
A New Method to Prepare Stable Polyaniline Dispersions for Highly Loaded Cathodes of All-Polymer Li-Ion Batteries
 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Polyaniline via Acid-Assisted Polymerization (PANI-a)
2.3. Synthesis of Polyaniline via Common Chemical Polymerization (PANI-c)
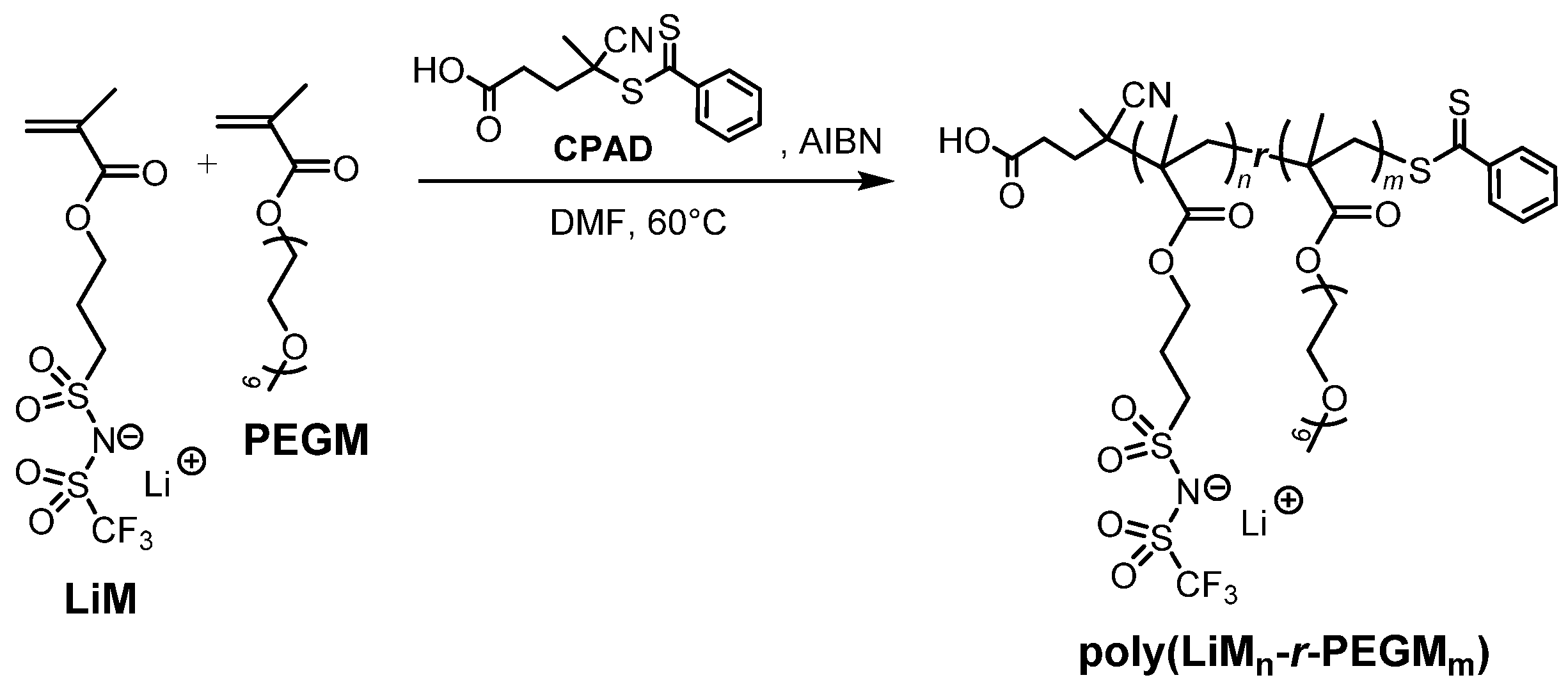
2.4. Synthesis of Lithium 1-[3-(methacryloyloxy)propylsulfonyl]-1-(trifluoromethanesulfonyl)imide (LiM)
2.5. Synthesis of Random Poly(LiM13-r-PEGM81) Copolymer
