Low-Temperature-Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinker: A Passive Smart Material with Potential as Viscoelastic Coupling. Part II—Viscoelastic and Rheological Properties

 , , ,
, , ,
Abstract
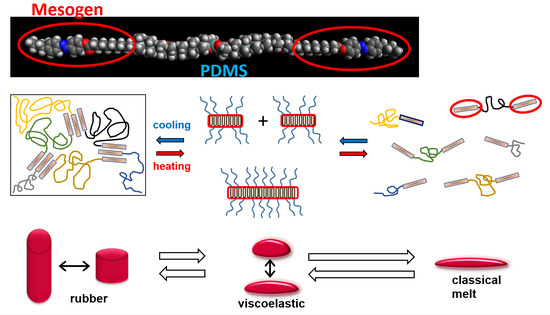
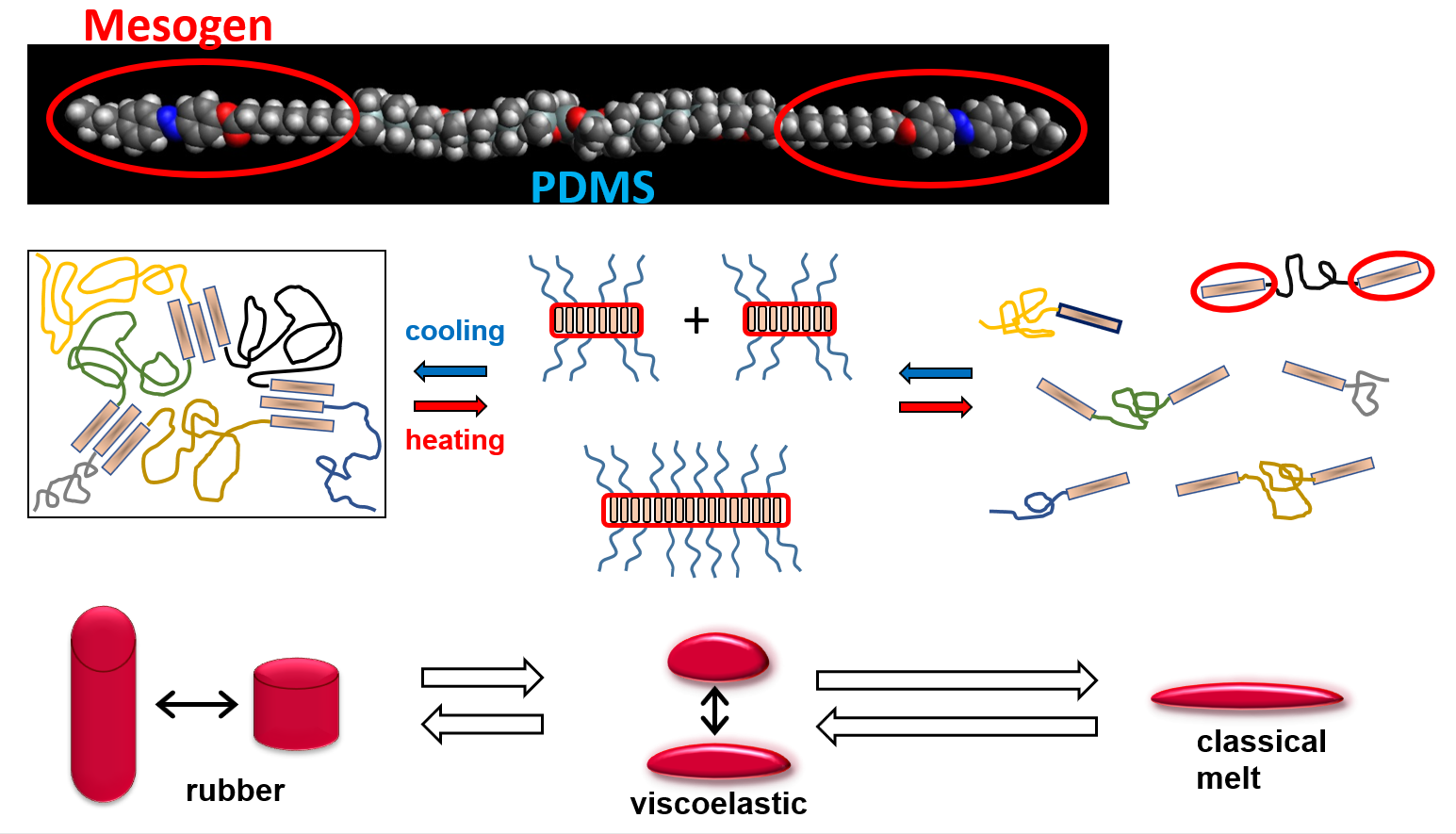
:
1. Introduction
1.1. Physically Crosslinked Polydimethylsiloxane
1.2. Rheology of Liquid Crystals and of Liquid-Crystalline Polymers (LCPs)
1.2.1. Models of Flow Behavior of LCPs
1.2.2. Effect of LCP Architecture
1.3. Rheology-Related Studies on α,ω-LC-Terminated Polymers
