3.4.1. Thermal Stability: TGA
The
stability of the prepared vulcanized rubber composites
against oxidation, as well as against
anaerobic thermolysis, was investigated using
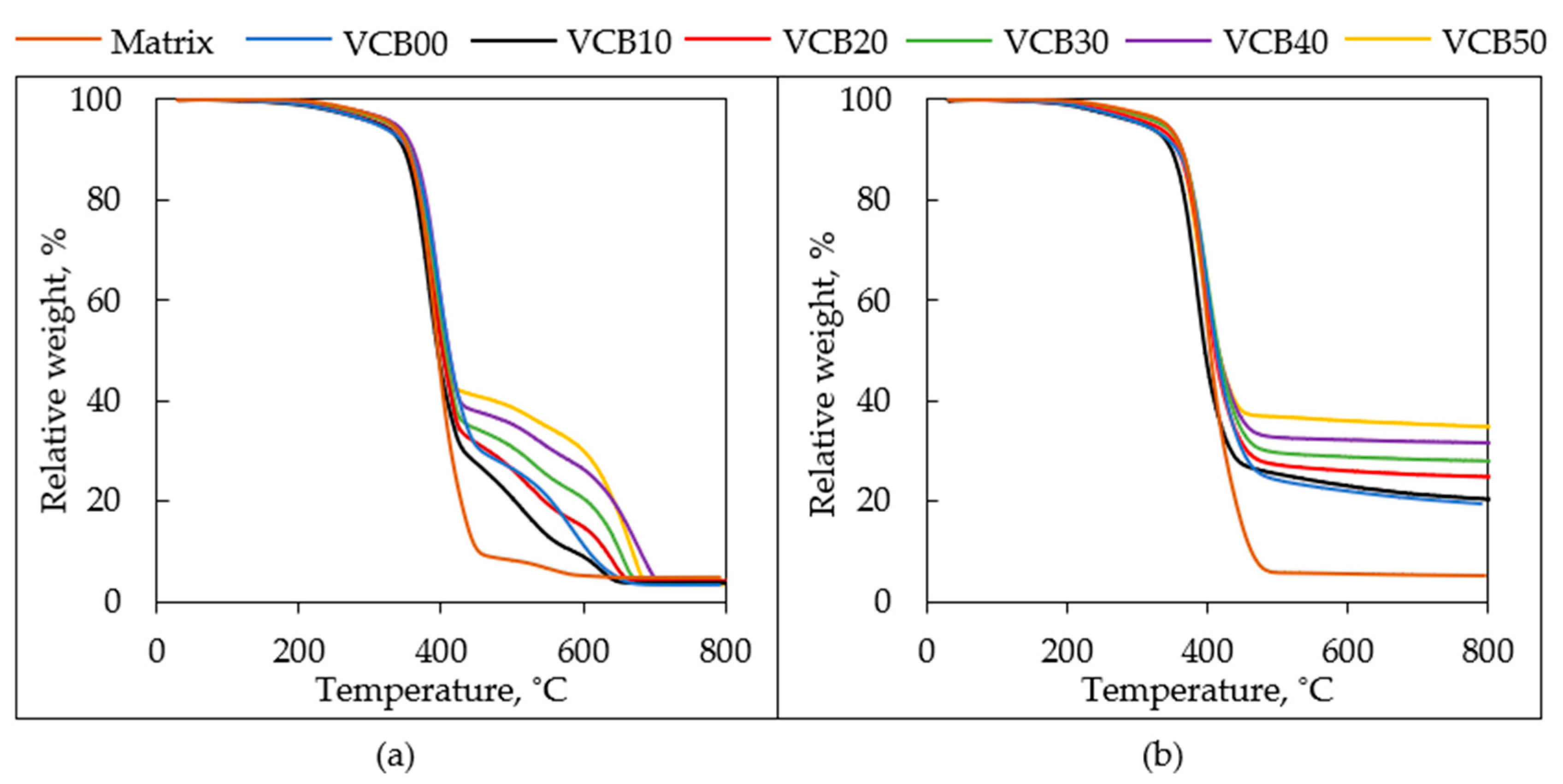
thermogravimetric analysis in air and in nitrogen, respectively, at the heating rate of 10 °C/min. The temperature dependence of the relative sample mass (‘TGA proper’) and of its derivative (dTGA) are presented in
Figure 7 and
Figure 8, respectively, for both types of TGA tests (in air and N
2). It can be summarized that the variation of the CB/HC ratio in the fillers phase only has a
modest effect on the temperatures of the onset and maximal rate of
decomposition, both in air and in nitrogen. Nevertheless, there appears to be a
detectable synergic stabilizing effect of the combined fillers, which in air is maximal at 10 and 20 phr of HC (VCB40 and VCB30), and in nitrogen at 20 and 30 phr of HC (VCB30 and VCB20), respectively. Even stronger is the anti-oxidizing effect of HC if it is used as the exclusive filler in VCB00. This composite has the highest temperatures of maximum decomposition (peaks in
Figure 8), both in air and nitrogen. Even more importantly, the intermediate char fraction (forming above 400 °C), which is observed in VCB00, is distinctly higher than in VCB10 and approximately equal to the one in VCB20. In nitrogen, the permanent char residue of VCB00 also is higher than would be expected from the trends. Apart from the
anomalous behavior of VCB00, a distinct effect of the
increasing HC fraction is the
decrease of the temporary (in air) or permanent (in nitrogen)
char fraction. The strong stabilizing effect of HC in VCB00, as well as the significant synergic stabilization in VCB20–VCB40, can be attributed to the
reactivity of HC (to its abovementioned organic character, characterized in detail in [
27]), which later, at the highest temperatures, leads to faster oxidation or pyrolysis of the previously stabilized char fraction. Interestingly, the chemical interaction of HC with CB leads to a less efficient stabilization in VCB20, especially in VCB10. Details of the observed TGA trends are discussed below.
The course of the weight loss curves: the
degradation process in both atmospheres
always starts at about 200 °C in all the samples (see
Figure 7a,b), while
5% of weight loss is reached between 310 and 350 °C. Near
390–400 °C, all the studied samples in both atmospheres
rapidly lose a substantial amount of their mass. This ‘step’ in the curves corresponds to the nearly complete
degradation of the polymeric matrix (natural rubber). Because of nearly identical (at the same phr of CB) decomposition temperatures as well as curve courses in both air and nitrogen, the degradation seems to have an analogous first mechanistic step, both in air and in N
2, namely rubber thermolysis, rather than oxidation (which follows as a subsequent process in air). Distinct differences in the TGA curves are noted at higher temperatures;
above 420 °C, a temporary (in air) or permanent (in nitrogen)
char fraction is observed, formed via thermal fragmentation, followed by oxidative or anaerobic crosslinking. In both atmospheres, the char fraction is the highest if the filler phase consists exclusively of carbon black (sample VCB50), and it decreases with an increasing fraction of HC in the fillers phase (due to the organic character of HC). However, as mentioned above, in the case of VCB00 (filled exclusively by 50 phr of HC), the char step is anomalously high, which suggests a strong anti-oxidizing and radical reactivity of HC (the effect is moderated by the addition of small amounts of CB, although the latter also has a stabilizing effect).
In air, the
oxidative degradation of temporarily formed char becomes fast near 650 °C. Above 700 °C, all organic residues are degraded in the air atmosphere. There
remains an
ash residue of ca. 3 to 4%, which does not undergo any changes until 800 °C (see
Table S1). This small but measurable ash fraction can be attributed to zinc oxide (ZnO) which was added to the rubbers (4 phr corresponding to 2.5 wt.% in the recipe in
Table 1). Its eventual partial reaction with oxidized sulfur (partial transformation into ZnSO
4) and also the small contribution of mineral ash from hydrochar (0.42 wt.% in the HC filler, as determined in previous work [
27]) could be responsible for the higher ash content, than that corresponding to pure ZnO from the rubber recipe.
