
Poly(imidazolium) Carbosilane Dendrimers: Synthesis, Catalytic Activity in Redox Esterification of α,β-Unsaturated Aldehydes and Recycling via Organic Solvent Nanofiltration

 , , , , and
, , , , and
Abstract
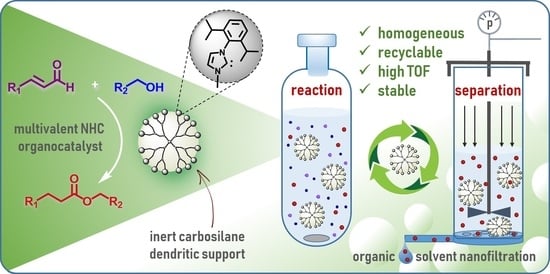
:
1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
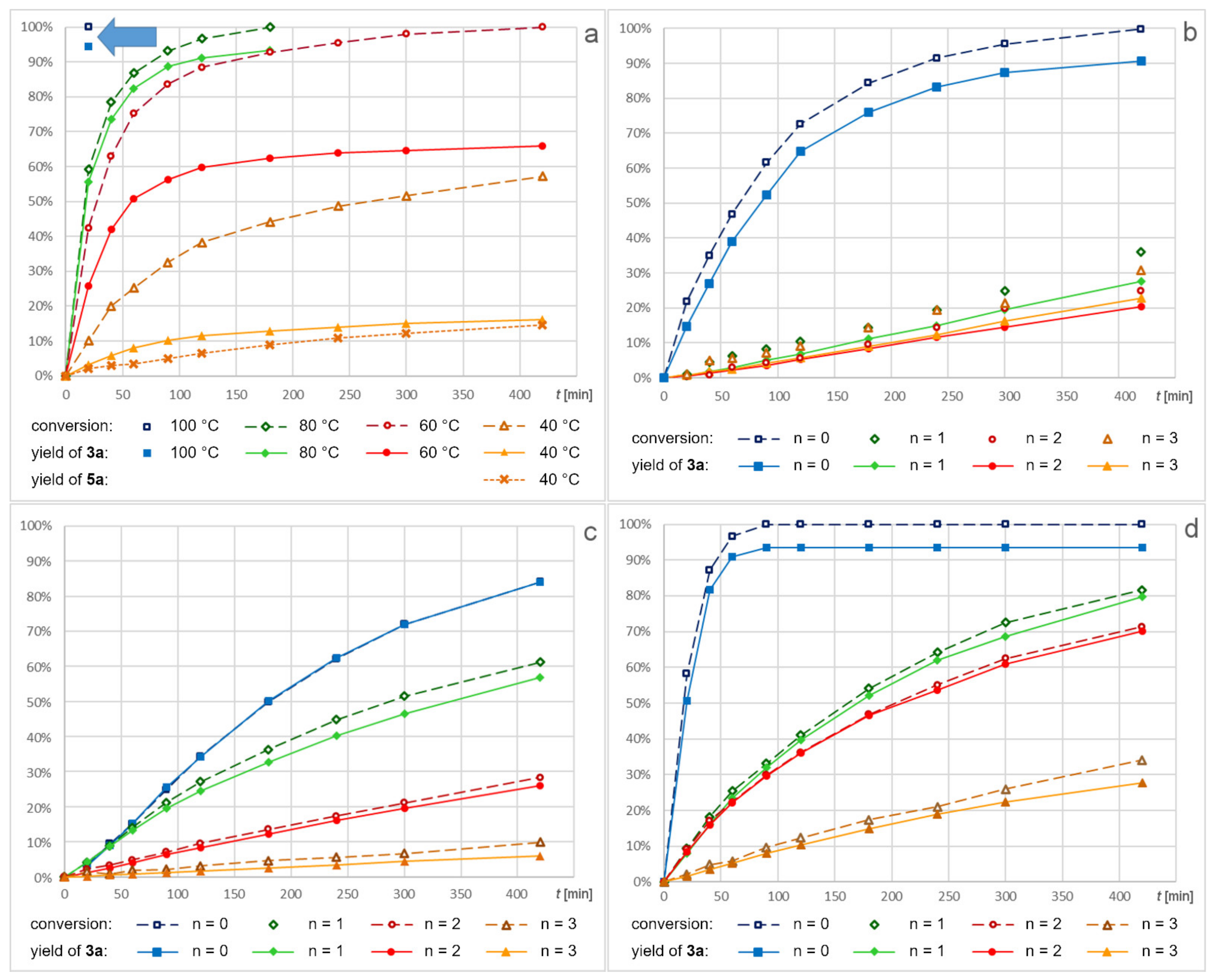
2.2. Screening of Catalysts
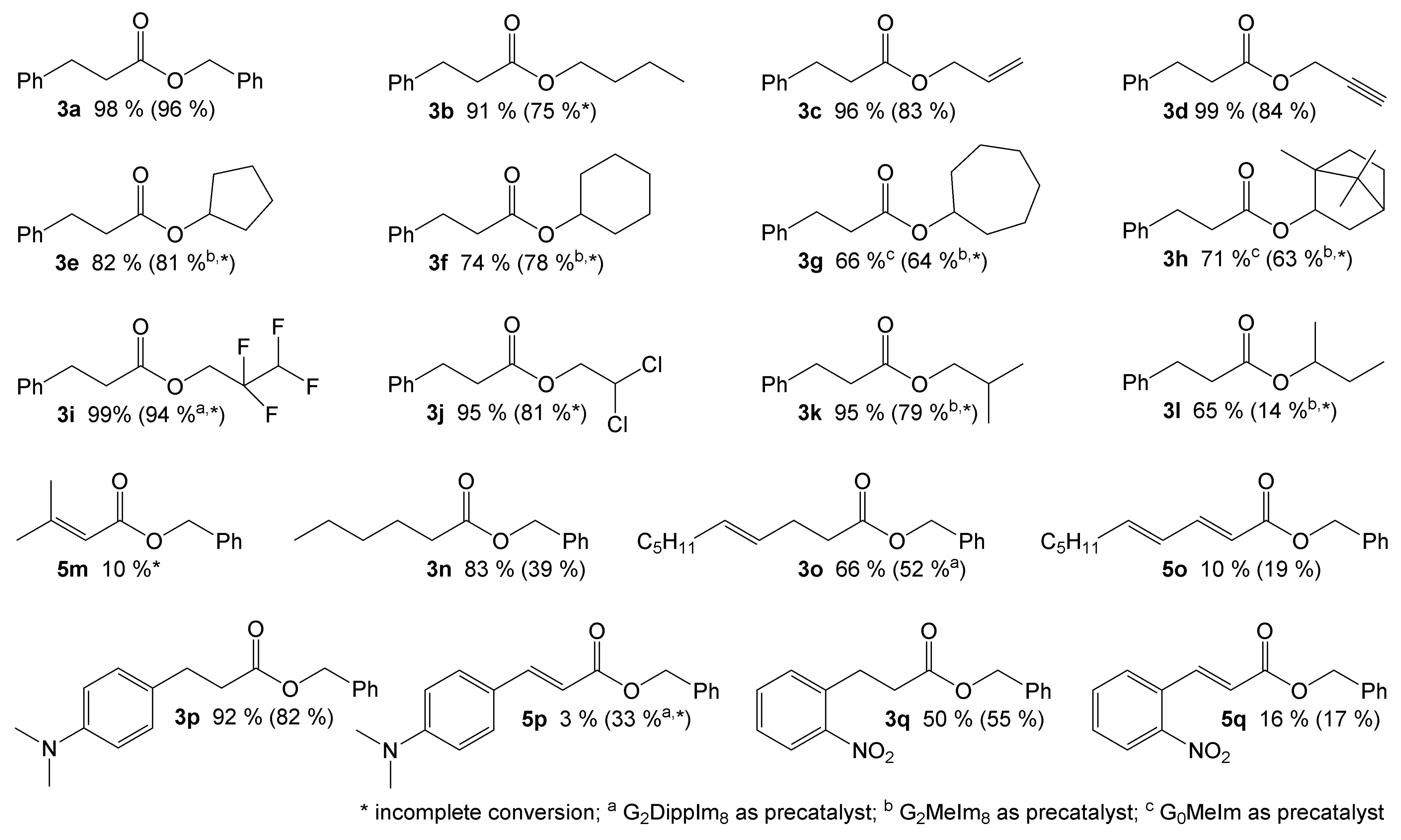
2.3. Screening of Substrates
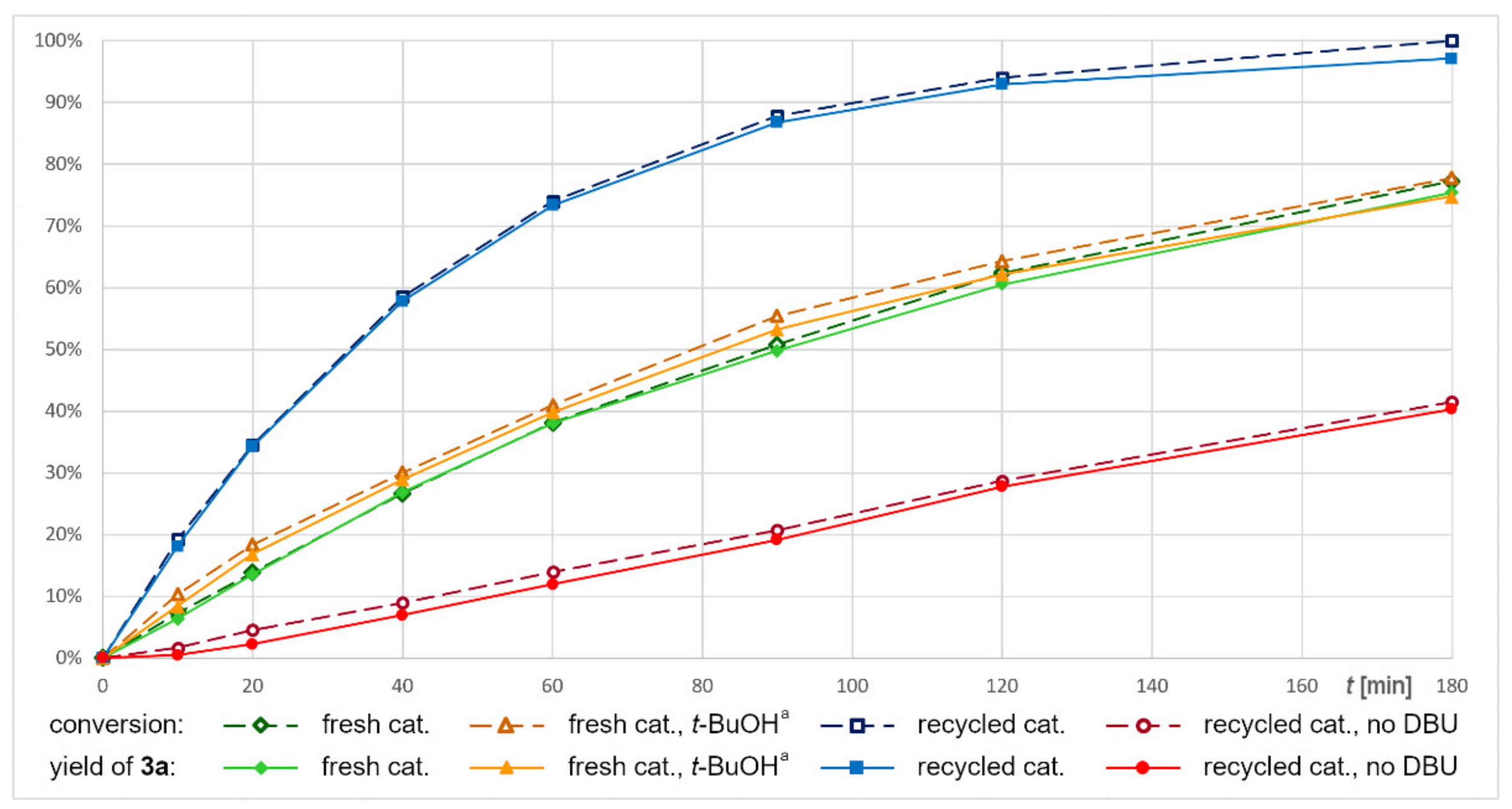
2.4. Recycling of Dendritic Catalysts
3. Materials and Methods
3.1. General Remarks
3.2. Synthesis of Model Imidazolium Salts and Poly(imidazolium) Carbosilane Dendrimers
3.3. Redox Esterification of Cinnamaldehyde
3.4. Organic Solvent Nanofiltration
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xin, B.; Hao, J. Imidazolium-Based Ionic Liquids Grafted on Solid Surfaces. Chem. Soc. Rev. 2014, 43, 7171–7187. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, J.; Zhang, Y.; Deng, Y. Nanoconfined Ionic Liquids. Chem. Rev. 2017, 117, 6755–6833. [Google Scholar] [CrossRef] [PubMed]
- Fournier, A.H.; de Robillard, G.; Devillers, C.H.; Plasseraud, L.; Andrieu, J. Imidazolium and Potassium Hydrogen Carbonate Salts as Ecofriendly Organocatalysts for Oxazolidinone Synthesis. Eur. J. Org. Chem. 2016, 2016, 3514–3518. [Google Scholar] [CrossRef]
- Chakraborty Ghosal, N.; Santra, S.; Das, S.; Hajra, A.; Zyryanov, G.V.; Majee, A. Organocatalysis by an Aprotic Imidazolium Zwitterion: Regioselective Ring-Opening of Aziridines and Applicable to Gram Scale Synthesis. Green Chem. 2016, 18, 565–574. [Google Scholar] [CrossRef]
- Ghosh, D.; Lee, J.-Y.; Liu, C.-Y.; Chiang, Y.-H.; Lee, H.M. Direct C-H Arylations of Unactivated Arenes Catalyzed by Amido-Functionalized Imidazolium Salts. Adv. Synth. Catal. 2014, 356, 406–410. [Google Scholar] [CrossRef]
