
Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis

 , , , , and
, , , , and 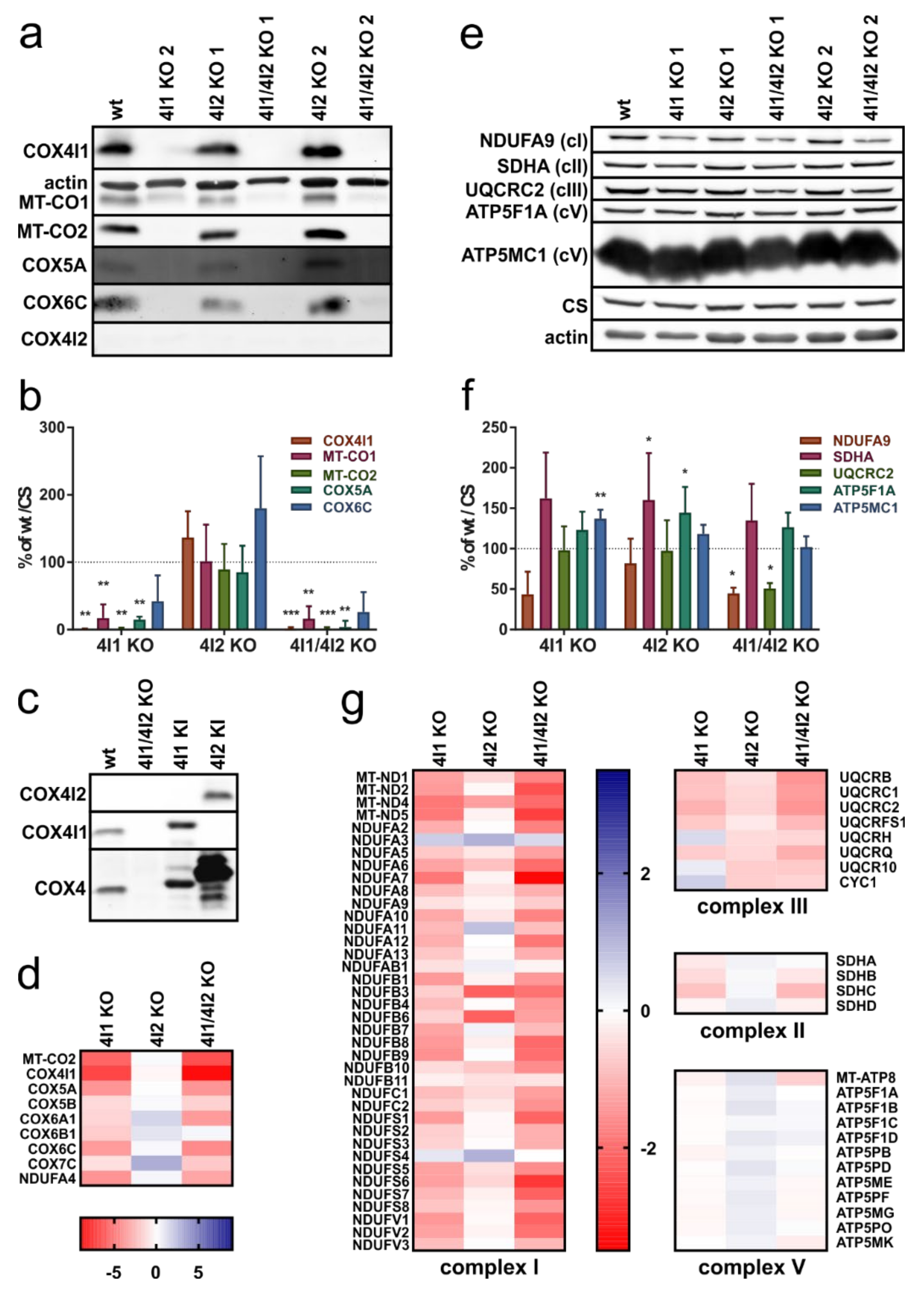{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Models
2.2. SDS-PAGE
2.3. Native Electrophoresis
2.4. Western Blot (WB) and Immunodetection
2.5. Mass Spectrometry—Label-Free Quantification (MS LFQ)
2.6. Stable Isotope Labeling Using Amino Acids in Cell Culture (SILAC)/Complexome Profiling
2.7. Mitochondrial Protein Synthesis Pulse-Chase Analysis
2.8. Measurement of Metabolic Fluxes
3. Results
3.1. Decreased Steady-State Content of cIV and cI Subunits in Cells Lacking COX4I1
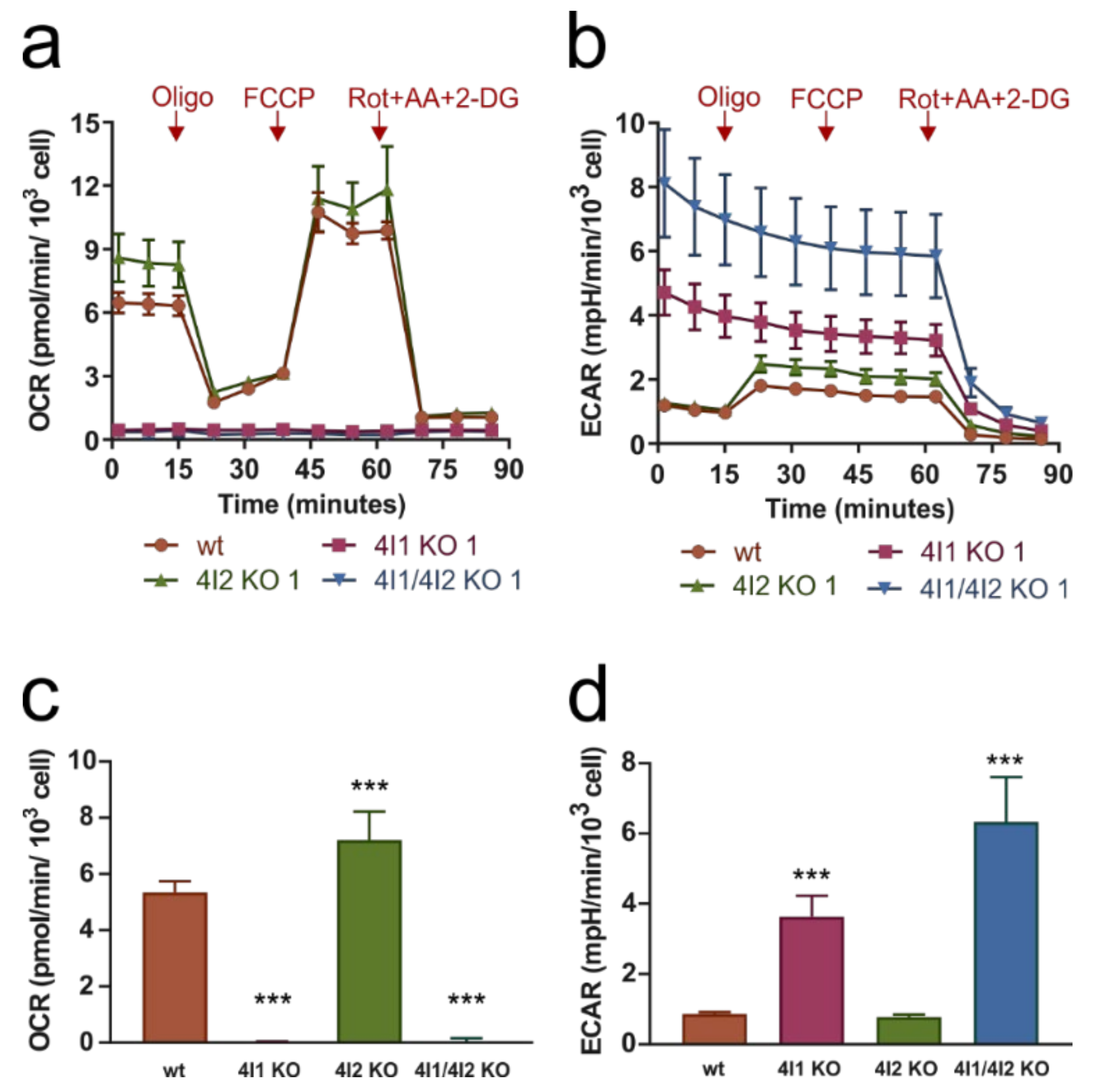
3.2. Compromised OXPHOS Function in COX4I1-Lacking Cells Was Compensated by Increased Glycolysis
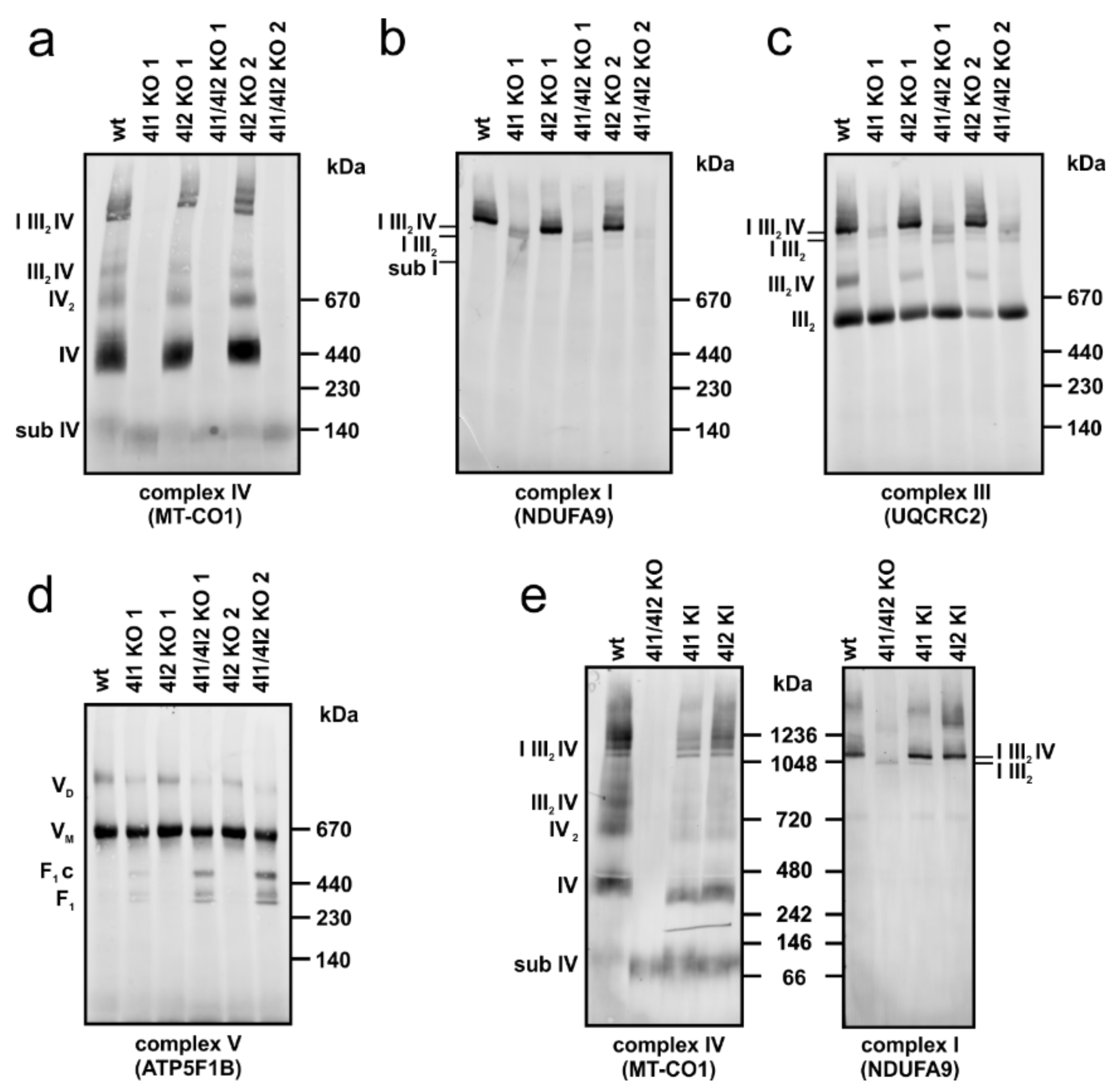
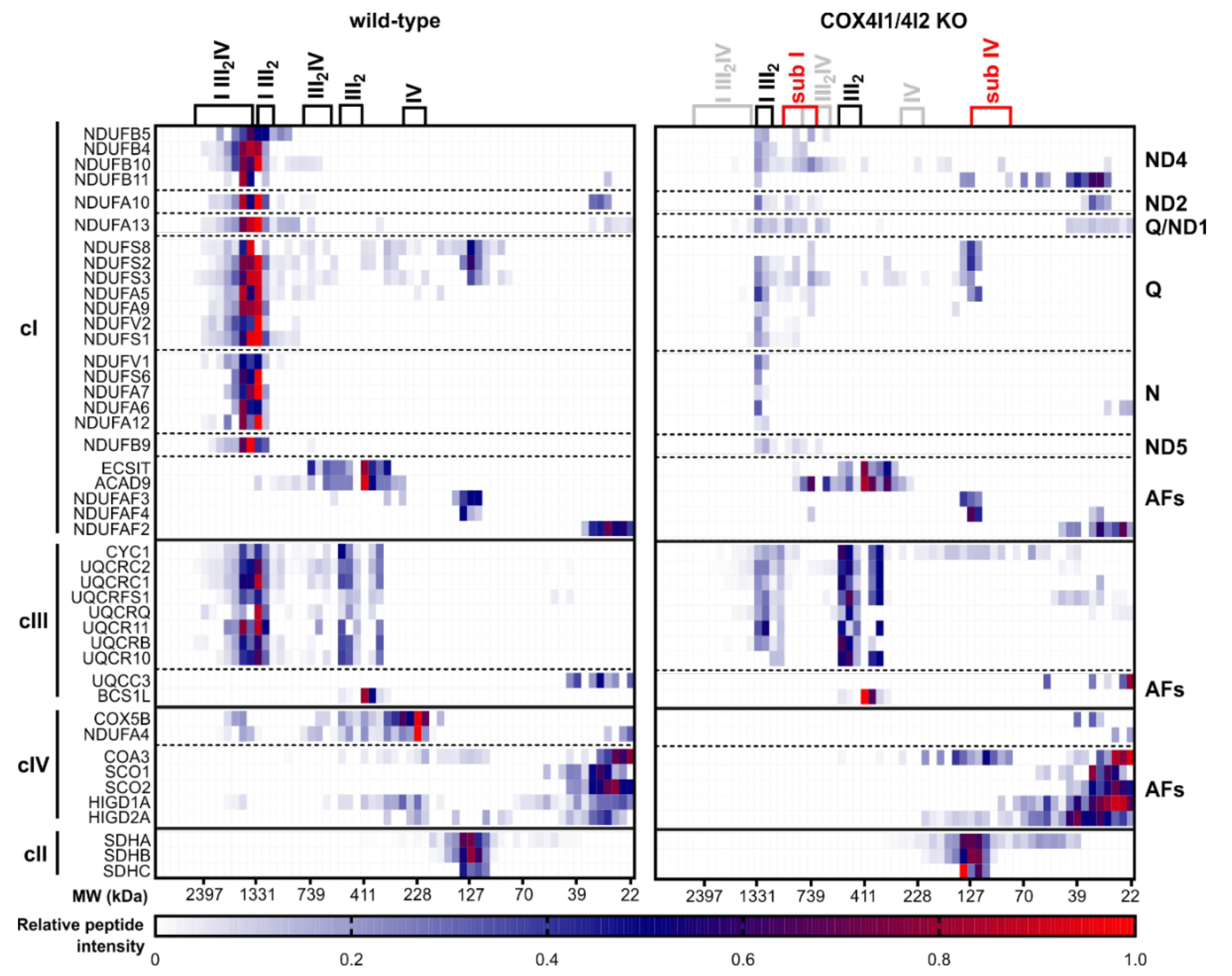
3.3. Effect of COX4 KO on OXPHOS Complexes Content and Assembly Status
3.4. Decreased Rate of Mitochondrial Protein Synthesis in COX4I1-Lacking Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hallberg, B.M.; Larsson, N.G. Making proteins in the powerhouse. Cell Metab. 2014, 20, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Caballero, L.; Guerrero-Castillo, S.; Nijtmans, L. Unraveling the complexity of mitochondrial complex I assembly: A dynamic process. Biochim. Biophys Acta BBA Bioenerg. 2016, 1857, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Signes, A.; Fernandez-Vizarra, E. Assembly of mammalian oxidative phosphorylation complexes I–V and supercomplexes. Essays Biochem. 2018, 62, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Stroud, D.A.; Surgenor, E.E.; Formosa, L.E.; Reljic, B.; Frazier, A.E.; Dibley, M.G.; Osellame, L.D.; Stait, T.; Beilharz, T.H.; Thorburn, D.R.; et al. Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 2016, 538, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg-Bord, M.; Schuldiner, M. Ground control to major TOM: Mitochondria-nucleus communication. FEBS J. 2017, 284, 196–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontanesi, F.; Soto, I.C.; Horn, D.; Barrientos, A. Assembly of mitochondrial cytochrome c-oxidase, a complicated and highly regulated cellular process. Am. J. Physiol. Cell Physiol. 2006, 291, C1129–C1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schagger, H.; Pfeiffer, K. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, L.A. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nature 2016, 537, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landazuri, M.O.; Enriquez, J.A. NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [Green Version]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunatova, K.; Reguera, D.P.; Houstek, J.; Mracek, T.; Pecina, P. Role of cytochrome c oxidase nuclear-encoded subunits in health and disease. Physiol. Res. 2020, 69, 947–965. [Google Scholar] [CrossRef]
- Sommer, N.; Huttemann, M.; Pak, O.; Scheibe, S.; Knoepp, F.; Sinkler, C.; Malczyk, M.; Gierhardt, M.; Esfandiary, A.; Kraut, S.; et al. Mitochondrial Complex IV Subunit 4 Isoform 2 Is Essential for Acute Pulmonary Oxygen Sensing. Circ. Res. 2017, 121, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Pajuelo Reguera, D.; Cunatova, K.; Vrbacky, M.; Pecinova, A.; Houstek, J.; Mracek, T.; Pecina, P. Cytochrome c Oxidase Subunit 4 Isoform Exchange Results in Modulation of Oxygen Affinity. Cells 2020, 9, 443. [Google Scholar] [CrossRef] [Green Version]
- Nijtmans, L.G.; Taanman, J.W.; Muijsers, A.O.; Speijer, D.; Van den Bogert, C. Assembly of cytochrome-c oxidase in cultured human cells. Eur. J. Biochem. 1998, 254, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennerlein, S.; Rehling, P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J. Cell Sci. 2015, 128, 833–837. [Google Scholar] [CrossRef] [Green Version]