In
nitrogen, the
char content reaches final values already near 550 °C in most samples (finished carbonization). The
final char residues at 800 °C are
between 19 and 34 wt.% (see
Figure 7b, except the filler-free matrix, where the residue is just 5.1 wt.%, slightly more than the ZnO content), depending on the CB/HC ratio (see
SI—Table S2). For comparison, the content of the combined filler phases was always equal to 31.45 wt.% (≡50 phr in the rubber recipe in
Table 1 in
Section 2. Materials and Methods), to which the CB component contributed by 0 to 31.45 wt.%, and HC by 31.45 wt.% down to 0 wt.%. In the case of VCB50 (see
SI—Table S2), only 3.2 wt.% in the final char (34.65 wt.%) originates from components other than the original amount of CB (31.45 wt.%). These 3.2 wt.% well correspond to the mentioned content of ZnO in the rubber recipe in
Table 1 (2.52 wt.%; thus, the additional residue other than ZnO and CB amounted to just 0.68 wt.%). Hence, the thermolysis of natural rubber to gaseous products can be considered practically quantitative in nitrogen at T > 450 °C; if hydrochar (HC) is incorporated, the final char residue increases (see
Figure 7b), but less so than would correspond to the content of the HC filler. According to calculations in
Table S2 (subtraction of inert CB and ZnO), an additional residue ranging between 3.93 wt.% (in VCB40) and 16.78 wt.% (in VCB00) can be attributed to HC, whose content in the intact sample was 6.29 to 31.45 wt.%, however. Hence,
only 53–62 wt.%
of the hydrochar mass survived the thermolysis in nitrogen as a consequence of the organic character of HC (its carbon content was only 71%, as found in [
27]). Due to the mentioned anomaly of VCB00 (very strong anti-oxidizing properties of pure HC), the smallest carbonized fraction of HC was found in VCB10 (46% of the original mass).
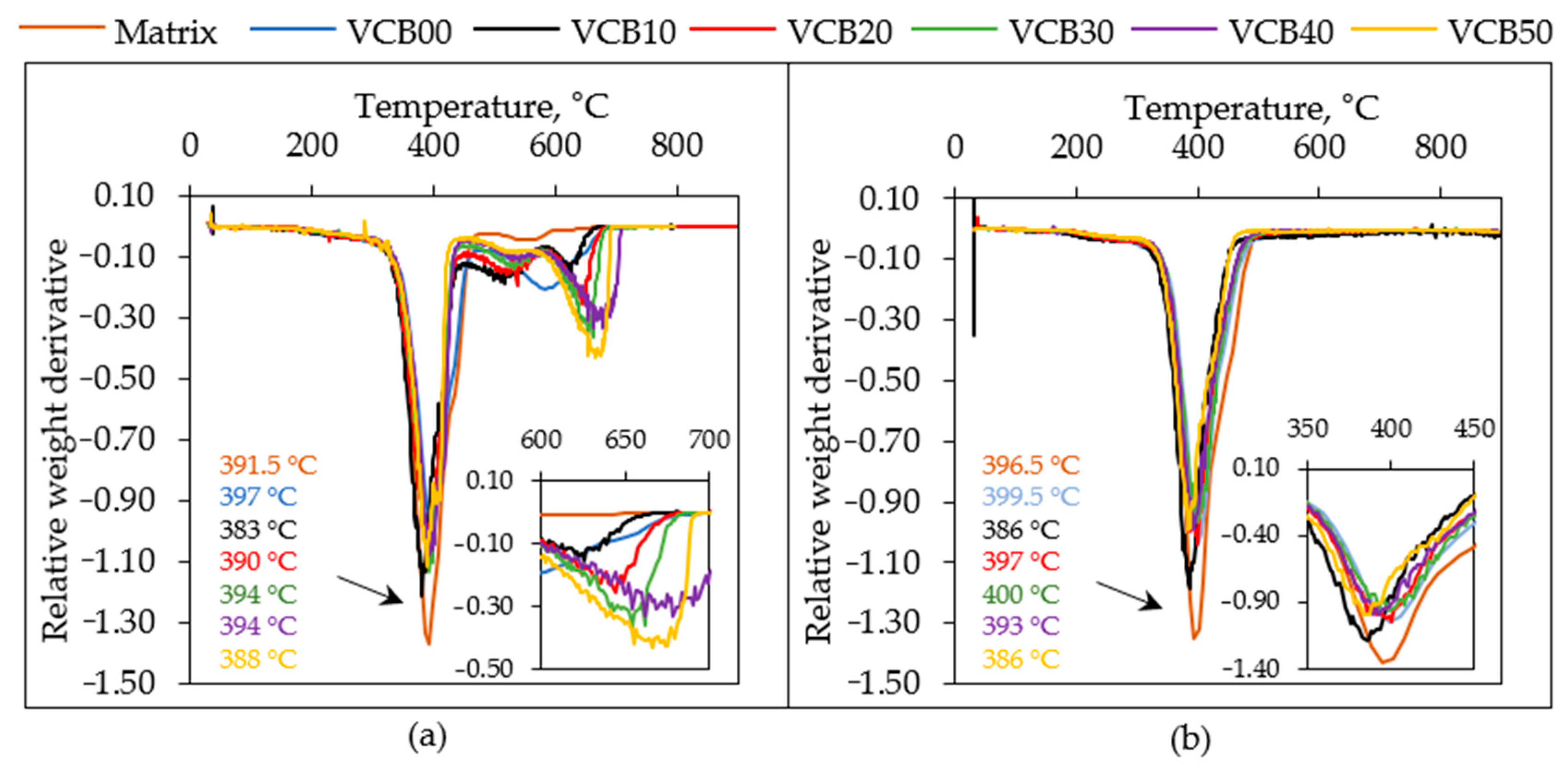
Figure 8 shows the
derivative analysis (dTGA) of the above TGA results. The negative peaks of the “relative weight derivative” indicate temperature points at which degradation rates achieve local maxima (rapid weight losses) and signalize different decomposition processes.
Both in air and in nitrogen, the abovementioned
dominant degradation process, the
thermolysis of the
natural rubber polymer, is signalized by the intense dTGA peak near
T = 390 °C. This process corresponds to the main step in the TGA curves in
Figure 7. The main degradation peaks of all the studied samples, both in air and in nitrogen, display very similar positions and also nearly identical intensities. In air, the peak slightly shifts from 391.5 °C to 397, 383, to 390, 394, 394, and then back to 388 °C if going from the neat matrix via VCB00 to VCB50. This trend in the temperatures of decomposition maxima confirms the abovementioned
strong anti-oxidizing effect of CB-free HC, as well as the synergic stabilizing effect of HC and CB at small to medium HC concentrations (VCB40, VCB30, and VCB20). The low temperature of the main peak of VCB10 (
Tdec = 383 °C) indicates that at this co-filler ratio, the chemical effect of HC is being moderated by the CB co-filler, as already suggested by the mechanical analysis of the swollen samples (effect on crosslinking during vulcanization). In the nitrogen atmosphere, the trends of the main decomposition peak are nearly identical to those in air, including the shifting temperatures of the decomposition maxima, namely 396.5, 399.5, 386, 397, 400, 393, and 386 °C, if going from the neat matrix via VCB00 to VCB50. VCB10 displays an analogous instability in nitrogen to that in air. The above results suggest that
hydrochar (HC) exerts an analogous chemical influence on rubber thermolysis during aerobic and anaerobic degradation. Finally, it can be noted that the neat matrix displays the most intense main decomposition peak, both in air and in nitrogen. This is because this sample contains only one significant component, the natural rubber itself.