1.3.1. Rheological Properties of Physically Crosslinked PDMS
1.3.2. Aim of This Work
2. Experimental Section
2.1. Materials
2.2. Rheological Characterization of the PDMS–BAFKU2 Copolymers
2.2.1. Equipment
2.2.2. Sample Geometry
2.2.3. Determination of Gel Points in Multi-Frequency Oscillatory Tests
2.2.4. Rate of Thermally Induced Physical Gelation
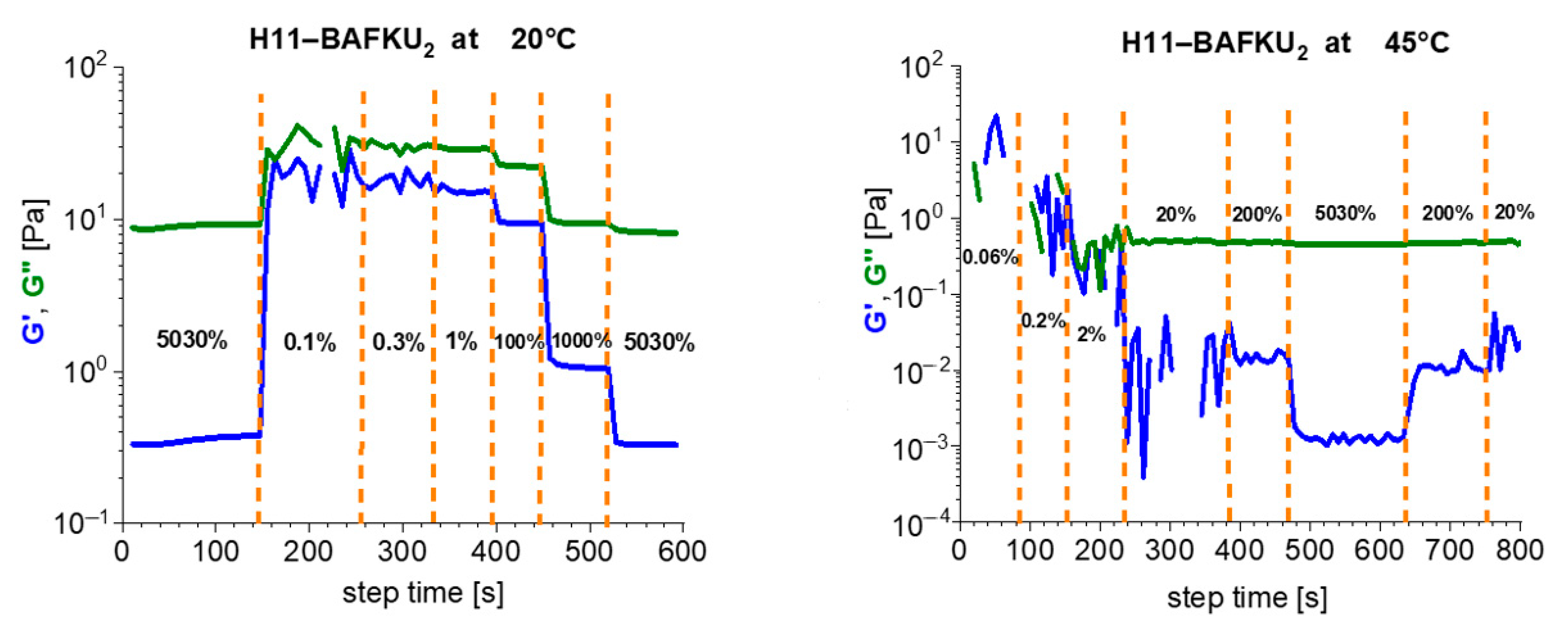
2.2.5. Analysis of Mechanical Disconnection of Crosslinks: Strain Sweep Tests
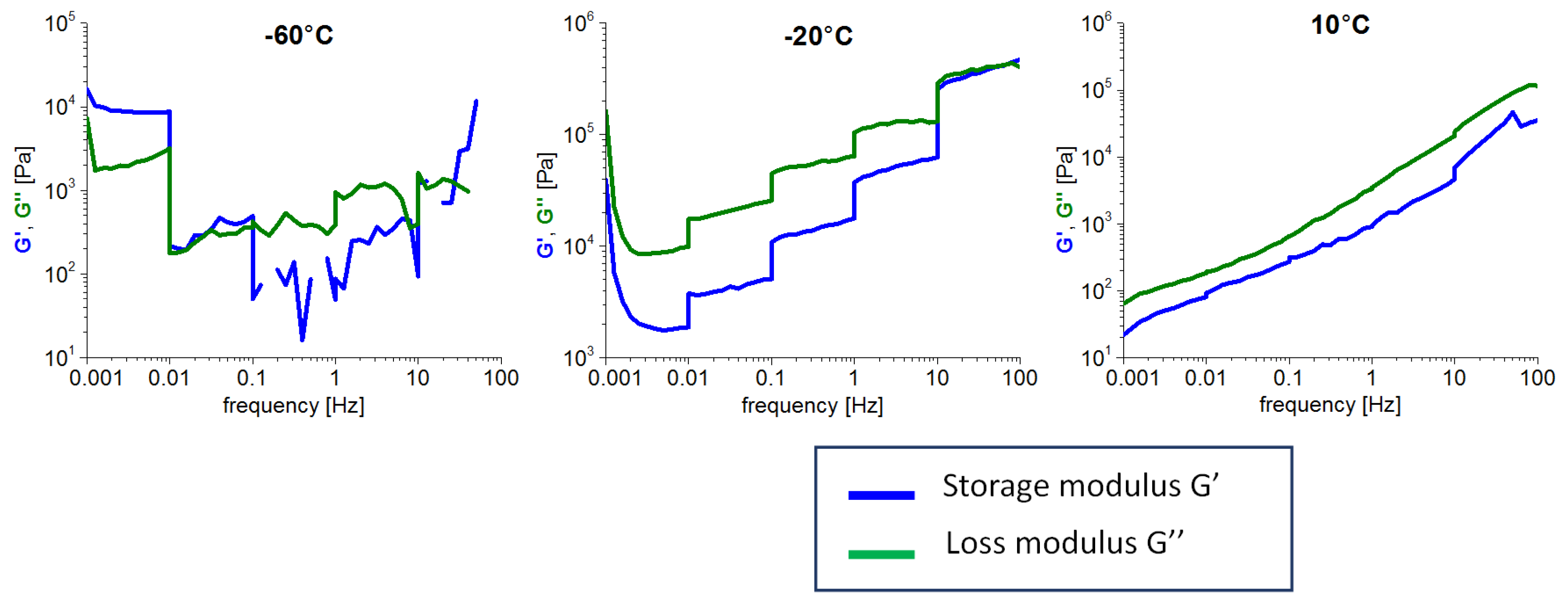