- Soto, I.C.; Fontanesi, F.; Liu, J.; Barrientos, A. Biogenesis and assembly of eukaryotic cytochrome c oxidase catalytic core. Biochim. Biophys Acta BBA Bioenerg. 2012, 1817, 883–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidoni, S.; Harbour, M.E.; Guerrero-Castillo, S.; Signes, A.; Ding, S.; Fearnley, I.M.; Taylor, R.W.; Tiranti, V.; Arnold, S.; Fernandez-Vizarra, E.; et al. MR-1S Interacts with PET100 and PET117 in Module-Based Assembly of Human Cytochrome c Oxidase. Cell Rep. 2017, 18, 1727–1738. [Google Scholar] [CrossRef] [Green Version]
- Mick, D.U.; Dennerlein, S.; Wiese, H.; Reinhold, R.; Pacheu-Grau, D.; Lorenzi, I.; Sasarman, F.; Weraarpachai, W.; Shoubridge, E.A.; Warscheid, B.; et al. MITRAC links mitochondrial protein translocation to respiratory-chain assembly and translational regulation. Cell 2012, 151, 1528–1541. [Google Scholar] [CrossRef] [Green Version]
- Stiburek, L.; Vesela, K.; Hansikova, H.; Pecina, P.; Tesarova, M.; Cerna, L.; Houstek, J.; Zeman, J. Tissue-specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem. J. 2005, 392, 625–632. [Google Scholar] [CrossRef] [Green Version]
- Aich, A.; Wang, C.; Chowdhury, A.; Ronsor, C.; Pacheu-Grau, D.; Richter-Dennerlein, R.; Dennerlein, S.; Rehling, P. COX16 promotes COX2 metallation and assembly during respiratory complex IV biogenesis. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Maghool, S.; Cooray, N.D.G.; Stroud, D.A.; Aragao, D.; Ryan, M.T.; Maher, M.J. Structural and functional characterization of the mitochondrial complex IV assembly factor Coa6. Life Sci. Alliance 