Separate oxidation of the co-fillers during degradation in air: In the case of the
dTGA curves of samples tested in air (see
Figure 8a), two smaller peaks can be observed in the temperature region from 400 to 700 °C. They correspond to the structured smaller step in the mass loss curves (see
Figure 7a) in the same temperature region. The peaks near 520 and 640 °C were assigned to the oxidation of hydrochar (together with an eventual charred fraction of the matrix) and of carbon black, respectively, as will be explained below.
CB oxidation (Tmax = ca. 640 °C): The height of the second (final) TGA sub-step in
Figure 7a, ranging from 550 to 700 °C (corresponding to the dTGA peak at 640 °C in
Figure 8a), which is part of the mentioned structured TGA mass loss step extending from 400 to 700 °C in
Figure 7a, clearly correlates with the CB content. The sum of the weight percentage of embedded CB plus the percentage of embedded ZnO well corresponds to the height of the mentioned 550–700 °C sub-step (see
SI—Table S1). In the example of the HC-free composite VCB50, it can be seen that the height of the 550–700 °C sub-step (here, the first, lower-temperature sub-step is practically absent) in
Figure 7a is ca. 35.0%, while the sum of the contents of ZnO + CB in that sample is 33.97%. Similar observations can be made for the other nanocomposites as well (see
SI—Table S1: e.g., VCB10: ca. 10.5% vs. 8.81%). The intensity of the dTGA peak associated with the oxidation of the CB filler increases with the CB content. Its position moderately shifts, as it is influenced by the previous oxidation of the hydrochar and charred matrix. If going from VCB10 to VCB50, the peak moves from 627 °C to 645, 656, 678, and finally slightly back to 669 °C.
Oxidation of Hydrochar + Charred Matrix (Tmax = ca. 520 °C): The ‘first’ TGA sub-step (region: 400–550 °C in
Figure 7a, corresponding to the peak at 520 °C in dTGA in
Figure 8a), which is part of the discussed structured TGA mass loss step at 400–700 °C in
Figure 7a, logically was assigned to the simultaneous oxidation of the charred matrix and of hydrochar, as the latter filler is less resistant to oxidation than CB, due to the organic nature of HC. The small but significant exclusive
contribution of the charred matrix (via oxidative crosslinking) is visible both in
Figure 7a (steps) and in
Figure 8a (small dTGA peak) in the case of VCB50, which contains no HC at all, but still displays a small ‘first sub-step’ (height ca. 7%; combined the 1st and 2nd steps: ca. 42%). In all samples, the combined height of the sub-steps at 400–550 °C and 550–700 °C roughly corresponds to the combined fractions of ZnO, CB, and hydrochar (see
SI—Table S1). The contribution of the 1st sub-step to the combined structured step expectedly increases with the fraction of the embedded HC. In VCB00 (with 50 phr of HC, where the second sub-step is absent), the height of the first sub-step is ca. 30.5%, which is slightly less than the HC content of 31.45%. However, at low hydrochar contents (e.g., in the mentioned VCB50 and VCB40), the 1st step is somewhat higher than the HC content due to the presence of a charred matrix, whose content is evaluated in the last column of
SI—Table S1. At high hydrochar contents (VCB00, VCB10, and less so VCB20), on the other hand, the 400–550 °C sub-step is lower than expected (negative values for charred matrix fraction in
Table S1), which means that
a part of the hydrochar degrades during the degradation of the natural rubber. The small dTGA peak near 520 °C, which corresponds to the simultaneous oxidation of hydrochar and the charred matrix, steadily decreases with decreasing content of hydrochar (the associated sub-step in
Figure 7a diminishes). The position of this dTGA peak shifts from 513 °C to 526, 534, 539, and back to 525 °C if going from VCB10 to VCB50. As noted in other chemistry-related properties, VCB00 (with HC as the sole filler) displays an anomalous anti-oxidizing stabilization. The peak of the oxidation of HC and the charred matrix is shifted to markedly higher temperatures, namely to 585 °C (between the dTGA peaks of the first and second sub-step). As also mentioned above (and as observed in other chemistry-related properties), in VCB40, which is close to VCB00 in its composition, the stabilizing effect of HC and CB is mutually canceled. Finally, the neat matrix also generates an intermediate char fraction upon oxidation (see
Figure 7a: nominal step height including ash 9.1%, char itself: 4.35%), whose decomposition maximum is somewhat higher than even that of VCB40, namely at 553 °C. The shifting degradation maxima of CB, as well as of HC + the charred matrix (near
Tmax = ca. 640 °C and
Tmax = ca. 520 °C, respectively), further confirm the finding
that CB-free HC displays a strong anti-oxidizing effect where moderate to medium contents of hydrochar (HC) in combination with CB exert a synergic chemical stabilizing effect and that the mentioned stabilization affects
both the oxidative and the anaerobic degradation processes.
3.4.2. Thermo-Oxidative Aging Test
The above-discussed analyses at elevated temperatures indicated that rapid thermal and/or oxidative degradation onsets above 200 °C (in the fast TGA analyses performed at 10 °C/min) or even near 150 °C (in DMTA analyses at 3 °C/min). Hence, the moderately elevated constant temperature of 180 °C was selected for performing thermo-oxidative aging tests ‘at harsh conditions’ by exposing the prepared samples to circulating air at this temperature (and in darkness) for different periods of time. Milder aging tests simulating real-use conditions were performed (see further below 3.4.3 Simulated weathering).
In the design of the thermo-oxidative aging test, the authors’ previous experience with the study of oxidation stability of poly(propylene oxide)-based polymer networks [
31] was useful, where differences between the samples led to marked effects in relatively short treatment times. The thermo-oxidative aging tests were performed using an oven with forced air circulation. Several specimens of each tested material were prepared. Each specimen was then subjected to a specific oxidation time at 180 °C, namely to 30, 60, 180, or 360 min. The mass of each specimen was recorded before and after the oxidation treatment. The
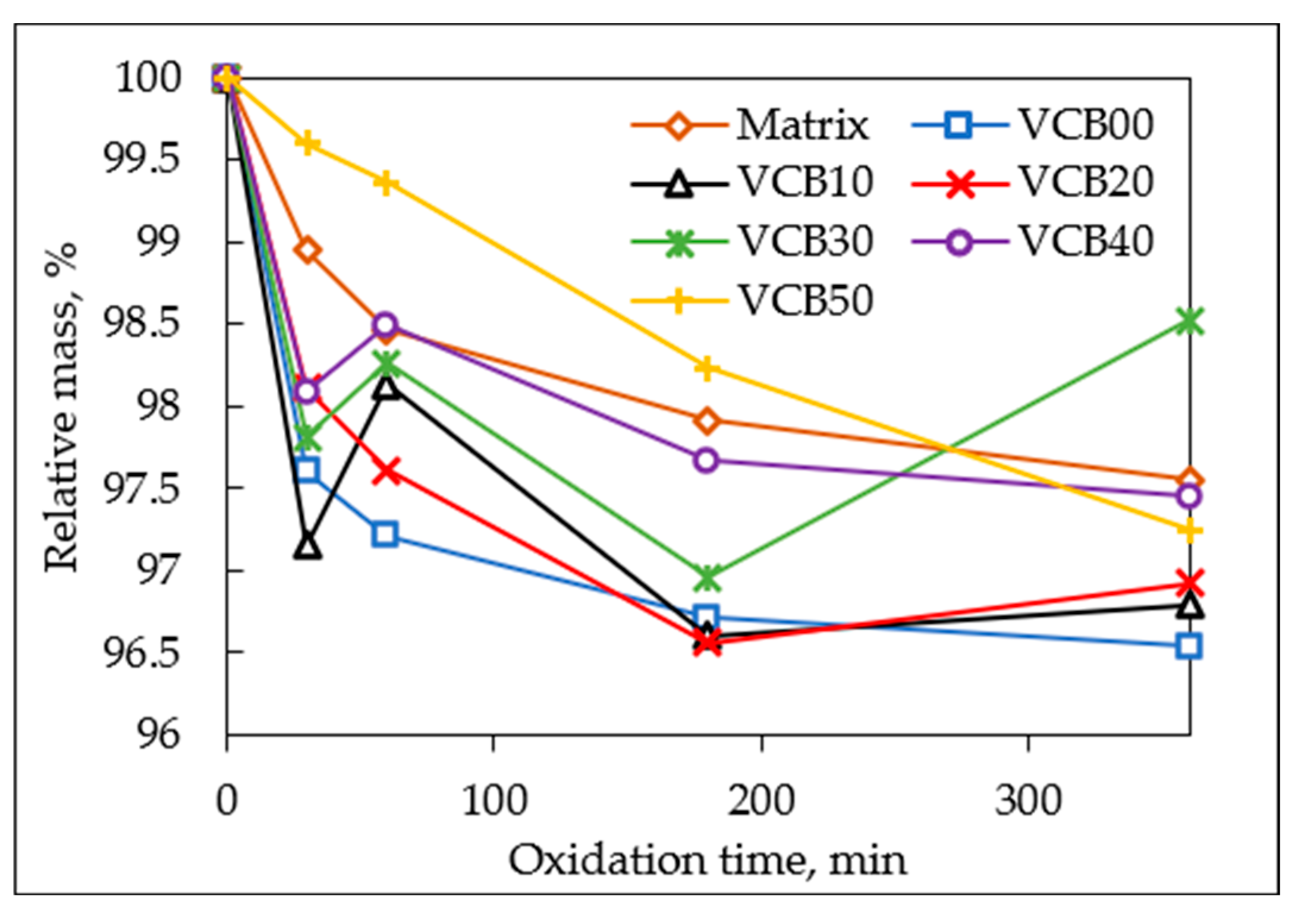
oxidation-time-dependent relative masses of all the tested materials are presented in
Figure 9 (the experimental data can be found in
SI—Table S3). Each oxidized specimen was subsequently subjected to thermo-mechanical analysis (DMTA) to evaluate oxidation-time-dependent changes in its properties (the results of these analyses are discussed further below).