2.2.6. Frequency Stiffening Tests
2.2.7. Creep and Creep Recovery Tests
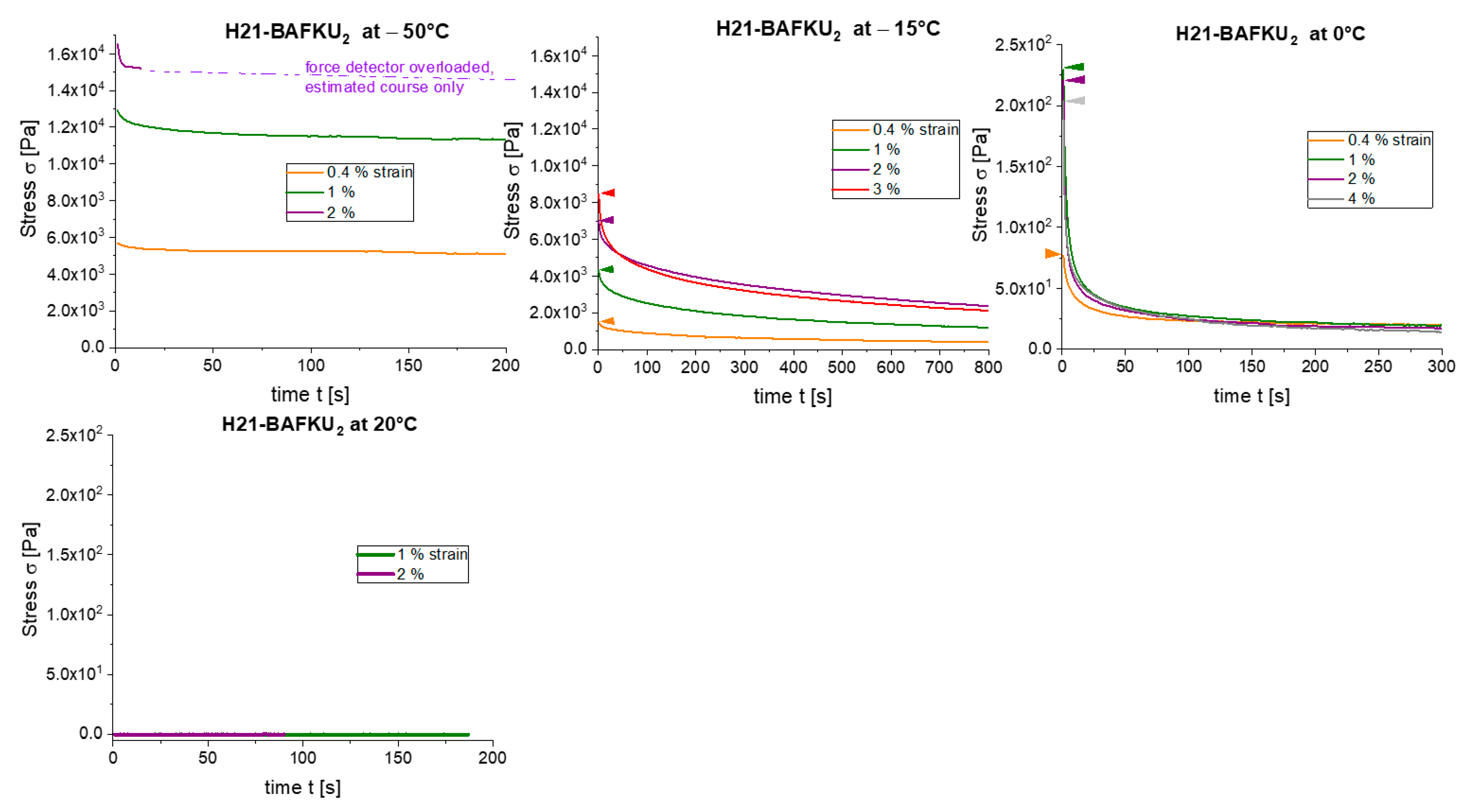
2.2.8. Stress Relaxation Tests
2.2.9. Simple Oscillatory Tests of Thixotropy
2.2.10. Thixotropic Loop Tests
3. Results and Discussion
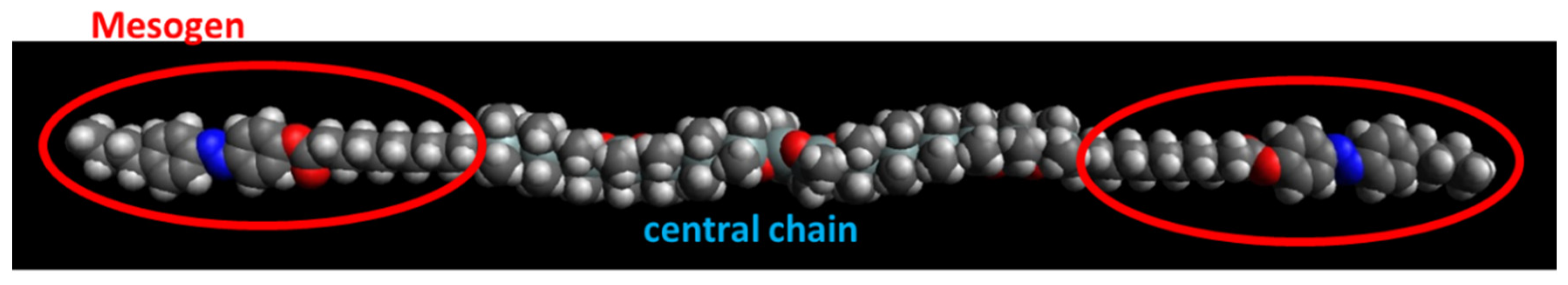
3.1. Description of the Studied Copolymers
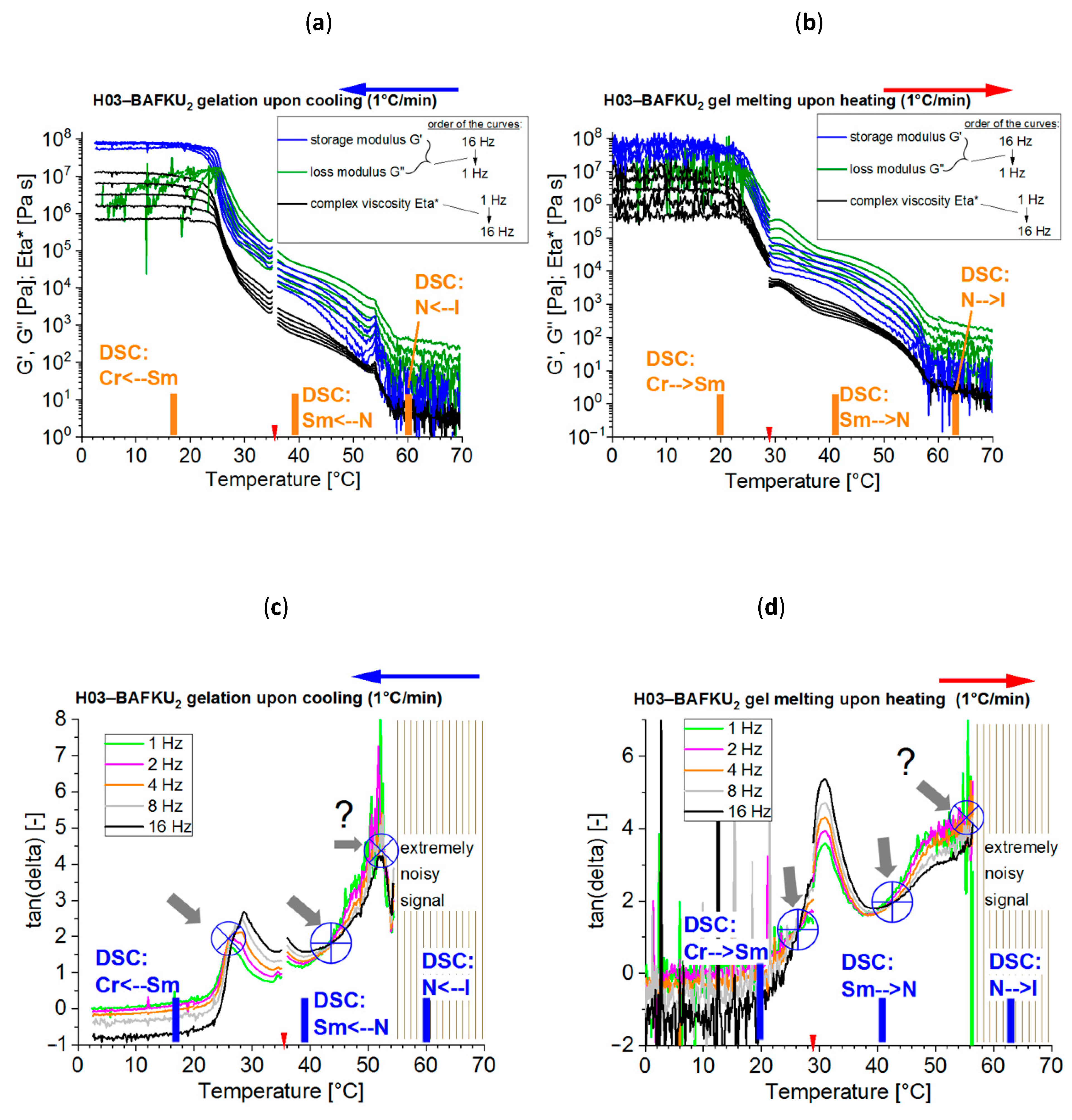
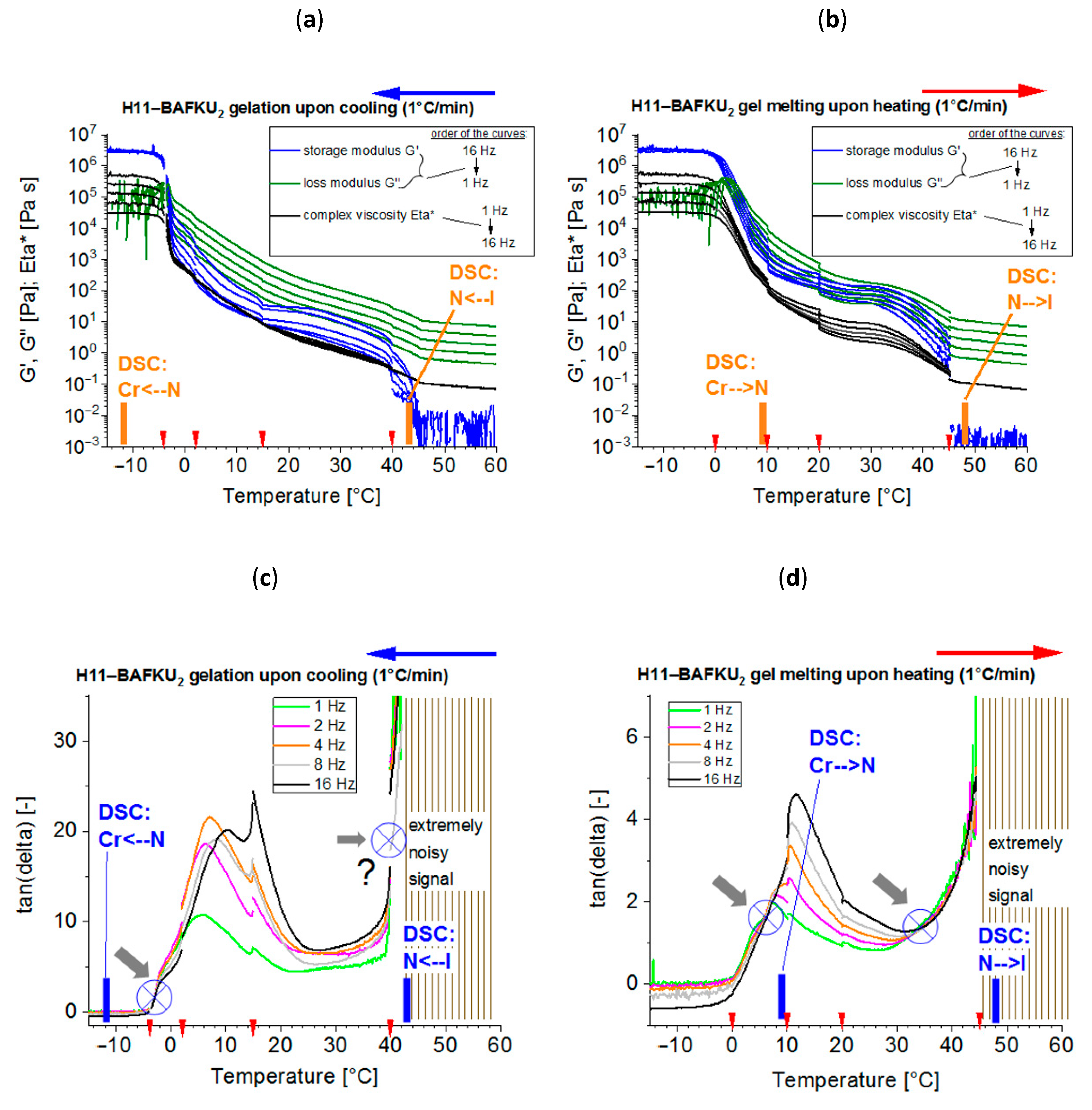
3.2. Reversible Gelation Processes Induced by Temperature Change
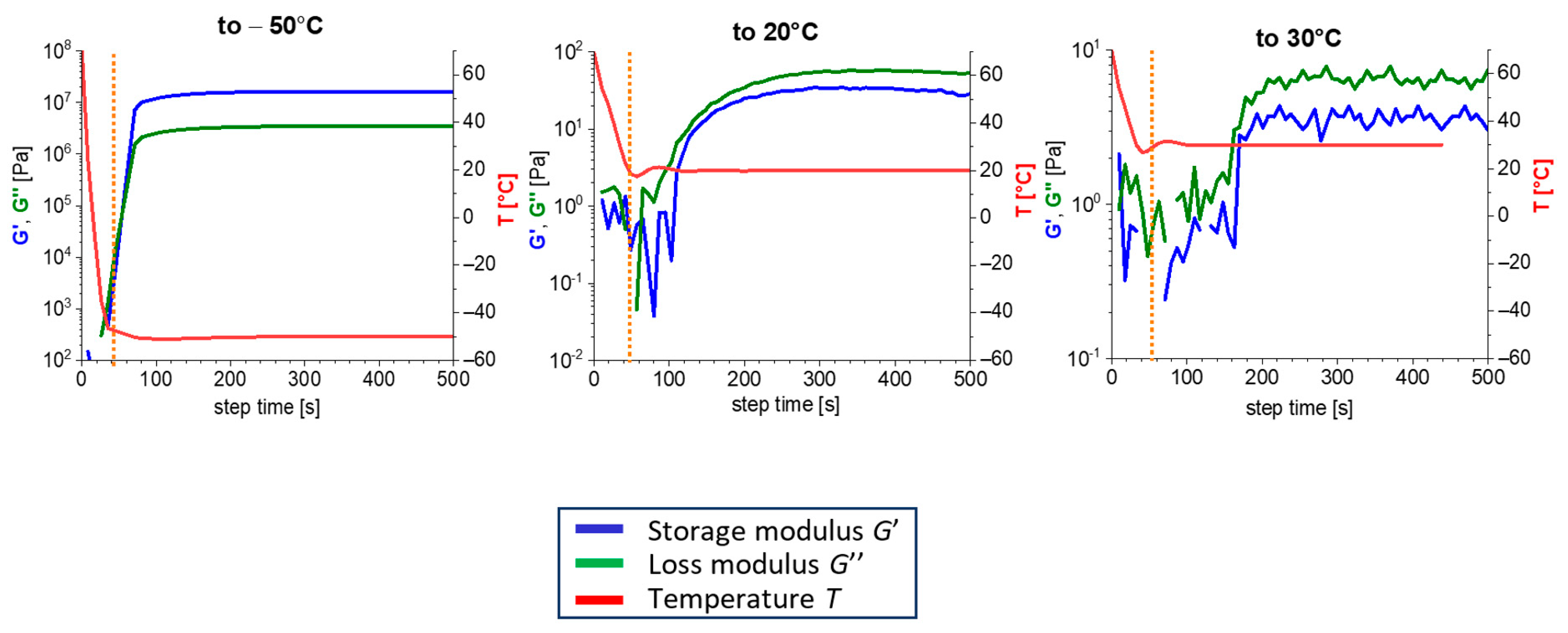