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Pacheu-Grau, D.; Wasilewski, M.; Oeljeklaus, S.; Gibhardt, C.S.; Aich, A.; Chudenkova, M.; Dennerlein, S.; Deckers, M.; Bogeski, I.; Warscheid, B.; et al. COA6 Facilitates Cytochrome c Oxidase Biogenesis as Thiol-reductase for Copper Metallochaperones in Mitochondria. J. Mol. Biol. 2020, 432, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Soma, S.; Morgada, M.N.; Naik, M.T.; Boulet, A.; Roesler, A.A.; Dziuba, N.; Ghosh, A.; Yu, Q.; Lindahl, P.A.; Ames, J.B.; et al. COA6 Is Structurally Tuned to Function as a Thiol-Disulfide Oxidoreductase in Copper Delivery to Mitochondrial Cytochrome c Oxidase. Cell Rep. 2019, 29, 4114–4126. [Google Scholar] [CrossRef] [Green Version]
- Hock, D.H.; Reljic, B.; Ang, C.S.; Muellner-Wong, L.; Mountford, H.S.; Compton, A.G.; Ryan, M.T.; Thorburn, D.R.; Stroud, D.A. HIGD2A is Required for Assembly of the COX3 Module of Human Mitochondrial Complex IV. Mol. Cell. Proteom. 2020, 19, 1145–1160. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial complex III deficiency associated with a homozygous mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Carossa, V.; Ghelli, A.; Tropeano, C.V.; Valentino, M.L.; Iommarini, L.; Maresca, A.; Caporali, L.; La Morgia, C.; Liguori, R.; Barboni, P.; et al. A novel in-frame 18-bp microdeletion in MT-CYB causes a multisystem disorder with prominent exercise intolerance. Hum. Mutat. 2014, 35, 954–958. [Google Scholar] [CrossRef]
- Feichtinger, R.G.; Brunner-Krainz, M.; Alhaddad, B.; Wortmann, S.B.; Kovacs-Nagy, R.; Stojakovic, T.; Erwa, W.; Resch, B.; Windischhofer, W.; Verheyen, S.; et al. Combined Respiratory Chain Deficiency and UQCC2 Mutations in Neonatal Encephalomyopathy: Defective Supercomplex Assembly in Complex III Deficiencies. Oxid. Med. Cell. Longev. 2017, 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Tucker, E.J.; Wanschers, B.F.