As seen in
Figure 9 (and in
SI—Table 3), the
mass losses of the tested specimens during the thermo-oxidative aging tests in the oven
were relatively modest (in spite of the released odor). After 6 h of these tests, the final mass losses ranged between 1.5% and 3.5% (
Figure 9), although an error margin of at least ±0.5% must be considered (because of manipulation with the samples). Due to the error margin, the small oscillations seen in the zoomed
Figure 9,
Figure 10,
Figure 11 and
Figure 12 should not be considered local trend reversals. Moreover, the TGA analysis did not indicate net mass uptake in any temperature region, e.g., via oxygen incorporation. If the trends (within error margins) are compared, it can be stated that the onset of oxidation-induced mass loss (in the first 30 min) is accelerated by the presence of hydrochar (HC), but later (3 h, 6 h) the mass loss practically stops in HC presence, while in its absence it continues (see trendline of VCB50 in
Figure 9). These results again confirm the
chemical effect of the hydrochar, which
accelerates the onset of degradation on the one hand (see the first 30 min; possibly due to the anti-oxidizing properties of HC, which could favor unhindered thermolysis) but later
stabilizes the composites (slowed-down mass loss in later stages in the samples VCB30, VCB40). An approximate trend (while still considering the significant error margin) can be observed. The smallest and slowest mass loss occurred in VCB50, while the largest and fastest one was in VCB00. The neat matrix itself degrades slightly more and also initially faster than VCB50. This trend is opposed to the trend in the below-discussed constancy of mechanical properties, where despite the mass loss trends, the sample VCB00 displays the distinctly highest constancy (the stabilizing effect of HC hence is connected with mass consumption).
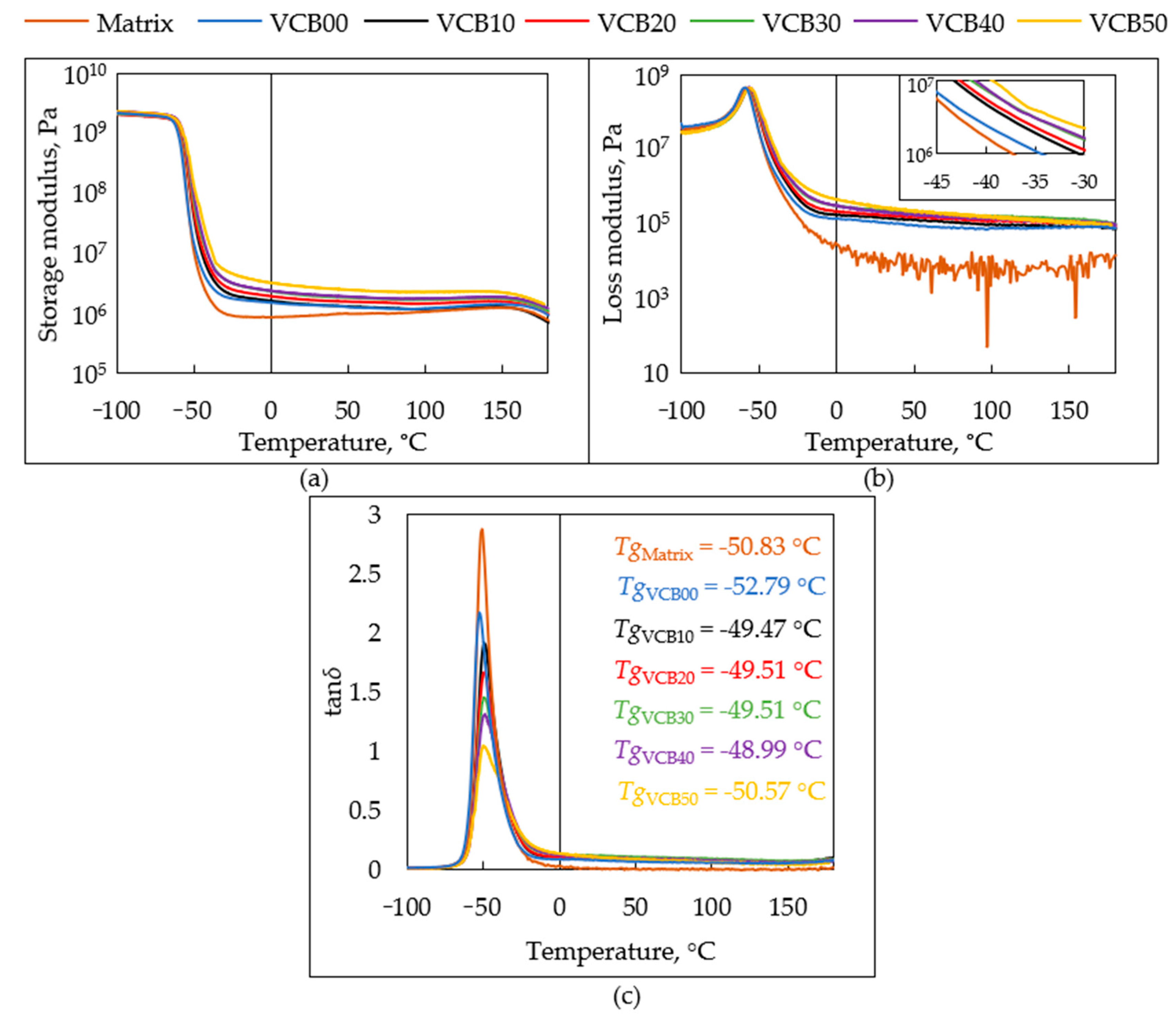
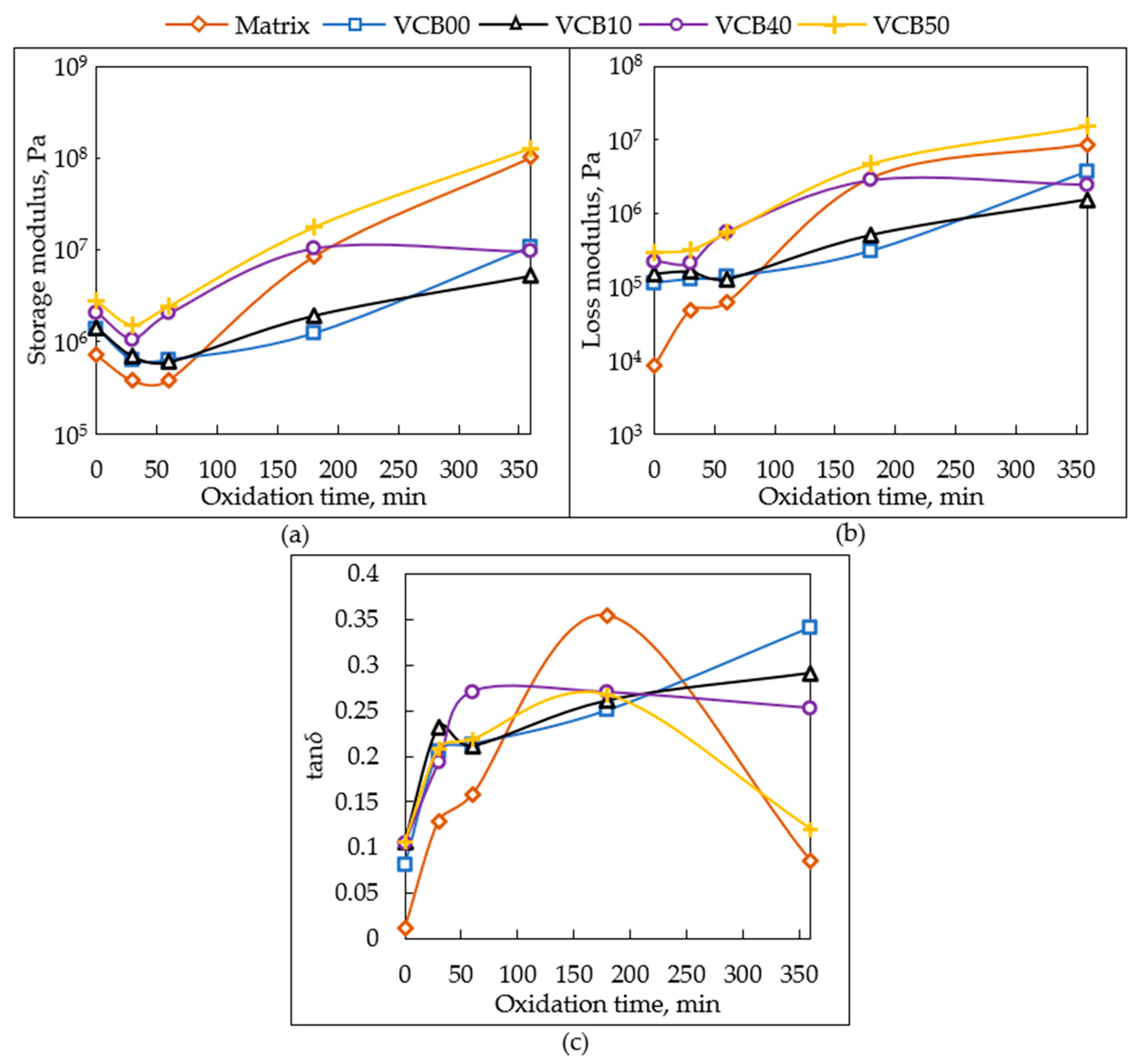
The
dynamic-mechanical thermal analysis (DMTA) of oxidized specimens yielded temperature-dependent plots of the storage modulus (
G′), of the loss modulus (
G″), and of the loss factor tan
δ. The results are analyzed in
Figure 10,
Figure 11 and
Figures S1–S3 and in
SI—Figures S1–S3: Overlaid DMTA curves of selected samples can be seen in
SI—Figures S1 and S2, in
Figure 11, and in
SI—Figure S3. Oxidation times, after which the different samples are compared in the latter figures, are 30, 60, 180, and 360 min, respectively. Additionally, in
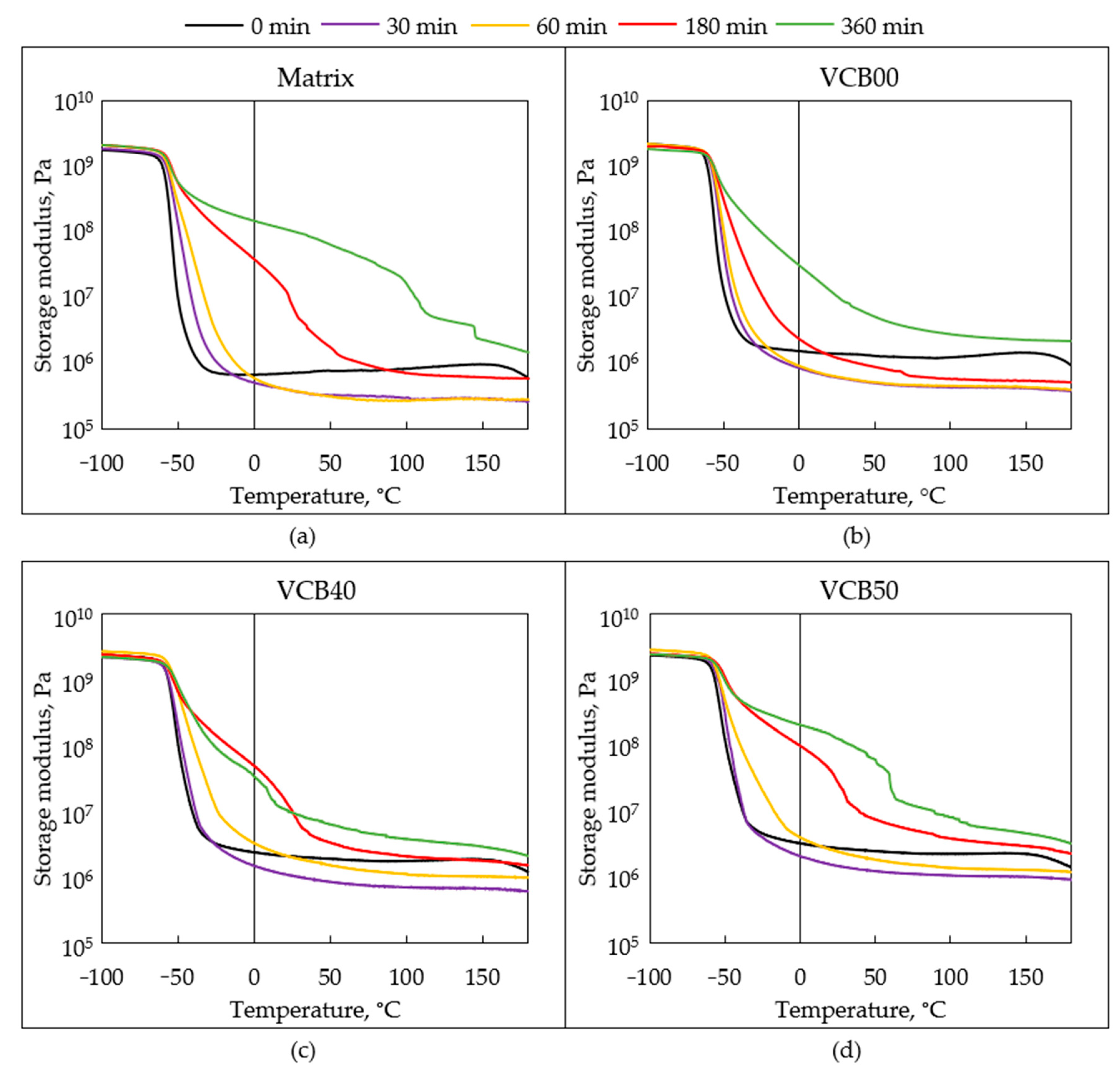
Figure 12, the effects of the progressing thermo-oxidative aging on the examples of selected samples are compared.
