER Stress. Cells. 2022; 11(3):555. https://doi.org/10.3390/cells11030555
Chicago/Turabian StyleKlebanovych, Anastasiya, Stanislav Vinopal, Eduarda Dráberová, Vladimíra Sládková, Tetyana Sulimenko, Vadym Sulimenko, Věra Vosecká, Libor Macůrek, Agustin Legido, and Pavel Dráber. 2022. "C53 Interacting with UFM1-Protein Ligase 1 Regulates Microtubule Nucleation in Response to ER Stress" Cells 11, no. 3: 555. https://doi.org/10.3390/cells11030555