2.6. Preparation of SPE/PANI-a@CC Composite Polymer Cathode
2.7. Measurements
3. Results and Discussions
3.1. Synthesis of Polyaniline via Formic Acid-Assisted Polymerization (PANI-a)
3.2. Study of PANI-a Suspension via Dynamic (DLS) and Static Light Scattering (SLS)
3.3. PANI-a Physical and Electrochemical Properties
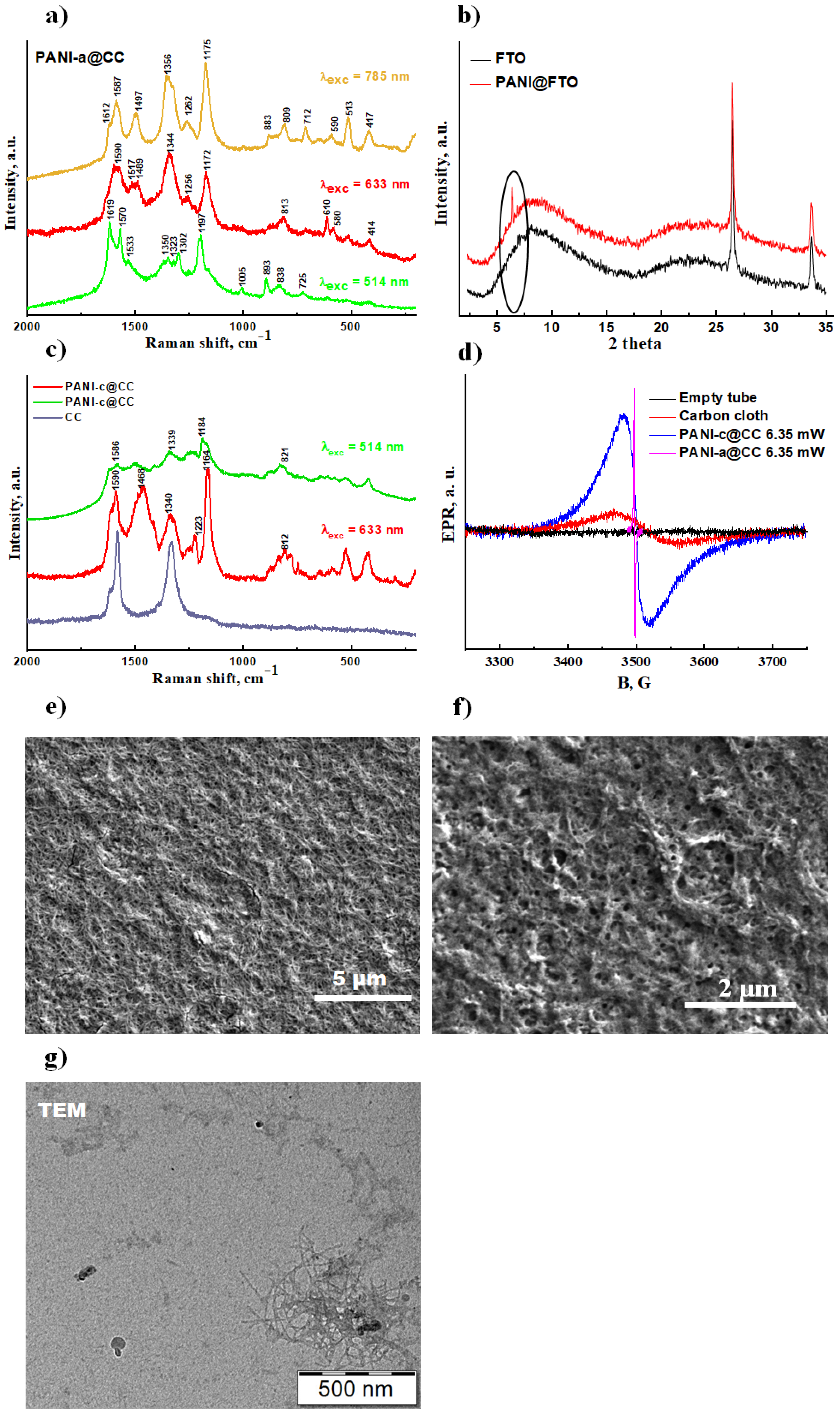
3.3.1. Spectroscopic Characterization
3.3.2. X-ray Diffraction Studies
3.3.3. Electronic Paramagnetic Resonance (EPR)
3.3.4. Scanning (SEM) and Transmission Electron (TEM) Microscopies