General trends: After oxidation times of 30 and 60 min, all the DMTA curves displayed practically identical shapes as in the case of the intact samples prior to oxidation, which can be seen in
Figure 4 further above (
Section 3.2.2. Thermo-mechanical properties (DMTA)). The only difference between the intact, 30-min, and 60-min oxidized samples was in the values of their moduli in the rubbery region. After 180 min of the thermo-oxidative aging test, the DMTA curves of the rubber composites are already markedly altered, as can be seen in
Figure 11 (similar curves after 360 min are shown in
Figure S3). The above
Figure 10 plots the
trends of the changes in dynamic-mechanical properties measured at room temperature, namely in
G′ (25 °C),
G″ (25 °C), and tan
δ (25 °C),
as a function of the oxidation time. The
changes in the storage modulus G′ (25 °C), which result
from the endured time of the thermo-oxidative aging test (
Figure 10a), supply interesting information about the crosslinking density in the aging samples. It can be seen that
G′ (and hence the crosslinking density) initially significantly decreases during the first 30 min, after which (at 60 min) the modulus (crosslinking density) either stagnates (matrix, VCB00, and VCB10), or it increases again (VCB40, VCB50). Next, a steady modulus increase is observed for most samples, but in VCB40, which together with VCB30 often was noted as displaying a synergic stabilizing effect of the co-fillers, the modulus increases only between 30 and 180 min, and after that (180–360 min) it stagnates or even slightly decreases. The trends of
G′ suggest that two processes occur during the thermo-oxidation tests.
At first (0 to 30 min), due to the presence of anti-oxidants in the commercial recipe of the studied rubbers, a slow
quasi-anaerobic thermolysis occurs. Oxygen is at least partly captured, and radical reactions are blocked by the anti-oxidants, while the polymer matrix network thermally degrades (polysulfide crosslinks can start to dissociate).
After 30 (or 60 min), the anti-oxidant additives are consumed, and the reactions between natural rubber and oxygen prevail and lead to
additional oxidative crosslinking, which causes the modulus to rise (the material becomes stiffer and becomes brittle). This oxidative crosslinking is the fastest in hydrochar-free VCB50 and in the neat matrix, while it is distinctly slower in the HC-rich samples VCB10 and VCB00 (see the smaller slope of the curve in
Figure 10a, between 60 and 360 min). The sample VCB40, which contains only 10 phr of HC but in which a stabilizing synergy effect of the combined filler pair was observed by several other methods, displays initially (from 0 to ca. 120 min) the same behavior with VCB50, but after 180 min, the oxidative crosslinking in VCB40 is wholly suppressed (slightly decreasing modulus instead of steady growth). This also correlates with the practically stopped mass loss of VCB40 after 180 min of the thermo-oxidative aging test (as discussed above,
Figure 9b).
Trends of G″ (25 °C) as a function of the oxidation time are very similar to the trends of
G′ (see
Figure 10b vs. a), but the range of values of
G″ is narrower if the behavior of the neat matrix is not considered. In the initial oxidation period (presence of the anti-oxidant additives), between 0 and 30 (60) min,
G″ just stagnates or only very slightly decreases in contrast to the strong decrease in
G′. In later stages,
G″ moderately increases in all samples except VCB40 (which displays synergic stabilization). The lowest
G″ values among the composites are observed for VCB00 and VCB10. The neat matrix, in contrast to the composites, displays a nearly steady increase of
G″ with the duration of the thermo-oxidative aging test. This increase markedly slows down after 3 h. The factors responsible for the
G″ trends are similar to the case of
G′. In the early stage, the thermolysis of crosslinks does not significantly influence the molecular friction responsible for
G″ in the case of the composites crosslinked by numerous bonds to the filler phase. Without the filler, however, the thermolysis generates an immediate increase of
G″ in the matrix. In the later stage of the thermo-oxidative aging test, from 30 (60) to 360 min, the oxidative crosslinking reactions produce not only network junctions but also branched structures in all the samples. The branched structures then raise molecular friction and hence
G″.
Trends of tan δ (25 °C) as a function of the oxidation time (see
Figure 10c) follow from the trends of
G′ and
G″, as well as from the precise values of both. Here it can be seen that in the
initial period (0 to 30 (60) min), the
anaerobic degradation markedly raises tan δ in all samples, while the
subsequent period of oxidative crosslinking practically leads to a
stagnation of the tan
δ value (VCB40), or to its modest (VCB10) or even distinct
further increase (VCB00). In VCB50 and in the neat matrix, where the additional crosslinking is very prominent in the late stages (see
Figure 10a), tan
δ markedly drops between 180 and 360 min, approximately to (VCB50), or close to (neat matrix) its starting value.
The glass transition temperatures (Tg), obtained from the tan
δ = f(
T) graphs in
Figures S1c, S2c,
Figure 11c and
Figure S3c, were defined here as temperatures of maxima of the tan
δ peaks in the mentioned graphs. In contrast to
G′ and
G″, the tan
δ = f(
T) graphs are much more sensitive indicators of endured oxidation and of the resulting structural changes. The intact rubber composites display very simple single tan δ peaks at their
Tg (see
Figure 4c further above). A special case is the intact VCB50, where an overlaid peak of an immobilized matrix fraction can be recognized as an asymmetry in the main transition peak, including a shoulder on the higher-temperature slope of the main
Tg peak. Nevertheless, the
Tg values of the intact rubber composites are practically identical.
After
30 min of the thermo-oxidative aging (
Figure S1c), the
Tg peaks are still very similar to that in the intact samples (in
Figure 4c), but the peak heights are significantly lower, and the values of tan δ in the post-
Tg plateau are higher. In the case of VCB50, the overlaid peak of the immobilized matrix nearly becomes quite visible. In the other samples, the symmetry of the peaks slightly varies (overlaid transitions) so that the nominal
Tg values oscillate. Additionally, the nominal
Tg values of all samples are slightly up-shifted (by 2 to 5 °C) compared to the intact state, indicating a somewhat more prominent role of the fraction of immobilized matrix.
After 60 min of thermo-oxidative aging, its effects on the Tg peaks become very distinct. The overlaid peak of the immobilized fraction in VCB50 becomes much more prominent (and broader). In VCB40, the transitions of free and immobilized matrix fractions are not yet separated, but their superposition yields a distinctly broadened and unsymmetrical single peak with a highly up-shifted (in T) main maximum (intact: −48.99 °C after 60 min: –29.35 °C). In VCB10, filled with 40 phr of HC (+ 10 phr of CB), the changes in the main transition peak are still modest after 60 min, but the nominal Tg value is up-shifted by several additional degrees (due to the immobilized fraction).
After 180 min of the thermo-oxidative aging test, the
Tg peaks in
Figure 11c are very different from the ones of the intact samples.