3.3. Rate of Physical Gelation upon Quenching the Melt
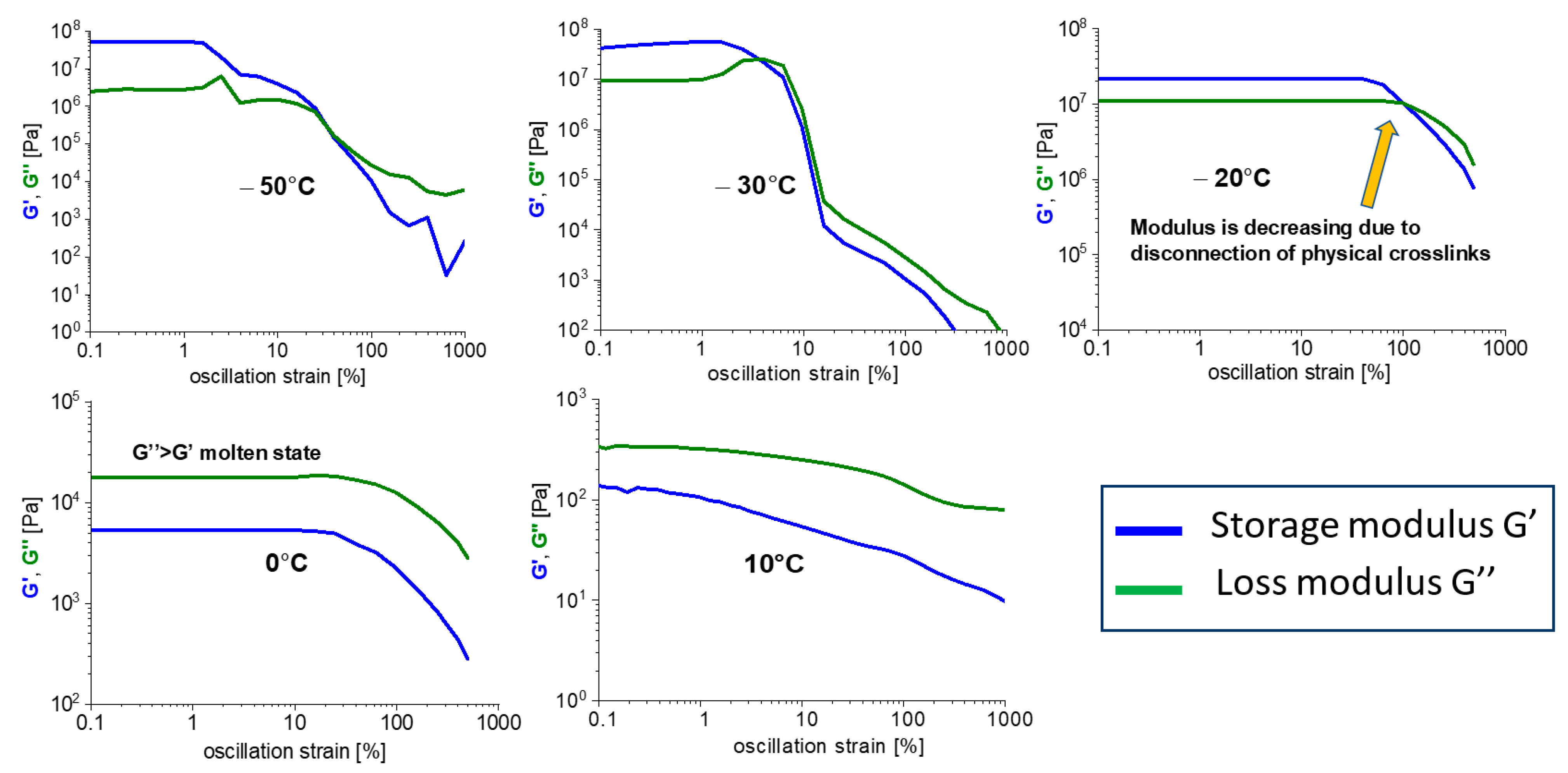
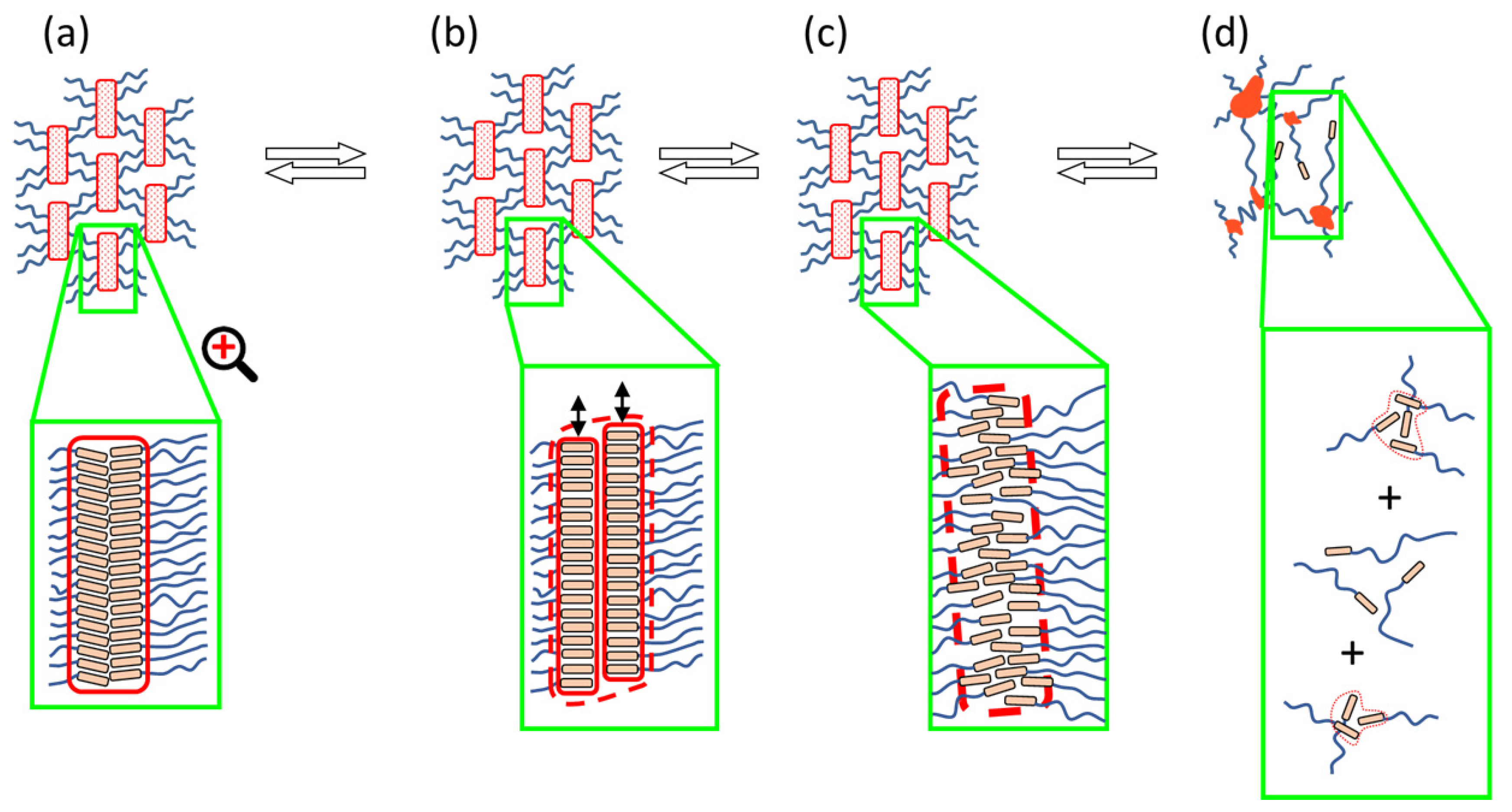
3.4. Mechanical Disconnection of the Physical Network
3.4.1. Strain Sweep Tests
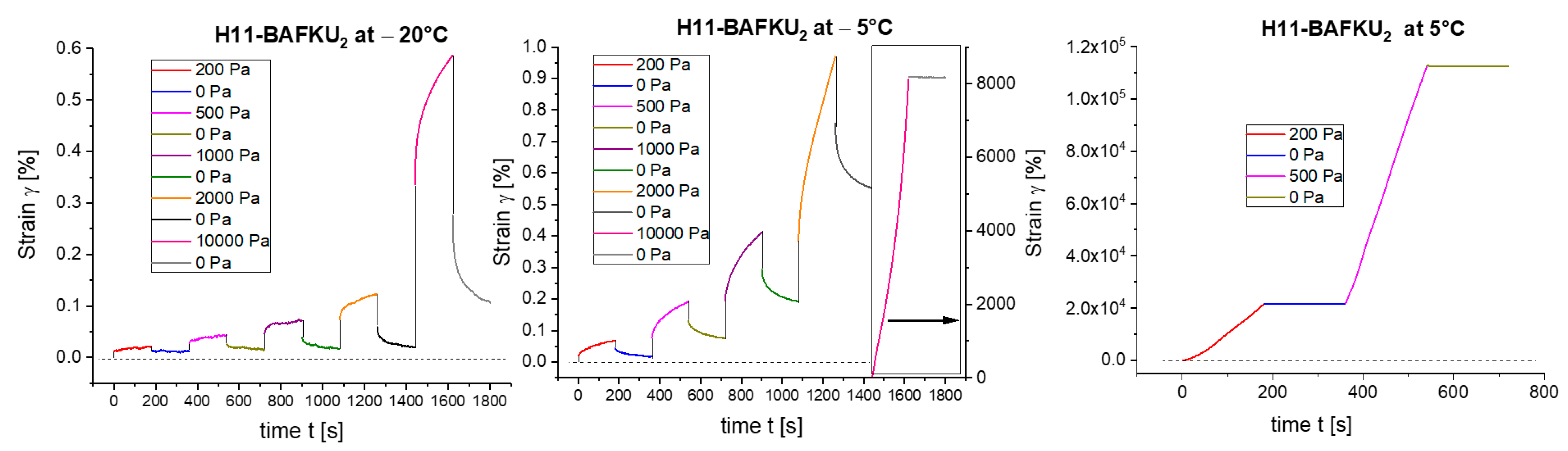
3.4.2. Creep Tests
3.4.3. Stress Relaxation
3.5. High-Frequency Stiffening and Self-Healing Effects
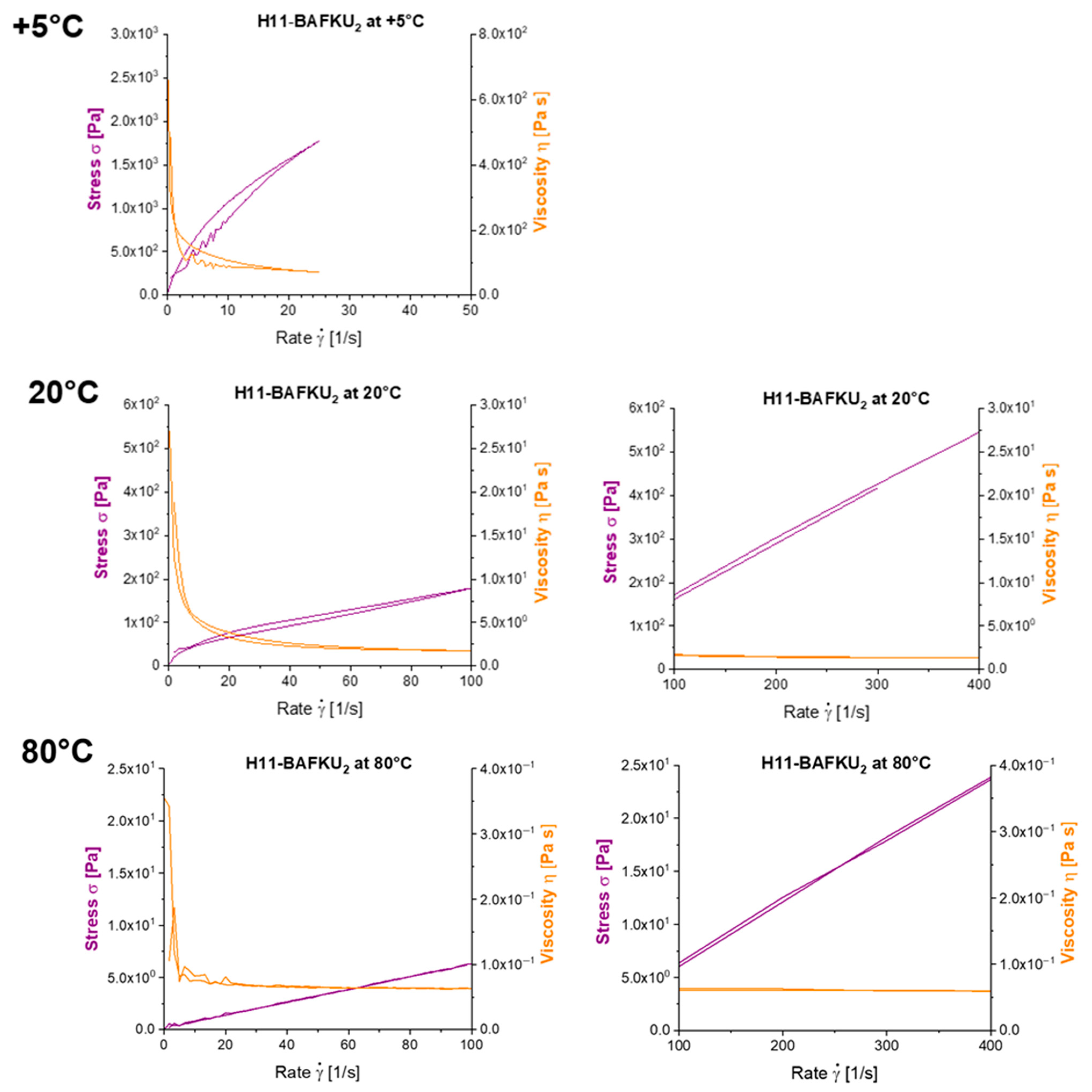
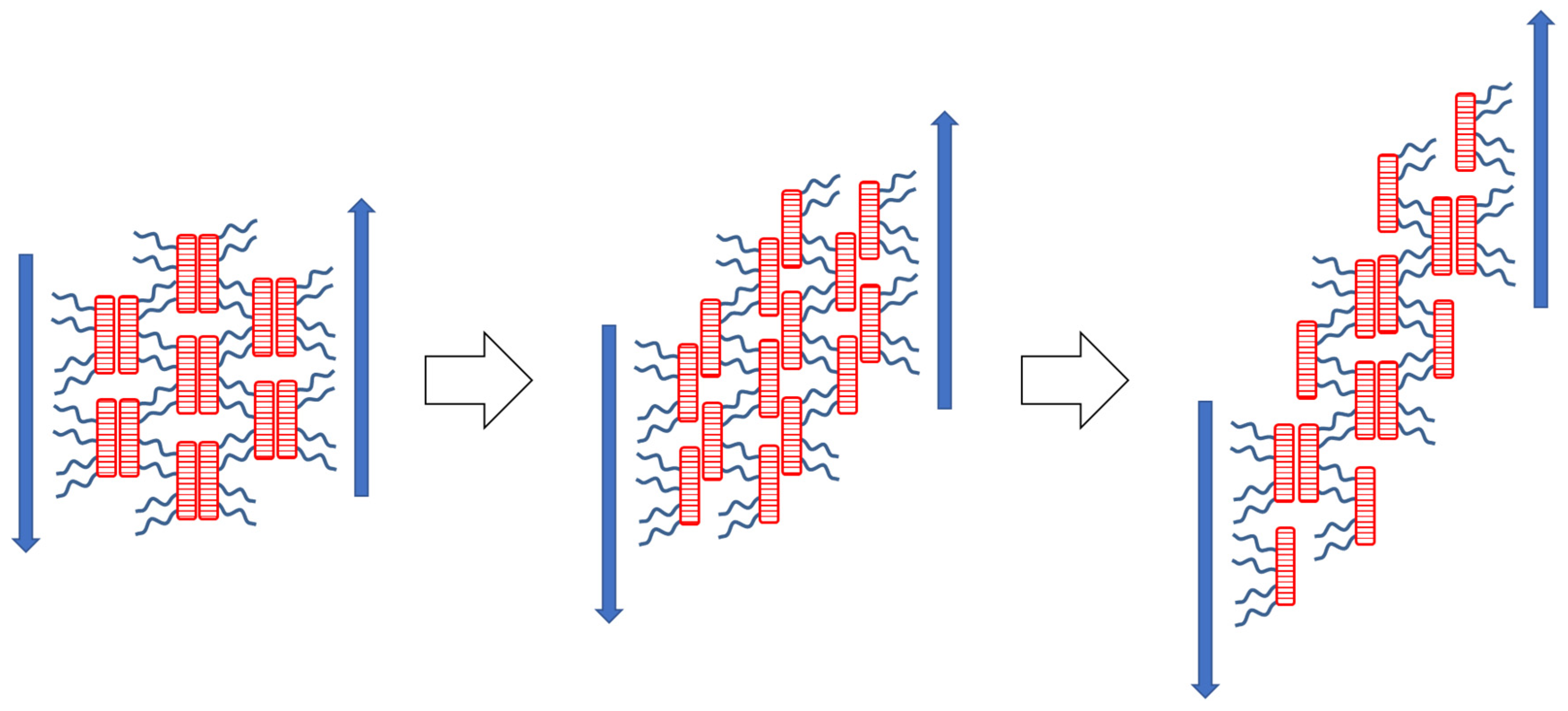
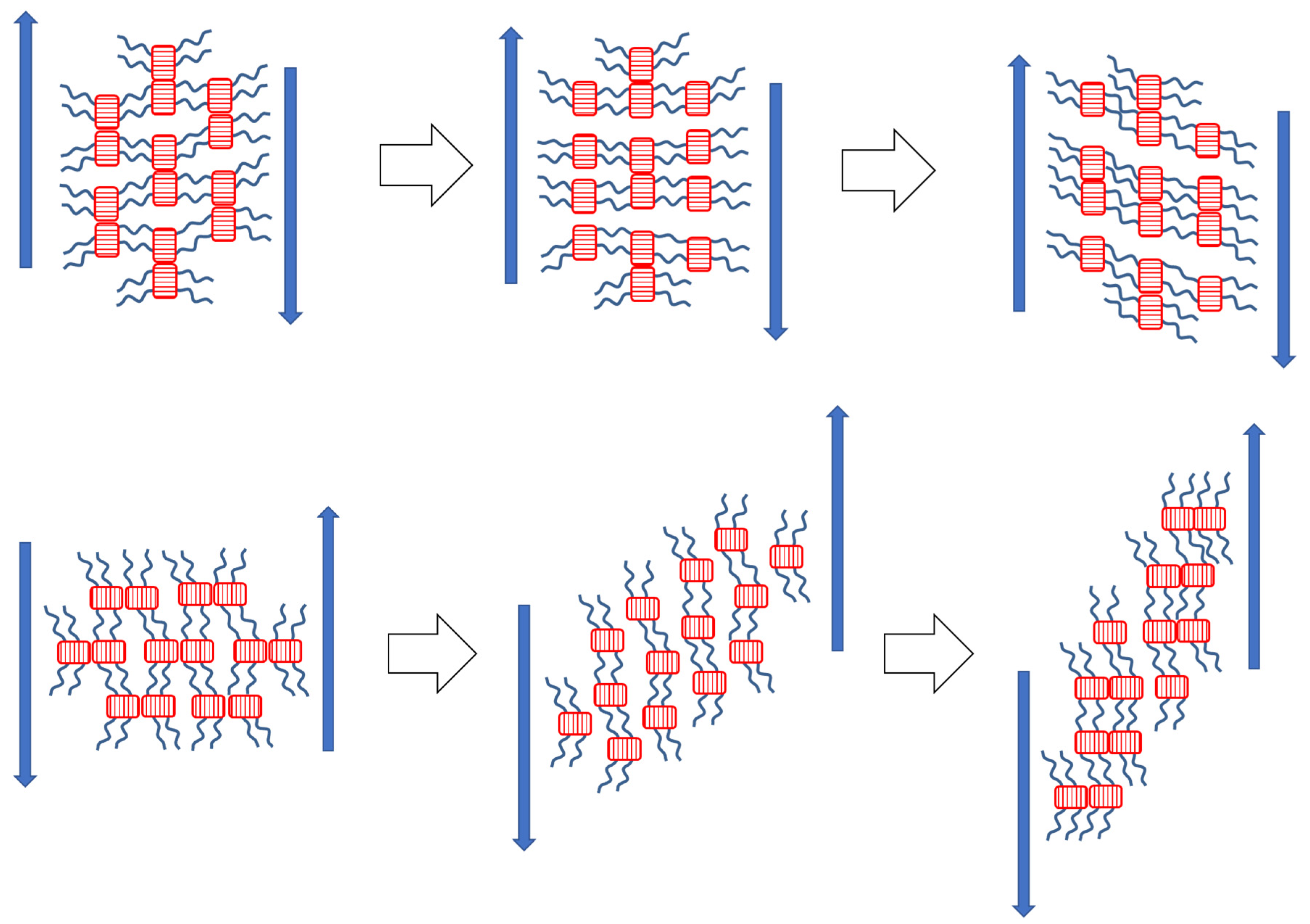
3.6. Thixotropy
3.6.1. Oscillatory Tests of Thixotropy and of Recovery of Viscoelasticity