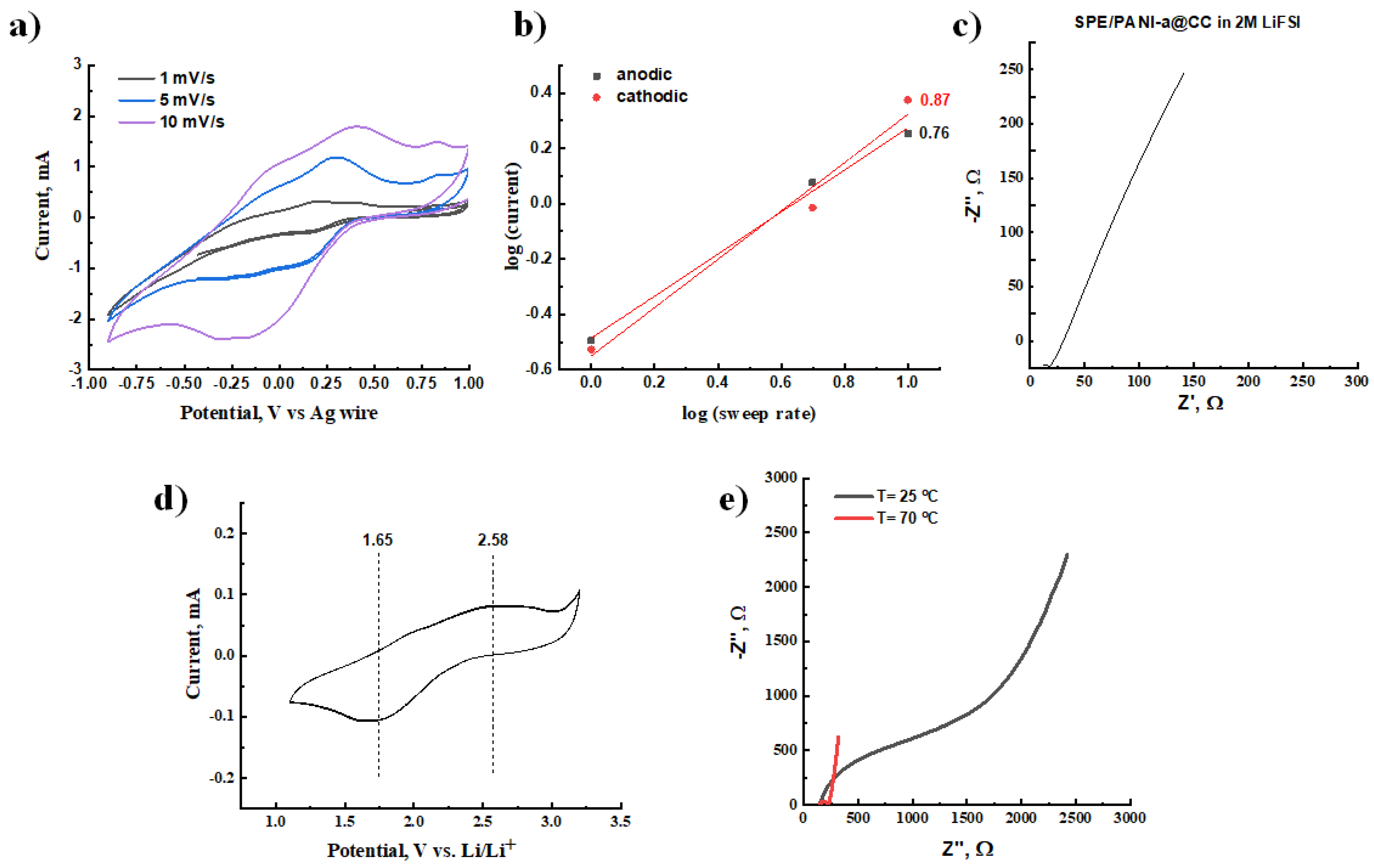
3.3.5. Cyclic Voltammetry (CV) and Electrochemical Impedance Spectroscopy (EIS)
3.4. Synthesis and Characterization of Solid Polymer Electrolyte (SPE)
3.5. Composite PANI-a/SPE Cathode Material: Preparation and Testing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ore, I.; Pigments, I.O.; Rock, P.; Crystal, Q.; Earths, R.; Ash, S. Mineral Commodity Summaries 2021; U.S. Geological Survey: Reston, VA, USA, 2021; ISBN 9781411343986.