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; van den Brand, M.A.; Leenders, A.M.; Rodenburg, R.J.; Reljic, B.; Compton, A.G.; et al. Mutations in the UQCC1-interacting protein, UQCC2, cause human complex III deficiency associated with perturbed cytochrome b protein expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Lastres, D.; Fontanesi, F.; Garcia-Consuegra, I.; Martin, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial complex I plays an essential role in human respirasome assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Perez-Perez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Penas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef] [PubMed]
- Diaz, F.; Fukui, H.; Garcia, S.; Moraes, C.T. Cytochrome c oxidase is required for the assembly/stability of respiratory complex I in mouse fibroblasts. Mol. Cell. Biol. 2006, 26, 4872–4881. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; D’Aurelio, M.; Deng, J.H.; Park, J.S.; Manfredi, G.; Hu, P.; Lu, J.; Bai, Y. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. J. Biol. Chem. 2007, 282, 17557–17562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hornig-Do, H.T.; Tatsuta, T.; Buckermann, A.; Bust, M.; Kollberg, G.; Rotig, A.; Hellmich, M.; Nijtmans, L.; Wiesner, R.J. Nonsense mutations in the COX1 subunit impair the stability of respiratory chain complexes rather than their assembly. EMBO J. 2012, 31, 1293–1307. [Google Scholar] [CrossRef] [Green Version]
- Timon-Gomez, A.; Nyvltova, E.; Abriata, L.A.; Vila, A.J.; Hosler, J.; Barrientos, A. Mitochondrial cytochrome c oxidase biogenesis: Recent developments. Semin. Cell Dev. Biol. 2018, 76, 163–178. [Google Scholar] [CrossRef] [PubMed]