Separate transition peaks are observed for the
free matrix fraction and for the prominent
immobilized fraction, even in the case of VCB10 (in which sample the peaks still partly overlap). The separate
Tg peaks indicate
micro-phase separation between domains of the free and immobilized matrix. The transition of the free matrix is slightly down-shifted (in
T) in VCB50 and VCB40 in comparison to intact samples (due to more irregular structure), while in VCB10, it is slightly up-shifted (in
T), but less so than after 60 or 30 min. The prominent immobilized phase displays relatively high
Tg values: −10 °C (VCB10), +15 °C (VCB20), +40 °C (VCB30), +23 °C (VCB40), and +27 °C (VCB50), respectively. The values of tan
δ in the post-
Tg plateau are relatively high. The
oxidized matrix displays the highest
Tg of the immobilized fraction (+32 °C), and the peak of this fraction is more prominent than the one of the intact matrix fraction (which is below the analogous peak of VCB50 in
Figure 11c). Hence, the
crosslinking in the neat matrix is most prominent. On the other hand,
VCB00, after 180 min, displays just a shoulder (centered on 0 °C and extending from ca. –20 to ca. +20 °C) on the broadened main transition peak, which indicates the
slowest progress of oxidative crosslinking in this sample. In VCB10, where the tan
δ = f(
T) curve displays a similar course to the one of VCB00, the peak of the immobilized fraction is already separated from the main transition.
After 360 min (6 h) of the thermo-oxidative aging (
Figure S3) the changes in the
Tg peaks intensify. The peaks of the immobilized phase get broader and shift/extend to even higher temperatures. Except in the HC-free sample VCB50, the
Tg peak of the ‘free’ matrix also broadens and becomes structured (indicating further chemical changes of the original ‘free matrix’).
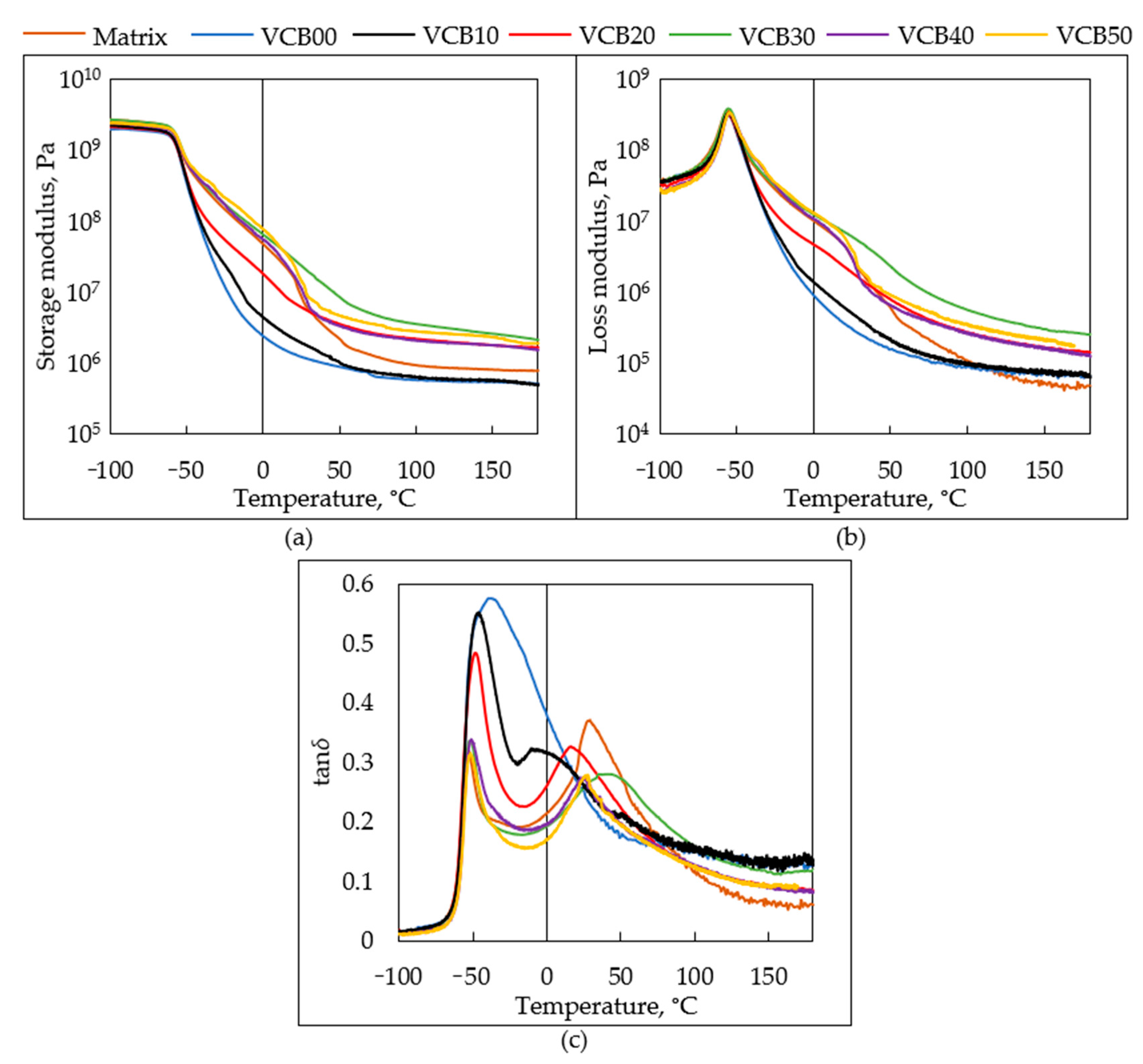
The
G′ = f(
T) and
G″ = f(
T)
curves of the strongly oxidized samples (see
Figure 11a,b: after 180 min, and
SI—Figure S3: after 360 min, as well as
Figure 12: general trends in the curves of
G′) display
distinctly different shapes if compared with the curves of the intact samples in
Figure 4a,b. In the case of both
G′ and
G″, an additional step appears at temperatures above the main glass transition (while the main
Tg is marked by the initial (low-temperature-) step in
G′ and by a peak in
G″). The step additional high-temperature step in
G′ is small in VCB10 and undistinguishable in VCB00 (here, the slope of the main step becomes less steep). On the other hand, the additional step is most prominent in the neat matrix (where the cracking of the specimen can be observed near 150 °C in
Figure 11c).
The
additional step in G′ results from
oxidative crosslinking, which yields an immobilized phase, while it also yields
branching and, hence, additional friction (smaller steps in
G″ in
Figure 11a,b and
SI—Figure S3). Interestingly, in the case of the sample VCB40, for which a stabilizing synergy effect of the co-fillers was observed in several different experiments, the additional steps in
G′ and
G″ decrease (simultaneously with the stopped weight loss) if the oxidation time is increased from 180 min to 360 min (see
Figure 11a,b and
SI—Figure S3a,b).
In the case of VCB40, the chemical
effect of hydrochar in synergy with CB
seems to partly reduce the micro-phase separation caused by oxidation.
Figure 12 focuses on
the changes in the G′ = f(T) curves which appear
with increasing duration of the thermo-oxidative aging test, by comparing the development in selected samples. It can be observed that the
oxidative crosslinking is fairly
efficiently blocked (no second step in
G′)
in the sample VCB00, filled exclusively by HC, especially in the first 3 h. Crosslinking in VB00 sets on between 3 and 6 h, but no second
G′ step appears even after 6 h. In
VCB40, where a
synergy effect of the fillers was observed already with other characterization methods, the formation of the second step in G′ is much less prominent than in VCB50 (filled exclusively by CB) and compared to the neat matrix. These findings further demonstrate the
stabilizing chemical effects of HC and HC/CB.
3.4.3. Simulated Weathering: Industrial ‘Florida Test’
The
weathering of the prepared rubber composites
in real-use conditions (less harsh than the thermo-oxidative aging test at 180 °C) was tested as “accelerated aging” according to the method PV 3930 (‘Florida Test’) developed by Volkswagen, Germany, which relies on the guideline ISO 4892-2. The Florida Test (setup: see Experimental Part) simulates a demanding, hot and humid climate, where the sunny weather is simulated by UV irradiation of the wavelength of 340 nm. One real ‘Florida day’ is simulated by a 120-min-treatment so that in one day (24 h) of the uninterrupted aging test, 12 real days are simulated. The results of the weathering tests are summarized in
Figure 13,
Figures S4 and S5.