- Meister, P.; Jia, H.; Li, J.; Klöpsch, R.; Winter, M.; Placke, T. Best Practice: Performance and Cost Evaluation of Lithium Ion Battery Active Materials with Special Emphasis on Energy Efficiency. Chem. Mater. 2016, 28, 7203–7217. [Google Scholar] [CrossRef]
- Canepa, P.; Sai Gautam, G.; Hannah, D.C.; Malik, R.; Liu, M.; Gallagher, K.G.; Persson, K.A.; Ceder, G. Odyssey of Multivalent Cathode Materials: Open Questions and Future Challenges. Chem. Rev. 2017, 117, 4287–4341. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Chen, X.; Cao, R.; Qian, J.; Xiang, H.; Zheng, J.; Zhang, J.G.; Xu, W. Enhanced performance of Li|LiFePO4 cells using CsPF6 as an electrolyte additive. J. Power Sources 2015, 293, 1062–1067. [Google Scholar] [CrossRef]
- Oz, E.; Altin, S.; Demirel, S.; Bayri, A.; Altin, E.; Baglayan, O.; Avci, S. Electrochemical effects and magnetic properties of B substituted LiCoO2: Improving Li-battery performance. J. Alloys Compd. 2016, 657, 835–847. [Google Scholar] [CrossRef]
- Zheng, X.; Li, X.; Wang, Z.; Guo, H.; Huang, Z.; Yan, G.; Wang, D. Investigation and improvement on the electrochemical performance and storage characteristics of LiNiO2-based materials for lithium ion battery. Electrochim. Acta 2016, 191, 832–840. [Google Scholar] [CrossRef]
- Chepurnaya, I.; Smirnova, E.; Karushev, M. Electrochemically Active Polymer Components in Next-Generation LiFePO4 Cathodes: Can Small Things Make a Big Difference? Batteries 2022, 8, 185. [Google Scholar] [CrossRef]
- Chung, Y.M.; Ryu, K.S. Surface coating and electrochemical properties of LiNi0.8Co0.15Al0.05O2/polyaniline composites as an electrode for Li-ion batteries. Bull. Korean Chem. Soc. 2009, 30, 1733–1737. [Google Scholar] [CrossRef]
- Kotz, R.; Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 2000, 45, 2483–2498. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, X.S. Carbon-based materials as supercapacitor electrodes. Chem. Soc. Rev. 2009, 38, 2520–2531. [Google Scholar] [CrossRef]
- Miller, J.; Burke, A. Electrochemical Capacitors: Challenges and Opportunities for Real-World Applications. Electrochem. Soc. Interface 2008, 17, 53. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Materials for electrochemical capacitors. Nat. Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef]
- Cheng, F.Y.; Tang, W.; Li, C.; Chen, J.; Liu, H.; Shen, P.; Dou, S. Conducting Poly(aniline) Nanotubes and Nanofibers: Controlled Synthesis and Application in Lithium/Poly(aniline) Rechargeable Batteries. Chem. Eur. J. 2006, 12, 3082–3088. [Google Scholar] [CrossRef]
- Rudge, A.; Raistnck, I.A.N.; Gottesfeld, S.; Ferraris, J. A Study of The Electrochemical Properties of Conducting Polymers for Application in Capacitors. Electrochim. Acta 1994, 39, 273–287. [Google Scholar] [CrossRef]
- Peng, C.; Hu, D.; Chen, G.Z. Theoretical specific capacitance based on charge storage mechanisms of conducting polymers: Comment on ‘Vertically oriented arrays of polyaniline nanorods and their super electrochemical properties’. Chem. Commun 2011, 47, 4105–4107. [Google Scholar] [CrossRef] [PubMed]