3.6.2. Thixotropic Loop Tests
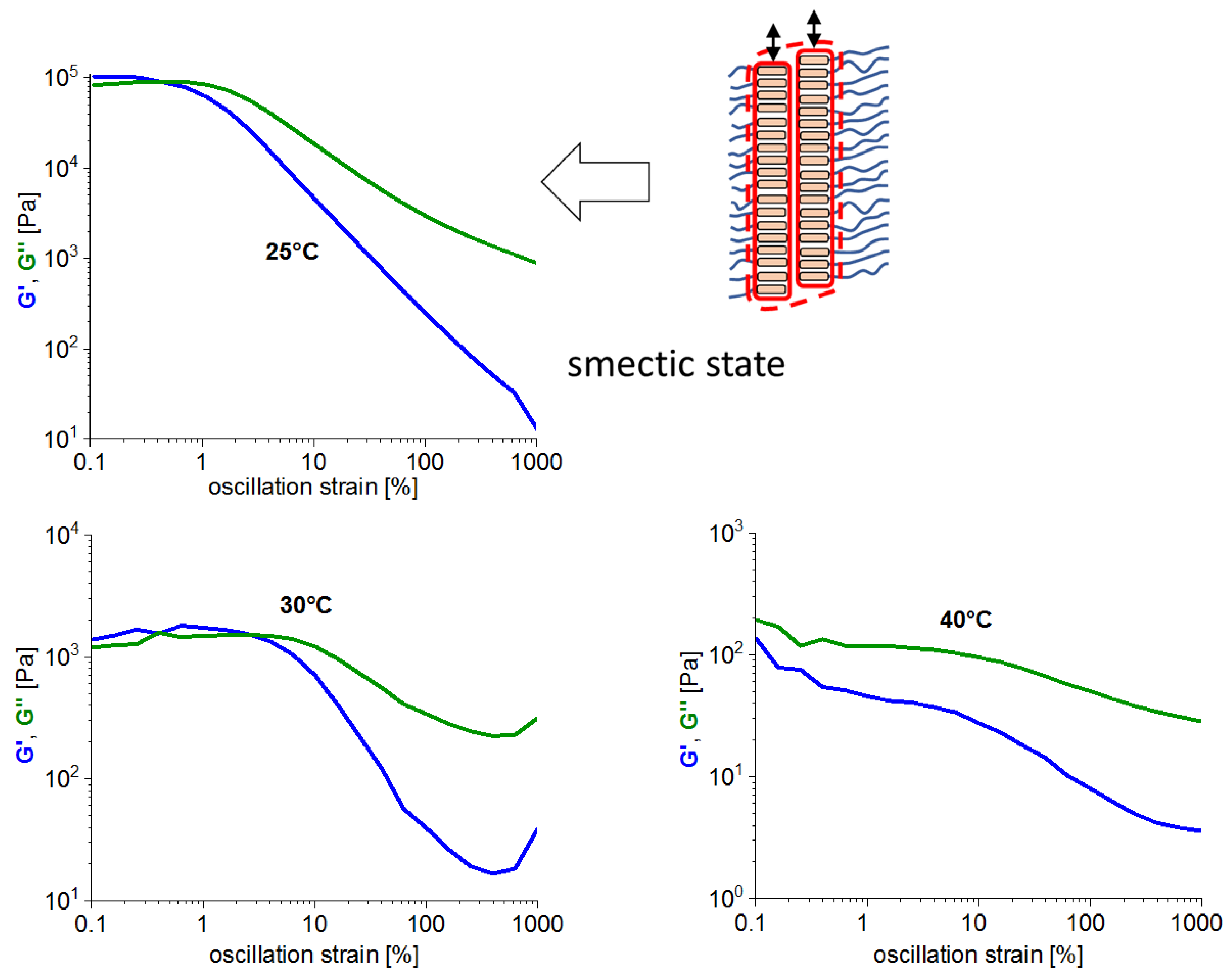
3.7. Comparison of Rheological Behavior of the Studied and of Classical LC Polymers
4. Conclusions
- −
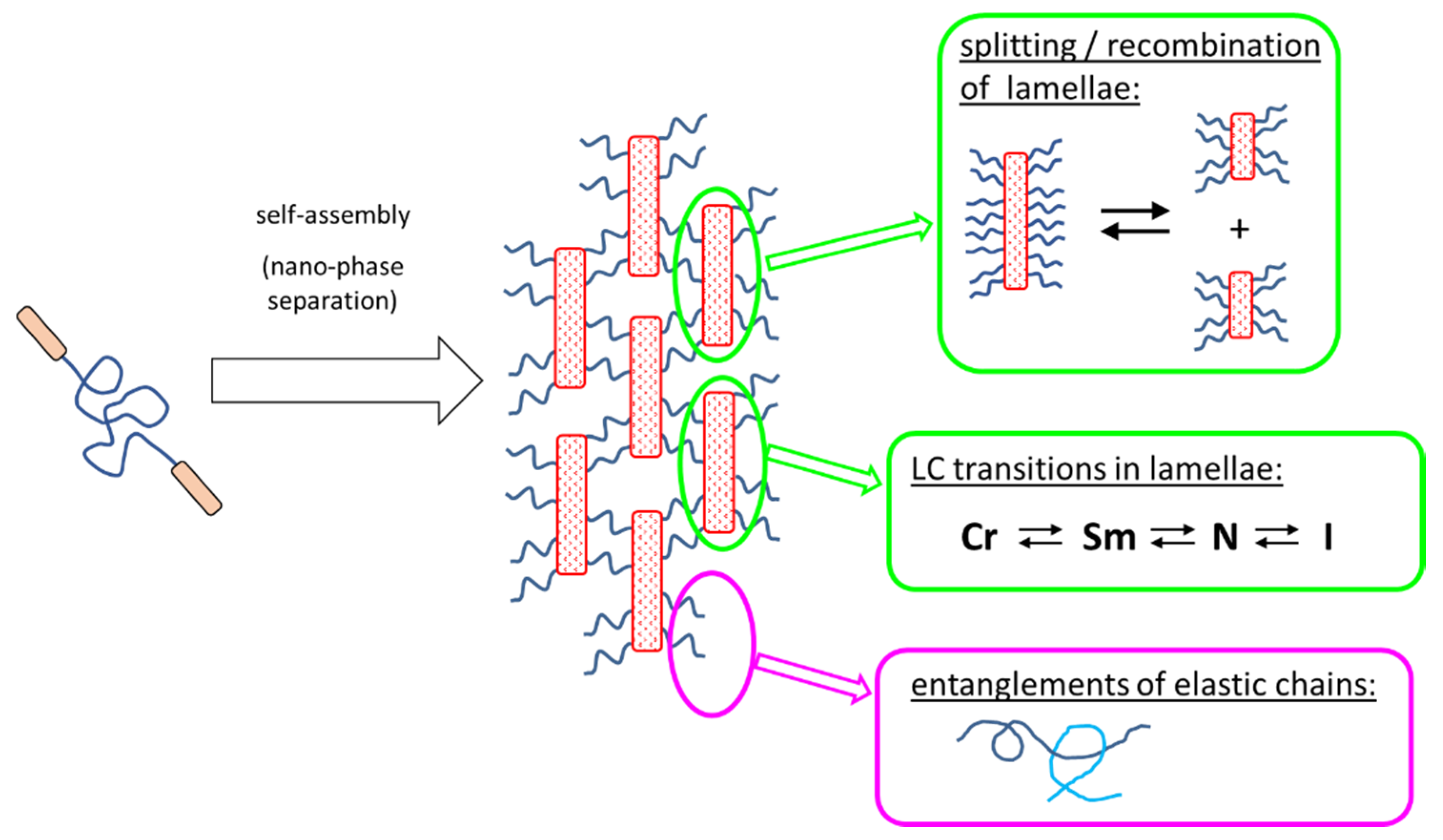
- Physically crosslinked thermo-reversible low-temperature-melting rubbers, based on linear polydimethylsiloxane (PDMS) capped in α,ω-positions with liquid-crystalline (LC) building blocks called “BAFKU”, were studied concerning their viscoelastic and rheological properties in melt, as well as in the rubbery state.