- Lobo-Jarne, T.; Perez-Perez, R.; Fontanesi, F.; Timon-Gomez, A.; Wittig, I.; Penas, A.; Serrano-Lorenzo, P.; Garcia-Consuegra, I.; Arenas, J.; Martin, M.A.; et al. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. EMBO J. 2020, 39, e103912. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Lin, C.-Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schägger, H. Tricine–SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Wittig, I.; Braun, H.-P.; Schägger, H. Blue native PAGE. Nat. Protoc. 2006, 1, 418–428. [Google Scholar] [CrossRef]
- Bentlage, H.A.C.M.; Wendel, U.; Schagger, H.; Ter Laak, H.J.; Janssen, A.J.M.; Trijbels, J.M.F. Lethal infantile mitochondrial disease with isolated complex I deficiency in fibroblasts but with combined complex I and IV deficiencies in muscle. Neurology 1996, 47, 243–248. [Google Scholar] [CrossRef] [Green Version]
- Hartmannova, H.; Piherova, L.; Tauchmannova, K.; Kidd, K.; Acott, P.D.; Crocker, J.F.; Oussedik, Y.; Mallet, M.; Hodanova, K.; Stranecky, V.; et al. Acadian variant of Fanconi syndrome is caused by mitochondrial respiratory chain complex I deficiency due to a non-coding mutation in complex I assembly factor NDUFAF6. Hum. Mol. Genet. 2016, 25, 4062–4079. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Ferrin, G.; Perez-Martos, A.; Fernandez-Silva, P.; Zeviani, M.; Enriquez, J.A. Isolation of mitochondria for biogenetical studies: An update. Mitochondrion 2010, 10, 253–262. [Google Scholar] [CrossRef]
- Heide, H.; Bleier, L.; Steger, M.; Ackermann, J.; Drose, S.; Schwamb, B.; Zornig, M.; Reichert, A.S.; Koch, I.; Wittig, I.; et al. Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 2012, 16, 538–549. [Google Scholar] [CrossRef] [Green Version]
- Van Strien, J.; Haupt, A.; Schulte, U.; Braun, H.-P.; Cabrera-Orefice, A.; Choudhary, J.S.; Evers, F.; Fernandez-Vizarra, E.; Guerrero-Castillo, S.; Kooij, T.W.A.; et al. CEDAR, an online resource for the reporting and exploration of complexome profiling data. bioRxiv 2020. [Google Scholar] [CrossRef]