- Tomšík, E.; Ivanko, I.; Kohut, O.; Hromádková, J. High-Rate Polyaniline/Carbon-Cloth Electrodes: Effect of Mass Loading on the Pseudocapacitive Performance. ChemElectroChem 2017, 4, 2884–2890. [Google Scholar] [CrossRef]
- Ismail, R.; Sedenkova, I.; Svoboda, J.; Lukesova, M.; Walterova, Z.; Tomsik, E. Acid-assisted polymerization: The novel synthetic route of sensing layers based on PANI films and chelating agents protected by non-biofouling layers for Fe2+ or Fe3+ potentiometric detection. J. Mater. Chem. B. 2023, 11, 1545–1556. [Google Scholar] [CrossRef]
- Tomšík, E.; Kohut, O.; Ivanko, I.; Pekárek, M.; Bieloshapka, I.; Dallas, P. Assembly and Interaction of Polyaniline Chains: Impact on Electro- and Physical-Chemical Behavior. J. Phys. Chem. C 2018, 122, 8022–8030. [Google Scholar] [CrossRef]
- Gospodinova, N.; Muat, V.; Kolev, H.; Romanova, J. New insight into the redox behavior of polyaniline. Synth. Met. 2011, 161, 2510–2513. [Google Scholar] [CrossRef]
- Shaplov, A.S.; Vlasov, P.S.; Armand, M.; Lozinskaya, E.I.; Ponkratov, D.O.; Malyshkina, I.A.; Vidal, F.; Okatova, O.V.; Pavlov, G.M.; Wandrey, C.; et al. Design and synthesis of new anionic “polymeric ionic liquids” with high charge delocalization. Polym. Chem. 2011, 2, 2609–2618. [Google Scholar] [CrossRef]
- Porcarelli, L.; Shaplov, A.S.; Salsamendi, M.; Nair, J.R.; Vygodskii, Y.S.; Mecerreyes, D.; Gerbaldi, C. Single-Ion Block Copoly(ionic liquid)s as Electrolytes for All-Solid State Lithium Batteries. ACS Appl. Mater. Interfaces 2016, 8, 10350–10359. [Google Scholar] [CrossRef]
- Lingua, G.; Grysan, P.; Vlasov, P.S.; Verge, P.; Shaplov, A.S.; Gerbaldi, C. Unique Carbonate-Based Single Ion Conducting Block Copolymers Enabling High-Voltage, All-Solid-State Lithium Metal Batteries. Macromolecules 2021, 54, 6911–6924. [Google Scholar] [CrossRef] [PubMed]
- Jakeš, J. Regularized Positive Exponential Sum (REPES) Program-A Way of Inverting Laplace Transform Data Obtained by Dynamic Light Scattering. Collect. Czechoslov. Chem. Commun. 1995, 60, 1781–1797. [Google Scholar] [CrossRef]
- Wu, J.; Zhang, Q.; Wang, J.; Huang, X.; Bai, H. A self-assembly route to porous polyaniline/reduced graphene oxide composite materials with molecular-level uniformity for high-performance supercapacitors. Energy Environ. Sci. 2018, 11, 1280–1286. [Google Scholar] [CrossRef]
- Wu, C.; Bein, T. Conducting Polyaniline Filaments in a Mesoporous Channel Host. Science 1994, 264, 1757–1759. [Google Scholar] [CrossRef]
- Sivakkumar, S.R.; Oh, J.S.; Kim, D.W. Polyaniline nanofibres as a cathode material for rechargeable lithium-polymer cells assembled with gel polymer electrolyte. J. Power Sources 2006, 163, 573–577. [Google Scholar] [CrossRef]
- Qiu, B.; Wang, J.; Li, Z.; Wang, X.; Li, X. Influence of Acidity and Oxidant Concentration on the Nanostructures and Electrochemical Performance of Polyaniline during Fast Microwave-Assisted Chemical Polymerization. Polymers 2020, 12, 310. [Google Scholar] [CrossRef]