Selected
samples, namely
VCB40, VCB10, and
VCB50, were subjected to two differently long
aging treatments:
7 days (corresponding to 2.75 real months in Florida, USA)
and 25 days (corresponding to 10 real months in Florida). After the Florida test, the samples were characterized using thermo-mechanical analysis (DMTA; see
Figure 13,
Figures S4 and S5) in order to assess the effects of aging. Generally,
only very small changes in thermomechanical properties were observed even after the longer Florida test (which simulated 10 months of aging), but specific
chemical effects of hydrochar nevertheless could be observed. In the case of the
CB-rich VCB40,
moderate additional crosslinking was detected, which was more extensive and followed a different kinetic course than in VCB50 filled exclusively by CB. In the case of the
HC-rich VCB10, a
nearly perfect stabilization of the properties was observed.
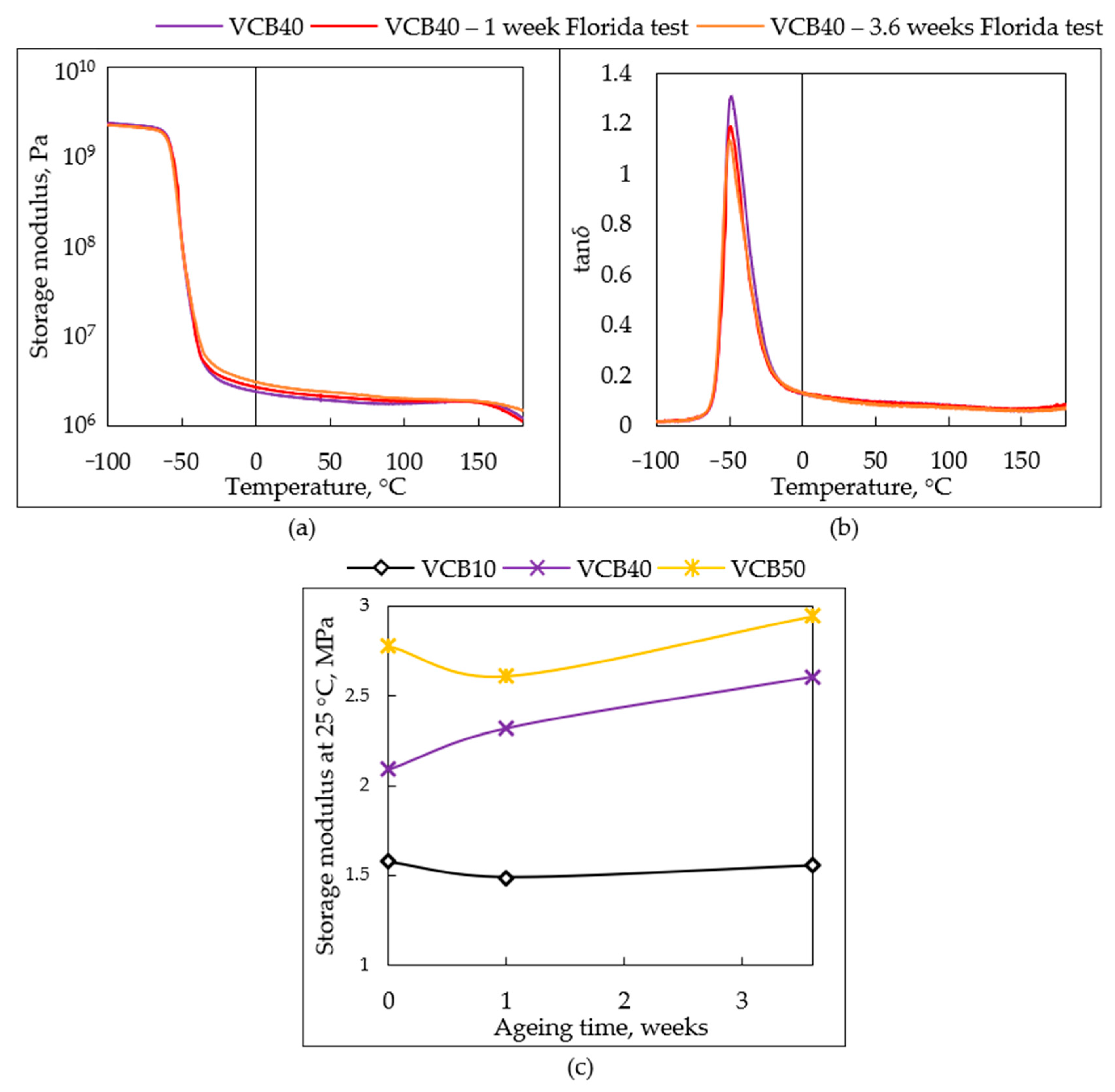
In
Figure 13a,b (
G′ =
f(
T) and tan
δ =
f(
T) curves), the small
changes in thermo-mechanical properties of VCB40 are presented as examples for all
the studied materials. The change of
G′ (25 °C) in dependence on the aging time is analyzed in
Figure 13c for all the tested samples.
VCB40: In
Figure 13a (
G′ =
f(
T) curves), it can be observed that the sample VCB40, which displayed the greatest final changes in mechanical properties during the Florida Test, undergoes slight additional crosslinking under the test conditions (see also trendline of
G′ (25 °C) in
Figure 13c). After 3.6 weeks (≡9.83 real months), the modulus
G′ (25 °C) rises by 24%. In the region of thermal and oxidative degradation in DMTA (at T > 150 °C), the modulus of the 3.6-weeks-aged VCB40 is still slightly higher than the modulus of the intact VCB40 (
Figure 13a). The shorter 1-week-Florida-Test (equivalent of 2.75 months at real conditions) also led to some additional crosslinking in VCB40:
G′ (25 °C) increased by 11% (in contrast to VCB50, where
G′ (25 °C) decreases after a 1 week of Florida Test). It can also be noted that between 1 and 3.6 weeks of aging (≡between 2.75 and 9.83 real months), the additional crosslinking in VCB40 was slightly slower than in VCB50.
The
hydrochar-rich sample VCB10 displays the smallest changes in DMTA as a result of Florida Test aging (see
SI—Figure S4a,b:
G′
= f(T), and tan
δ = f(T), and trendlines comparison in
Figure 13c). The modulus
G′ (25 °C) decreases by 5.7% after 1 week (≡2.75 real months), and then, after 3.6 weeks (≡9.83 real months), it increases again by 4.5%, so that the net modulus change relative to the intact sample is only −1.2% (a negligible decrease) after 3.6 weeks (≡9.83 real months). More visible is the modest decrease of
G′(VCB10) in the high-temperature region above 110 °C (see DMTA in
SI—Figure S4a). The value of tan
δ (25 °C) (see
Figure S4b) remains unchanged after 1 week (≡2.75 real months) and drops by 13.4% of its initial value after 3.6 weeks (≡9.83 real months), which means a slight increase in elastic character.
The
hydrochar-free rubber composite VCB50 displays a smaller net effect of the aging but comparably large incremental changes in properties during the course of the Florida Test, as VCB40 does. At first, after 1 week (≡2.75 real months), the modulus
G′ (25 °C) of VCB50 decreases by 6.3%. Subsequently, after 3.6 weeks (≡9.83 real months), it increases again by 12.4%, so a net modulus increase relative to the intact sample is achieved, specifically by 6.1% after 3.6 weeks. Above 110 °C, the modulus
G′ is altered only very slightly by the aging (see DMTA in
SI—Figure S4c). The value of tan
δ displays only minimal changes due to the aging of VCB50: it drops by 2.6% of the original tan
δ value after 3.6 weeks (≡9.83 real months).

 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}