- McKenzie, M.; Lazarou, M.; Ryan, M.T. Chapter 18 Analysis of respiratory chain complex assembly with radiolabeled nuclear- and mitochondrial-encoded subunits. Methods Enzymol. 2009, 456, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.D.; Jourdain, A.A.; Calvo, S.E.; Ballarano, C.A.; Doench, J.G.; Root, D.E.; Mootha, V.K. A Genome-wide CRISPR Death Screen Identifies Genes Essential for Oxidative Phosphorylation. Cell Metab. 2016, 24, 875–885. [Google Scholar] [CrossRef] [Green Version]
- Stiburek, L.; Hansikova, H.; Tesarova, M.; Cerna, L.; Zeman, J. Biogenesis of eukaryotic cytochrome c oxidase. Physiol. Res. 2006, 55 (Suppl. 2), S27–S41. [Google Scholar]
- Huttemann, M.; Klewer, S.; Lee, I.; Pecinova, A.; Pecina, P.; Liu, J.; Lee, M.; Doan, J.W.; Larson, D.; Slack, E.; et al. Mice deleted for heart-type cytochrome c oxidase subunit 7a1 develop dilated cardiomyopathy. Mitochondrion 2012, 12, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, R.; Zhang, H.; Kim, J.W.; Shimoda, L.; Dang, C.V.; Semenza, G.L. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell 2007, 129, 111–122. [Google Scholar] [CrossRef] [Green Version]
- Huttemann, M.; Lee, I.; Liu, J.; Grossman, L.I. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007, 274, 5737–5748. [Google Scholar] [CrossRef]
- Kaila, V.R.; Verkhovsky, M.I.; Wikstrom, M. Proton-coupled electron transfer in cytochrome oxidase. Chem. Rev. 2010, 110, 7062–7081. [Google Scholar] [CrossRef] [PubMed]