- Qiu, B.; Li, Z.; Wang, X.; Li, X.; Zhang, J. Exploration on the Microwave-Assisted Synthesis and Formation Mechanism of Polyaniline Nanostructures Synthesized in Different Hydrochloric Acid Concentrations. Polym. Chem. 2017, 55, 3357–3369. [Google Scholar] [CrossRef]
- Hambitzer, G.; Stassen, I. Polymerization of aniline in aqueous sulfuric acid -study by electrochemical thermospray mass spectrometry. Synth. Met. 1993, 57, 1045–1050. [Google Scholar] [CrossRef]
- Trchova, M.; Sedenkova, I.; Stejskal, J.; Bok, J. Polymerization of Aniline in the Solutions of Strong and Weak Acids: The Evolution of Infrared Spectra and Their Interpretation Using Factor Analysis. Appl. Spectrosc. 2007, 61, 1153–1162. [Google Scholar]
- Rao, P.S.; Sathyanarayana, D.N. Synthesis of electrically conducting copolymers of aniline with o/m -amino benzoic acid by an inverse emulsion pathway. Polymer 2002, 43, 5051–5058. [Google Scholar]
- Kuramoto, N.; Tomita, A. Aqueous polyaniline suspensions: Chemical oxidative polymerization of dodecylbenzene-sulfonic acid aniline salt. Polymer 1997, 38, 3055–3058. [Google Scholar] [CrossRef]
- Gomes, E.C.; Oliveira, M.A.S. Chemical Polymerization of Aniline in Hydrochloric Acid (HCl) and Formic Acid (HCOOH) Media. Differences Between the Two Synthesized Polyanilines. Am. J. Polym. Sci. 2012, 2, 5–13. [Google Scholar] [CrossRef]
- Wu, W.; Pan, D.; Li, Y.; Zhao, G.; Jing, L.; Chen, S. Facile fabrication of polyaniline nanotubes using the self-assembly behavior based on the hydrogen bonding: A mechanistic study and application in high-performance electrochemical supercapacitor electrode. Electrochim. Acta 2015, 152, 126–134. [Google Scholar] [CrossRef]
- Li, G.; Jiang, L.; Peng, H. One-dimensional polyaniline nanostructures with controllable surfaces and diameters using vanadic acid as the oxidant. Macromolecules 2007, 40, 7890–7894. [Google Scholar] [CrossRef]
- Sun, Z.; Geng, Y.; Li, J.; Wang, F. Chemical polymerization of aniline with hydrogen peroxide as oxidant. Synth. Met. 1997, 84, 99–100. [Google Scholar] [CrossRef]
- Zoromba, M.S.; Alghool, S.; Abdel-Hamid, S.M.S.; Bassyouni, M.; Abdel-Aziz, M.H. Polymerization of aniline derivatives by K2Cr2O7 and production of Cr2O3 nanoparticles. Polym. Adv. Technol. 2017, 28, 842–848. [Google Scholar] [CrossRef]
- Yasuda, A.; Shimidzu, T. Chemical Oxidative Polymerization of Aniline with Ferric Chloride. Polymer 1993, 25, 329–338. [Google Scholar] [CrossRef]
- Ding, Q.; Qian, R.; Jing, X.; Han, J.; Yu, L. Reaction of aniline with KMnO4 to synthesize polyaniline-supported Mn nanocomposites: An unexpected heterogeneous free radical scavenger. Mater. Lett. 2019, 251, 222–225. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, H.; Hou, C.; Qiao, M.; Chen, Y.; Zhang, H. Multidimensional polyaniline structures from micellar templates. J. Mater. Sci. 2017, 52, 2995–3002. [Google Scholar] [CrossRef]