- −
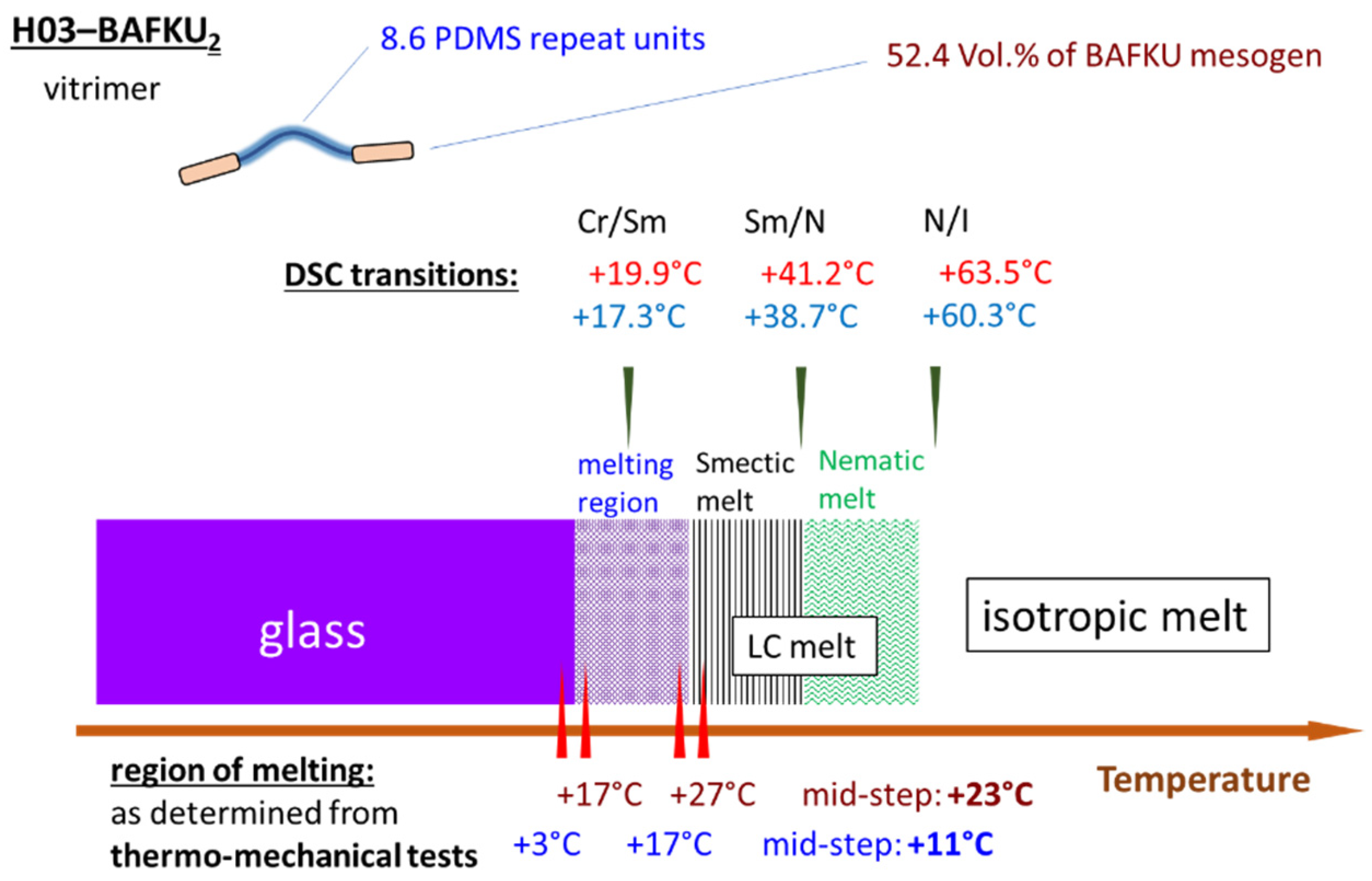
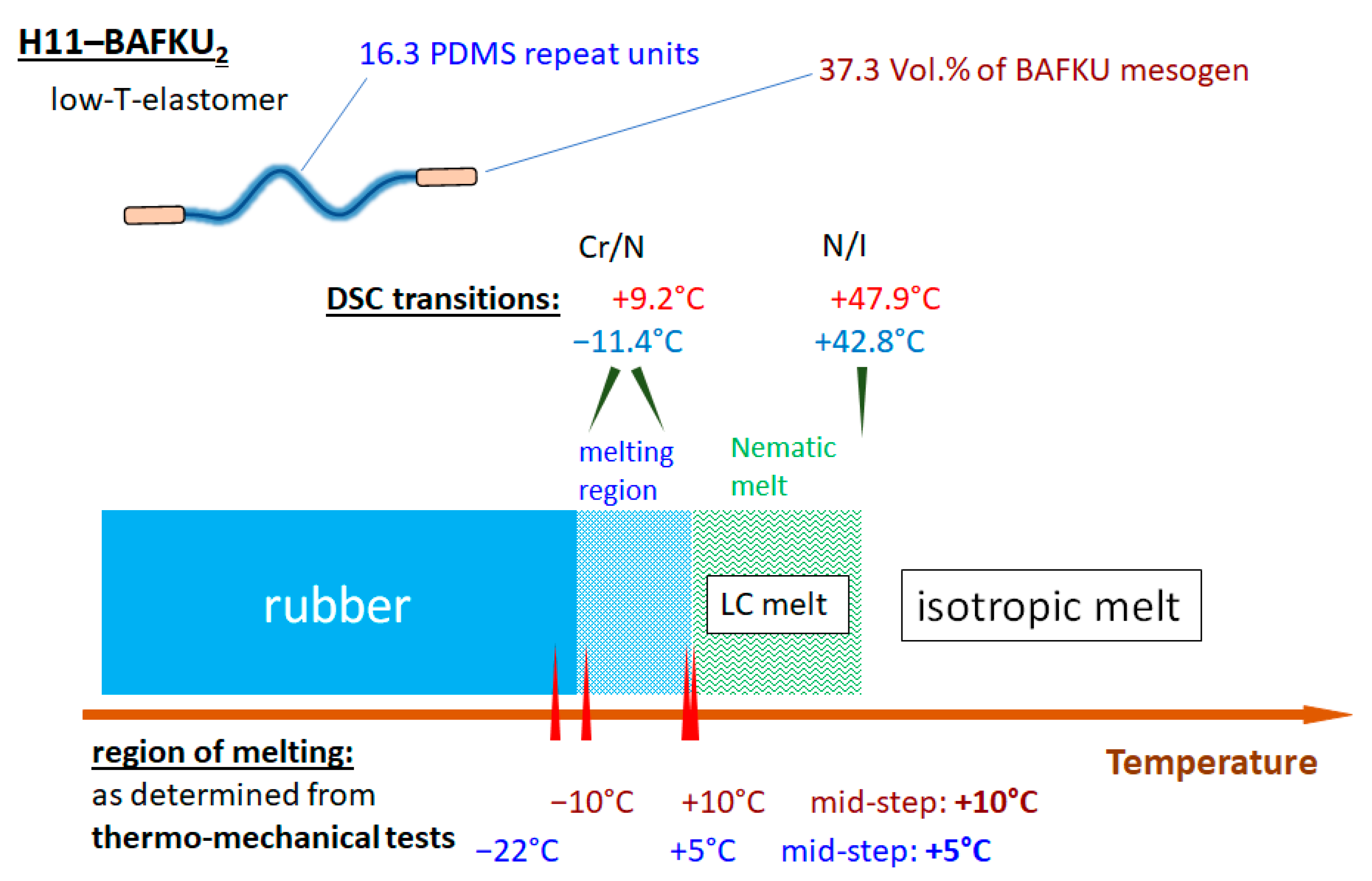
- The properties of three copolymers were compared, which differed in the length of the central PDMS chain, namely DMS H03 (8.6 dimethyl siloxane repeat units), DMS H11 (16.3-mer) and DMS H21 (64.4-mer).