- Stoldt, S.; Wenzel, D.; Kehrein, K.; Riedel, D.; Ott, M.; Jakobs, S. Spatial orchestration of mitochondrial translation and OXPHOS complex assembly. Nat. Cell Biol. 2018, 20, 528–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter-Dennerlein, R.; Oeljeklaus, S.; Lorenzi, I.; Ronsor, C.; Bareth, B.; Schendzielorz, A.B.; Wang, C.; Warscheid, B.; Rehling, P.; Dennerlein, S. Mitochondrial Protein Synthesis Adapts to Influx of Nuclear-Encoded Protein. Cell 2016, 167, 471–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Richter-Dennerlein, R.; Pacheu-Grau, D.; Liu, F.; Zhu, Y.; Dennerlein, S.; Rehling, P. MITRAC15/COA1 promotes mitochondrial translation in a ND2 ribosome-nascent chain complex. EMBO Rep. 2020, 21, e48833. [Google Scholar] [CrossRef]
- Quiros, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell Biol. 2017, 216, 2027–2045. [Google Scholar] [CrossRef] [PubMed]
- Fornuskova, D.; Stiburek, L.; Wenchich, L.; Vinsova, K.; Hansikova, H.; Zeman, J. Novel insights into the assembly and function of human nuclear-encoded cytochrome c oxidase subunits 4, 5a, 6a, 7a and 7b. Biochem. J. 2010, 428, 363–374. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čunátová, K.; Reguera, D.P.; Vrbacký, M.; Fernández-Vizarra, E.; Ding, S.; Fearnley, I.M.; Zeviani, M.; Houštěk, J.; Mráček, T.; Pecina, P. Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis. Cells 2021, 10, 369. https://doi.org/10.3390/cells10020369
Čunátová K, Reguera DP, Vrbacký M, Fernández-Vizarra E, Ding S, Fearnley IM, Zeviani M, Houštěk J, Mráček T, Pecina P. Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis. Cells. 2021; 10(2):369. https://doi.org/10.3390/cells10020369
Chicago/Turabian StyleČunátová, Kristýna, David Pajuelo Reguera, Marek Vrbacký, Erika Fernández-Vizarra, Shujing Ding, Ian M. Fearnley, Massimo Zeviani, Josef Houštěk, Tomáš Mráček, and Petr Pecina. 2021. "Loss of COX4I1 Leads to Combined Respiratory Chain Deficiency and Impaired Mitochondrial Protein Synthesis" Cells 10, no. 2: 369. https://doi.org/10.3390/cells10020369