- Stejskal, J.; Sapurina, I.; Trchová, M.; Konyushenko, E.N.; Holler, P. The genesis of polyaniline nanotubes. Polymer 2006, 47, 8253–8262. [Google Scholar] [CrossRef]
- Chao, D.; Lu, X.; Chen, J.; Zhang, W.; Wei, Y. Anthranilic acid assisted preparation of Fe3O4-Poly(aniline-co-o-anthranilic acid) nanoparticles. J. Appl. Polym. Sci. 2006, 102, 1666–1671. [Google Scholar] [CrossRef]
- Ji, J.; Li, R.; Li, H.; Shu, Y.; Li, Y.; Qiu, S.; He, C.; Yang, Y. Phytic acid assisted fabrication of graphene/polyaniline composite hydrogels for high-capacitance supercapacitors. Compos. Part B Eng. 2018, 155, 132–137. [Google Scholar] [CrossRef]
- Tomšík, E.; Ivanko, I.; Svoboda, J.; Šeděnková, I.; Zhigunov, A.; Hromádková, J.; Pánek, J.; Lukešová, M.; Velychkivska, N.; Janisová, L. Method of Preparation of Soluble PEDOT: Self-Polymerization of EDOT without Oxidant at Room Temperature. Macromol. Chem. Phys. 2020, 221, 2000219. [Google Scholar] [CrossRef]
- Kolouchova, K.; Groborz, O.; Skarkova, A.; Brabek, J.; Rosel, D.; Svec, P.; Starcuk, Z.; Slouf, M.; Hruby, M. Thermo- and ROS-Responsive Self-Assembled Polymer Nanoparticle Tracers for 19 F MRI Theranostics. Biomacromolecules 2021, 22, 2325–2337. [Google Scholar] [CrossRef] [PubMed]
- Morávková, Z.; Trchová, M.; Tomšík, E.; Čechvala, J.; Stejskal, J. Enhanced thermal stability of multi-walled carbon nanotubes after coating with polyaniline salt. Polym. Degrad. Stab. 2012, 97, 1405–1414. [Google Scholar] [CrossRef]
- Rakić, A.A.; Vukomanović, M.; Ćirić-Marjanović, G. Formation of nanostructured polyaniline by dopant-free oxidation of aniline in a water/isopropanol mixture. Chem. Pap. 2014, 68, 372–383. [Google Scholar] [CrossRef]
- Bláha, M.; Trchová, M.; Morávková, Z.; Humpolíček, P.; Stejskal, J. Semiconducting materials from oxidative coupling of phenylenediamines under various acidic conditions. Mater. Chem. Phys. 2018, 205, 423–435. [Google Scholar] [CrossRef]
- Kiefer, W. Recent Advances in linear and nonlinear Raman spectroscopy I. J. Raman Spectrosc. 2007, 38, 1538–1553. [Google Scholar] [CrossRef]
- El-Bashir, S.M.; Yahia, I.S.; Binhussain, M.A.; AlSalhi, M.S. Design of Rose Bengal/FTO optical thin film system as a novel nonlinear media for infrared blocking windows. Results Phys. 2017, 7, 1852–1858. [Google Scholar] [CrossRef]
- Murugesan, R.; Subramanian, E. Effect of organic dopants on electrodeposition and characteristics of polyaniline under the varying influence of H2SO4 and HClO4 electrolyte media. Mater. Chem. Phys. 2003, 80, 731–739. [Google Scholar] [CrossRef]
- Pouget, J.P.; Oblakowski, Z.; Nogami, Y.; Albouy, P.A.; Laridjani, M.; Oh, E.J.; Min, Y.; MacDiarmid, A.G.; Tsukamoto, J.; Ishiguro, T.; et al. Recent structural investigations of metallic polymers. Synth. Met. 1994, 65, 131–140. [Google Scholar] [CrossRef]
- Buron, C.C.; Lakard, B.; Monnin, A.F.; Moutarlier, V.; Lakard, S. Elaboration and characterization of polyaniline films electrodeposited on tin oxides. Synth. Met. 2011, 161, 2162–2169. [Google Scholar] [CrossRef]