- −
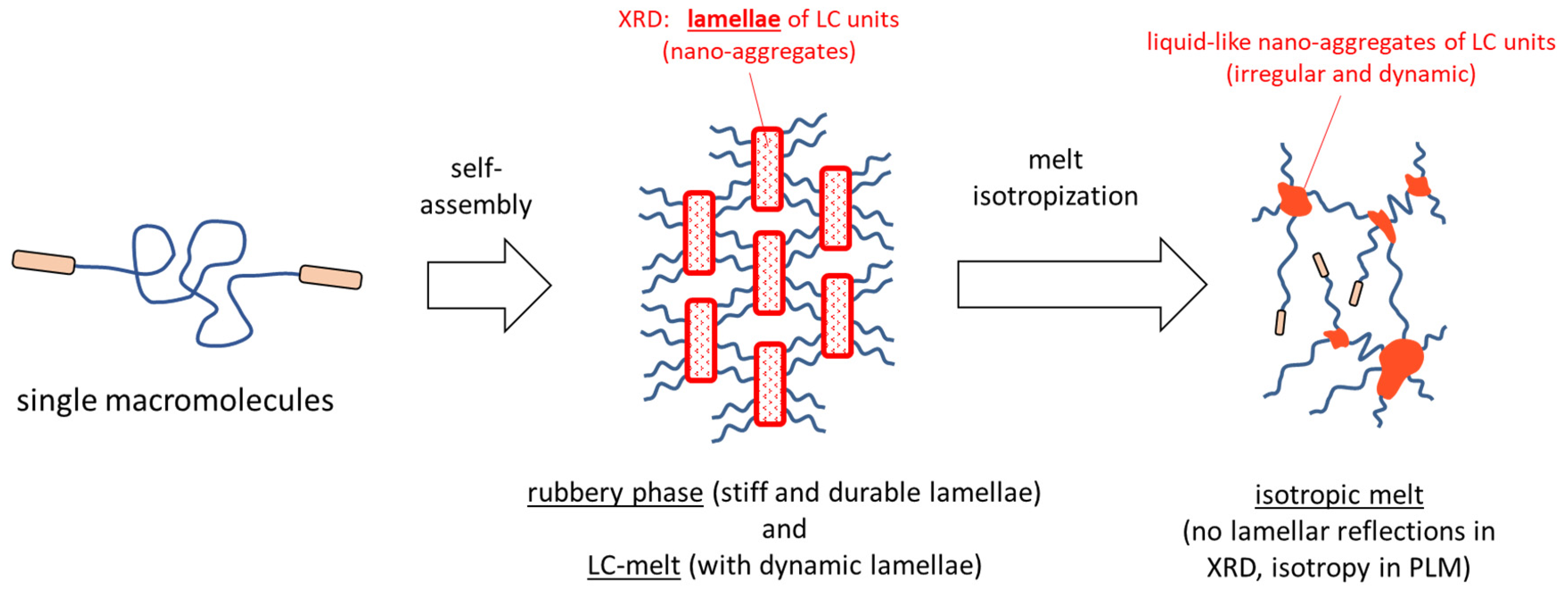
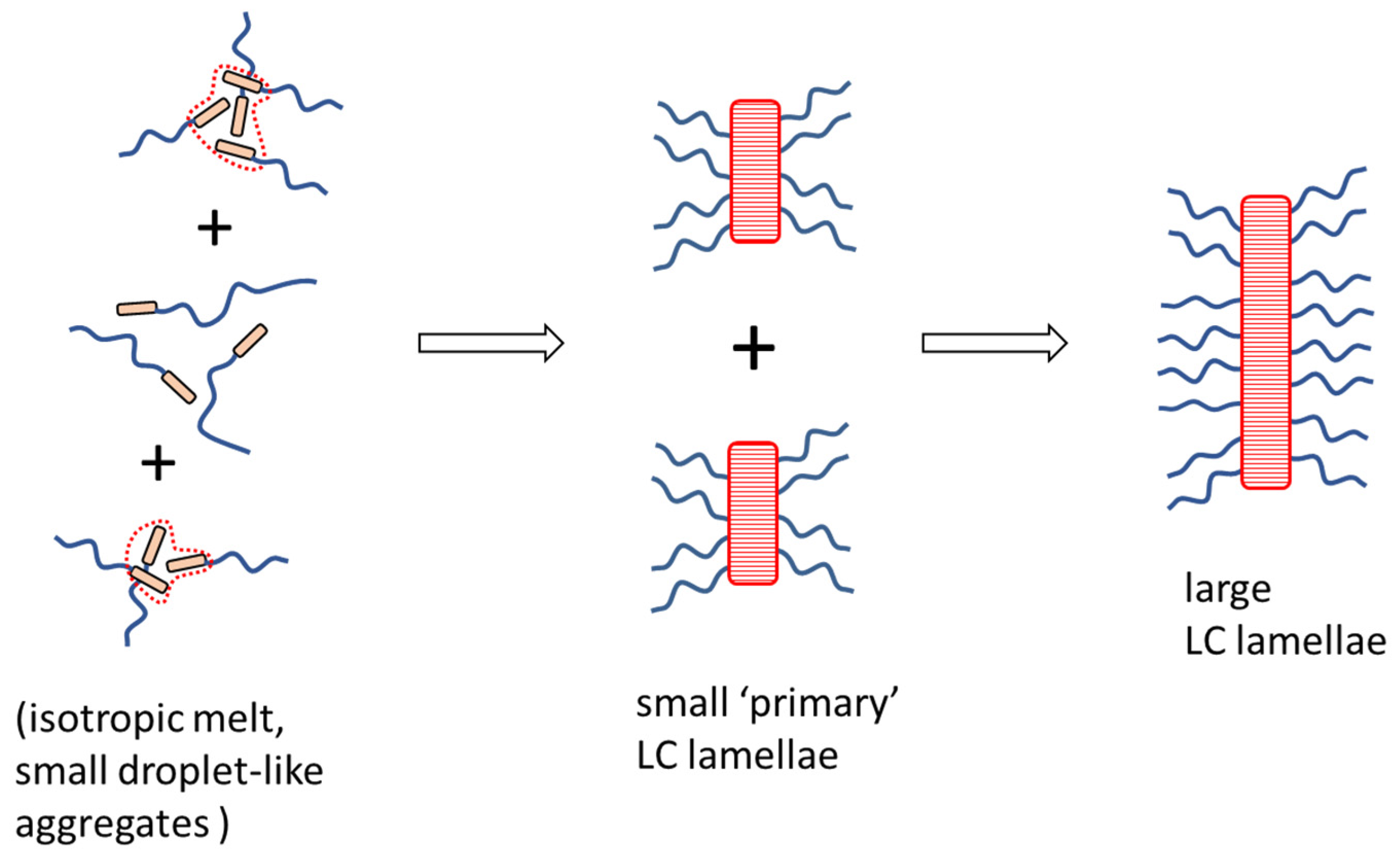
- Physical crosslinking via nano-aggregation of BAFKU units in all three tested copolymers was found to be fairly efficient and very large step-wise changes of elasticity and viscosity were observed, which correlated with the thermotropic properties of the crosslinks.
- −
- The rheological behavior of the copolymers was found to be controlled by an interplay of nano-phase separation of the LC end-groups (growth and splitting of their aggregates) and of the thermotropic transitions in these aggregates (which change their stiffness). Entanglements of the elastic PDMS chains also were found to play a role.
- −
- In contrast to LC-rich liquid crystalline (co)polymers (LCPs), the studied copolymers display viscosity increase if going form isotropic to nematic (or further to the smectic) state, because their viscoelasticity is controlled by the larger-scale morphology, which is responsible for the physical crosslinking. The latter in turn is controlled by nano-phase separation and by the strengthening or loosening of the aggregates of LC units via thermotropic transitions.
- −
- The copolymers display up to three gel points, if a temperature scan (in both directions) is performed in the range from rubbery state to isotropic melt. The gel points correlate with the thermotropic transitions (I/N, N/Sm and Sm/Cr in the shortest copolymer and I/N, N/Cr in the longer ones) and with the associated changes in stiffness and dynamic size of LC nano-aggregates.
- −
- The kinetics of (physical) gelation to network is fairly fast if induced by temperature (abrupt melt cooling): between 0.5 and 3 min (typically ca. 1 min). It slows down at higher final temperatures. This kinetics is controlled by temperature-dependent nano-phase-separation dynamics.
- −
- The physical crosslinks can be reversibly disconnected by large mechanical strain in the rubbery state and in the melt (thixotropy in the latter case). The kinetics of subsequent re-generation was found to be extremely fast: ca. 1 s in oscillatory tests.
- −
- Thixotropic loop tests, in which the samples were subjected to continuous flow, also indicate a very fast regeneration of destroyed physical crosslinks, so that only very small hysteresis is observed in these tests, in spite of very strong shear-thinning tendency in all the copolymers in wide temperature ranges. Nevertheless, in contrast to oscillation experiments, the crosslink regeneration in continuous flow is not immediate.
- −
- The ‘warmer’ rubbery phase of the longer copolymers can be relatively easily transformed to liquid by strong shear.
- −
- Frequency-stiffening was observed in the rubbery state, as well as in the melt. Such a behavior is characteristic of classical elastomers and linear polymers.
- −
- The studied low-temperature elastomers might be of interest as passive smart materials for advanced applications such as viscoelastic coupling for example, in soft robotics (transitions melt/viscoelastic melt/rubber) but also as damping materials (energy absorption via physical crosslink disconnection). Additionally, the incorporated LC building blocks of azo type open the possibility of reversible UV-light-induced switching of material properties.
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jaunich, M.; Stark, W.; Wolff, D. Comparison of low temperature properties of different elastomer materials investigated by a new method for compression set measurement. Polym. Test. 2012, 31, 987–992. [Google Scholar] [CrossRef]
- Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Strachota, B.; Šlouf, M.; Zhigunov, A.; Vyroubalová, M.; Kaňková, D.; Netopilík, M.; Walterová, Z. Low-temperature-meltable elastomers based on linear polydimethylsiloxane chains alpha,omega-terminated with mesogenic groups as physical crosslinker: A passive smart material with potential as viscoelastic coupling. Part I: Synthesis and phase behaviour. Polymers 2020, 12, 2476. [Google Scholar] [CrossRef]
- Strachota, A.; Kroutilová, I.; Kovářová, J.; Matějka, L. Epoxy Networks Reinforced with Polyhedral Oligomeric Silsesquioxanes (POSS). Thermomechanical Properties. Macromolecules 2004, 37, 9457–9464. [Google Scholar] [CrossRef]
- Rodzeń, K.; Strachota, A.; Ribot, F.; Matějka, L.; Kovářová, J.; Trchová, M.; Šlouf, M. Reactivity of the tin homolog of POSS, butylstannoxane dodecamer, in oxygen-induced crosslinking reactions with an organic polymer matrix: Study of long-time behaviour. Polym. Degrad. Stab. 2015, 118, 147–166. [Google Scholar] [CrossRef]
- Mossety-Leszczak, B.; Strachota, B.; Strachota, A.; Steinhart, M.; Šlouf, M. The orientation-enhancing effect of diphenyl aluminium phosphate nanorods in a liquid-crystalline epoxy matrix ordered by magnetic field. Eur. Polym. J. 2015, 72, 238–255. [Google Scholar] [CrossRef]
- Strachota, A.; Whelan, P.; Kříž, J.; Brus, J.; Urbanová, M.; Šlouf, M.; Matějka, L. Formation of nanostructured epoxy networks containing polyhedral oligomeric silsesquioxane (POSS) blocks. Polymer 2007, 48, 3041–3058. [Google Scholar] [CrossRef]
- Colombani, O.; Barioz, C.; Bouteiller, L.; Chaneac, C.; Fomperie, L.; Lortie, F.; Montes, H. Attempt toward 1D Cross-Linked Thermoplastic Elastomers: Structure and Mechanical Properties of a New System. Macromolecules 2005, 38, 1752–1759. [Google Scholar] [CrossRef]
- Botterhuis, N.E.; van Beek, D.J.M.; van Gemert, G.M.L.; Bosman, A.W.; Sijbesma, R.P. Self-Assembly and Morphology of Polydimethylsiloxane Supramolecular Thermoplastic Elastomers. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 3877–3885. [Google Scholar] [CrossRef]
- Ślęczkowski, M.L.; Meijer, E.W.; Palmans, A.R.A. Cooperative Folding of Linear Poly(dimethyl siloxane)s via Supramolecular Interactions. Macromol. Rapid Commun. 2017, 38, 1700566. [Google Scholar] [CrossRef]
- Rambarran, T.; Bertrand, A.; Gonzaga, F.; Boisson, F.; Bernard, J.; Fleury, E.; Ganachaud, F.; Brook, M.A. Sweet supramolecular elastomers from a,x-(b-cyclodextrin terminated) PDMS. Chem. Commun. 2016, 52, 6681–6684. [Google Scholar] [CrossRef]
- Strachota, A.; Rodzeń, K.; Ribot, F.; Trchová, M.; Steinhart, M.; Starovoytova, L.; Pavlova, E. Behavior of Tin-Based “Super-POSS” Incorporated in Different Bonding Situations in Hybrid Epoxy Resins. Macromolecules 2014, 47, 4266–4287. [Google Scholar] [CrossRef]
- Fawcett, A.S.; Brook, M.A. Thermoplastic Silicone Elastomers through Self-Association of Pendant Coumarin Groups. Macromolecules 2014, 47, 1656–1663. [Google Scholar] [CrossRef]
- Lamers, B.A.G.; Graf, R.; de Waal, B.F.M.; Vantomme, G.; Palmans, A.R.A.; Meijer, E.W. Polymorphism in the Assembly of Phase-Segregated Block Molecules: Pathway Control to 1D and 2D Nanostructures. J. Am. Chem. Soc. 2019, 141, 15456–15463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamers, B.A.G.; Ślęczkowski, M.L.; Wouters, F.; Engels, T.A.P.; Meijer, E.W.; Palmans, A.R.A. Tuning polymer properties of non-covalent crosslinked PDMS by varying supramolecular interaction strength. Polym. Chem. 2020, 11, 2847–2854. [Google Scholar] [CrossRef] [Green Version]
- Vasilev, V.G.; Pryakhina, T.A.; Shragin, D.I.; Kononevich, Y.N.; Papkov, V.S.; Muzafarov, A.M. Formation of a Physical Crosslinked Structure in Polydimethylsiloxanes Modified with Long-Chain Hydrocarbon Substituents with Polar Fragments. Polym. Sci. Ser. 2017, 59, 320–327. [Google Scholar] [CrossRef]
- Dollase, T.; Spiess, H.W.; Gottlieb, M.; Yerushalmi-Rozen, R. Crystallization of PDMS: The effect of physical and chemical crosslinks. Europhys. Lett. 2002, 60, 390–396. [Google Scholar] [CrossRef]
- Petr, M.; Katzman, B.; DiNatale, W.; Hammond, P.T. Synthesis of a New, Low-Tg Siloxane Thermoplastic Elastomer with a Functionalizable Backbone and Its Use as a Rapid, Room Temperature Photoactuator. Macromolecules 2013, 46, 2823–2832. [Google Scholar] [CrossRef]
- Dodge, L.; Chen, Y.; Brook, M.A. Silicone Boronates Reversibly Crosslink Using Lewis Acid–Lewis Base Amine Complexes. Chem. Eur. J. 2014, 20, 9349–9356. [Google Scholar] [CrossRef] [PubMed]
- Li, C.H.; Wang, C.; Keplinger, C.; Zuo, J.L.; Jin, L.; Sun, Y.; Zheng, P.; Cao, Y.; Lissel, F.; Linder, C.; et al. A highly stretchable autonomous self-healing elastomer. Nat. Chem. 2016, 8, 618–624. [Google Scholar] [CrossRef]