- Gospodinova, N.; Ivanov, D.A.; Anokhin, D.V.; Mihai, I.; Brun, S.; Romanova, J.; Tadjer, A. Unprecedented Route to Ordered Polyaniline: Direct Synthesis of Highly Crystalline Fibrillar Films with Strong p-p Stacking Alignment. Macromol. Rapid Commun. 2009, 30, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Tadyszak, K.; Strzelczyk, R.; Coy, E.; Ma, M.; Augustyniak-jab, M.A. Size effects in the conduction electron spin resonance of anthracite and higher anthraxolite. Magn. Reson. Chem 2016, 54, 239–245. [Google Scholar] [CrossRef]
- Dyson, F. Electron Spin Resonance Absorption in Metals. II. Theory of Electron Diffusion and the Skin Effect. Phys. Rev. 1955, 98, 349–358. [Google Scholar] [CrossRef]
- Wertz, J.; Bolton, J. Electron Spin Resonance; Chapman and Hall: New York, NY, USA; London, UK, 1986; ISBN 9789401083072. [Google Scholar]
- Popovych, V.; Bester, M.; Stefaniuk, I.; Kuzma, M. Dyson line and modified Dyson line in the EPR measurements. Nukleonika 2015, 60, 385–388. [Google Scholar] [CrossRef]
- Lozinskaya, E.I.; Ponkratov, D.O.; Malyshkina, I.A.; Grysan, P.; Lingua, G.; Gerbaldi, C.; Shaplov, A.S.; Vygodskii, Y.S. Self-assembly of Li single-ion-conducting block copolymers for improved conductivity and viscoelastic properties. Electrochim. Acta 2022, 413, 140126. [Google Scholar] [CrossRef]
- Kozarenko, O.A.; Dyadyun, V.S.; Papakin, M.S.; Posudievsky, O.Y.; Koshechko, V.G.; Pokhodenko, V.D. Effect of potential range on electrochemical performance of polyaniline as a component of lithium battery electrodes. Electrochim. Acta 2015, 184, 111–116. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RH,0 (nm) 1 | Rg,app (nm) | Mw app × 108 (g mol−1) | Dapp × 1015 (cm2 s−1) | v |
|---|---|---|---|---|
| 56.8 ± 11.4 | 97.6 ± 19.5 | 7.3 ± 1.5 | 2.89 ± 0.075 | 1.72 |
| Sample | A, (arb. u.) | w, (G) | b, (abr. u.) | A/|B| | x0, (G) |
|---|---|---|---|---|---|
| Carbon cloth | 0.16 | 73.7 | 0.33 | 1.6 | 3503.9 |
| PANI-c | 1.19 | 34.7 | 0.11 | 1.08–1.34 | 3497.9 |
| PANI-a | 14.10 | 0.87 | 0.23 | 1.33–1.43 | 3497.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomšík, E.; Nosov, D.R.; Ivanko, I.; Pokorný, V.; Konefał, M.; Černochová, Z.; Tadyszak, K.; Schmidt, D.F.; Shaplov, A.S. A New Method to Prepare Stable Polyaniline Dispersions for Highly Loaded Cathodes of All-Polymer Li-Ion Batteries. Polymers 2023, 15, 2508. https://doi.org/10.3390/polym15112508
Tomšík E, Nosov DR, Ivanko I, Pokorný V, Konefał M, Černochová Z, Tadyszak K, Schmidt DF, Shaplov AS. A New Method to Prepare Stable Polyaniline Dispersions for Highly Loaded Cathodes of All-Polymer Li-Ion Batteries. Polymers. 2023; 15(11):2508. https://doi.org/10.3390/polym15112508
Chicago/Turabian StyleTomšík, Elena, Daniil R. Nosov, Iryna Ivanko, Václav Pokorný, Magdalena Konefał, Zulfiya Černochová, Krzysztof Tadyszak, Daniel F. Schmidt, and Alexander S. Shaplov. 2023. "A New Method to Prepare Stable Polyaniline Dispersions for Highly Loaded Cathodes of All-Polymer Li-Ion Batteries" Polymers 15, no. 11: 2508. https://doi.org/10.3390/polym15112508