- Li, X.; Zhang, D.; Xiang, K.; Huang, G. Synthesis of polyborosiloxane and its reversible physical crosslinks. RSC Adv. 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Seetapan, N.; Fuongfuchat, A.; Sirikittikul, D.; Limparyoon, N. Unimodal and bimodal networks of physically crosslinked polyborodimethylsiloxane: Viscoelastic and equibiaxial extension behaviors. J. Polym. Res. 2013, 20, 183. [Google Scholar] [CrossRef]
- Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Šlouf, M.; Zhigunov, A.; Vyroubalová, M.; Kaňková, D.; Netopilík, M. Meltable copolymeric elastomers based on polydimethylsiloxane with multiplets of pendant liquid-crystalline groups as physical crosslinker: A self-healing structural material with a potential for smart applications. Eur. Polym. J. 2020, 137. [Google Scholar] [CrossRef]
- Wissbrun, K.F. Rheology of Rod-like Polymers in the Liquid Crystalline State. J. Rheol. 1981, 25, 619–662. [Google Scholar] [CrossRef]
- Marrucci, G. Rheology of liquid crystalline polymers. Pure Appl. Chem. 1985, 57, 1545–1552. [Google Scholar] [CrossRef]
- Guskey, S.M.; Winter, H.H. Transient shear behavior of a thermotropic liquid crystalline polymer in the nematic state. J. Rheol. 1991, 35, 1191–1207. [Google Scholar] [CrossRef]
- Marrucci, G.; Greco, F. Flow Behavior of Liquid Crystalline Polymers. In Advances in Chemical Physics; Prigogine, I., Rice, S.A., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 1993; Volume 86, pp. 331–404. ISBN 9780471598459; 9780470141458. [Google Scholar] [CrossRef]
- Jamieson, A.M.; Gu, D.; Chen, F.L.; Smith, S. Viscoelastic behavior of nematic monodomains containing liquid crystal polymers. Prog. Polym. Sci. 1996, 21, 981–1033. [Google Scholar] [CrossRef]
- Kiss, G.; Porter, R.S. Rheology of concentrated solutions of poly(γ-benzyl-glutamate). J. Polym. Sci. Polym. Symp. 1978, 65, 193–211. [Google Scholar] [CrossRef]
- Kiss, G.; Porter, R.S. Rheology of concentrated solutions of helical polypeptides. J. Polym. Sci. Polym. Phys. Ed. 1980, 18, 361–388. [Google Scholar] [CrossRef]
- Moldenaers, P.; Mewis, J. On the nature of viscoelasticity in polymeric liquid crystals. J. Rheol. 1993, 37, 367–380. [Google Scholar] [CrossRef]
- Asada, T.; Muramatsu, H.; Watanabe, R.; Onogi, S. Rheooptical Studies of Racemic Poly(γ-benzyl glutamate) Liquid Crystals. Macromolecules 1980, 13, 867–871. [Google Scholar] [CrossRef]
- Onogi, S.; Asada, T. Rheology and Rheo-Optics of Polymer Liquid Crystals. In Rheology; Astarita, G., Marrucci, G., Nicolais, L., Eds.; Springer: Boston, MA, USA, 1980; Volume 1, pp. 127–147. ISBN 978-1-4684-3742-3; 978-1-4684-3740-9. [Google Scholar] [CrossRef]
- Doi, M. Molecular dynamics and rheological properties of concentrated solutions of rodlike polymers in isotropic and liquid crystalline phases. J. Polym. Sci. Polym. Physics Ed. 1981, 19, 229–243. [Google Scholar] [CrossRef]
- Doi, M.; Edwards, S.F. The Theory of Polymer Dynamics; Oxford University Press: Oxford, UK, 1986; ISBN 0198519761; 9780198519768. [Google Scholar]
- Larson, R.G.; Doi, M. Mesoscopic domain theory for textured liquid crystalline polymers. J. Rheol. 1991, 35, 539–563. [Google Scholar] [CrossRef]
- Larson, R.G. The Structure and Rheology of Complex Fluids; Oxford University Press: New York, NY, USA, 1999; ISBN 019512197X; 978-0195121971. [Google Scholar]
- Burghardt, W.R. Molecular orientation and rheology in sheared lyotropic liquid crystalline polymers. Macromol. Chem. Phys. 1998, 199, 471–488. [Google Scholar] [CrossRef]
- Baek, S.G.; Magda, J.J.; Larson, R.G. Rheological differences among liquid-crystalline polymers. I. The first and second normal stress differences of PBG solutions. J. Rheol. 1993, 37, 1201–1224. [Google Scholar] [CrossRef]
- Baek, S.G.; Magda, J.J.; Larson, R.G.; Hudson, S.D. Rheological differences among liquid-crystalline polymers. II. Disappearance of negative N1 in densely packed lyotropes and thermotropes. J. Rheol. 1994, 38, 1473–1503. [Google Scholar] [CrossRef]
- Ugaz, V.M.; Burghardt, W.R.; Zhou, W.; Kornfield, J.A. Transient molecular orientation and rheology in flow aligning thermotropic liquid crystalline polymers. J. Rheol. 2001, 45, 1029–1063. [Google Scholar] [CrossRef]
- Azoug, A.; Vasconcellos, V.; Dooling, J.; Saed, M.; Yakacki, C.M.; Nguyen, T.D. Viscoelasticity of the polydomain-monodomain transition in main-chain liquid crystal elastomers. Polymer 2016, 98, 165–171. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.M.; Han, C.D. Rheology of Nematic Side-Chain Liquid-Crystalline Polymer: Comparison with Main-Chain Liquid-Crystalline Polymer. Macromolecules 2002, 35, 6263–6273. [Google Scholar] [CrossRef]
- Kim, S.S.; Han, C.D. Effect of Thermal History on the Rheological Behavior of a Thermotropic Liquid-Crystalline Polymer. Macromolecules 1993, 26, 3176–3186. [Google Scholar] [CrossRef]
- Colby, R.H.; Gillmor, J.R.; Galli, G.; Laus, M.; Ober, C.K.; Hall, E. Linear viscoelasticity of side chain liquid crystal polymer. Liq. Cryst. 1993, 13, 233–245. [Google Scholar] [CrossRef]
- Berghausen, J.; Fuchs, J.; Richtering, W. Rheology and Shear Orientation of a Nematic Liquid Crystalline Side-Group Polymer with Laterally Attached Mesogenic Units. Macromolecules 1997, 30, 7574–7581. [Google Scholar] [CrossRef]
- Chang, S.; Han, C.D. Effect of Flexible Spacers on the Rheological Behavior of Main-Chain Thermotropic Liquid-Crystalline Polymers Having Bulky Pendent Side Groups. Macromolecules 1997, 30, 2021–2034. [Google Scholar] [CrossRef]
- Lee, K.M.; Han, C.D. Effect of Flexible Spacer Length on the Rheology of Side-Chain Liquid-Crystalline Polymers. Macromolecules 2003, 36, 8796–8810. [Google Scholar] [CrossRef]
- Wewerka, A.; Viertler, K.; Vlassopoulos, D.; Stelzer, F. Structure and rheology of model side-chain liquid crystalline polymers with varying mesogen length. Rheol. Acta 2001, 40, 416–425. [Google Scholar] [CrossRef]
- Yang, I.K.; Chang, S.H. The Smectic Rheology of a Polysiloxane Side Chain Liquid Crystalline Polymer. J. Polym. Res. 2002, 9, 163–168. [Google Scholar] [CrossRef]
- Hoshio, H.; Jin, J.I.; Lenz, R.W. Liquid crystalline behavior of polymeric glycols terminated with aromatic diester and diacid mesogenic groups. J. Appl. Polym. Sci. 1984, 29, 547–554. [Google Scholar] [CrossRef]
- Chien, J.C.W.; Zhou, R.; Lillya, C.P. Liquid-crystalline compounds and polymers from promesogens. Macromolecules 1987, 20, 2340–2344. [Google Scholar] [CrossRef]
- Lin, Y.G.; Zhou, R.; Chien, J.C.W.; Winter, H.H. Rheology of a Twin Liquid Crystalline Polymer. Macromolecules 1988, 21, 2014–2018. [Google Scholar] [CrossRef] [Green Version]
- Oppermann, W.; Braatz, K.; Finkelmann, H.; Gleim, W.; Kock, H.J.; Rehage, G. Viscoelastic properties of silicone polymers with liquid crystalline behaviour. Rheol. Acta 1982, 21, 423–426. [Google Scholar] [CrossRef]
- Stukenbroeker, T.; Wang, W.; Winne, J.M.; Du Prez, F.E.; Nicolaÿ, R.; Leibler, L. Polydimethylsiloxane quenchable vitrimers. Polym. Chem. 2017, 8, 6590–6593. [Google Scholar] [CrossRef]
- Meng, Y.; Xu, W.; Newman, M.R.; Benoit, D.S.W.; Anthamatten, M. Thermoreversible Siloxane Networks: Soft Biomaterials with Widely Tunable Viscoelasticity. Adv. Funct. Mater. 2019, 29, 1903721. [Google Scholar] [CrossRef]
- Winter, H.H.; Chambon, F. Analysis of linear viscoelasticity of a crosslinking polymer at the gel point. J. Rheol. 1986, 30, 367–382. [Google Scholar] [CrossRef]






















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horodecka, S.; Strachota, A.; Mossety-Leszczak, B.; Kisiel, M.; Strachota, B.; Šlouf, M. Low-Temperature-Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinker: A Passive Smart Material with Potential as Viscoelastic Coupling. Part II—Viscoelastic and Rheological Properties. Polymers 2020, 12, 2840. https://doi.org/10.3390/polym12122840
Horodecka S, Strachota A, Mossety-Leszczak B, Kisiel M, Strachota B, Šlouf M. Low-Temperature-Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinker: A Passive Smart Material with Potential as Viscoelastic Coupling. Part II—Viscoelastic and Rheological Properties. Polymers. 2020; 12(12):2840. https://doi.org/10.3390/polym12122840
Chicago/Turabian StyleHorodecka, Sabina, Adam Strachota, Beata Mossety-Leszczak, Maciej Kisiel, Beata Strachota, and Miroslav Šlouf. 2020. "Low-Temperature-Meltable Elastomers Based on Linear Polydimethylsiloxane Chains Alpha, Omega-Terminated with Mesogenic Groups as Physical Crosslinker: A Passive Smart Material with Potential as Viscoelastic Coupling. Part II—Viscoelastic and Rheological Properties" Polymers 12, no. 12: 2840. https://doi.org/10.3390/polym12122840