- Bobbink, F.D.; Dyson, P.J. Synthesis of Carbonates and Related Compounds Incorporating CO2 using Ionic Liquid-Type Catalysts: State-of-the-Art and Beyond. J. Catal. 2016, 343, 52–61. [Google Scholar] [CrossRef]
- Büttner, H.; Longwitz, L.; Steinbauer, J.; Wulf, C.; Werner, T. Recent Developments in the Synthesis of Cyclic Carbonates from Epoxides and CO2. Top. Curr. Chem. 2017, 375, 1–56. [Google Scholar] [CrossRef]
- Marion, N.; Díez-González, S.; Nolan, S.P. N-Heterocyclic Carbenes as Organocatalysts. Angew. Chem. Int. Ed. 2007, 46, 2988–3000. [Google Scholar] [CrossRef]
- Ryan, S.J.; Candish, L.; Lupton, D.W. Acyl Anion Free N-Heterocyclic Carbene Organocatalysis. Chem. Soc. Rev. 2013, 42, 4906–4917. [Google Scholar] [CrossRef]
- Grossmann, A.; Enders, D. N-Heterocyclic Carbene Catalyzed Domino Reactions. Angew. Chem. Int. Ed. 2012, 51, 314–325. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Inesi, A. Advances in the Knowledge of N-Heterocyclic Carbenes Properties. The Backing of the Electrochemical Investigation. Catalysts 2016, 6, 178. [Google Scholar] [CrossRef]
- Menon, R.S.; Biju, A.T.; Nair, V. Recent Advances in Employing Homoenolates Generated by N-Heterocyclic Carbene (NHC) Catalysis in Carbon-Carbon Bond-Forming Reactions. Chem. Soc. Rev. 2015, 44, 5040–5052. [Google Scholar] [CrossRef] [Green Version]
- De Sarkar, S.; Biswas, A.; Samanta, R.C.; Studer, A. Catalysis with N-Heterocyclic Carbenes under Oxidative Conditions. Chemistry 2013, 19, 4664–4678. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Toy, P. Self-Supported N-Heterocyclic Carbenes and Their Use as Organocatalysts. Molecules 2016, 21, 1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cywar, R.M.; Wang, L.; Chen, E.Y.-X. Thermally Regulated Recyclable Carbene Catalysts for Upgrading of Biomass Furaldehydes. ACS Sustain. Chem. Eng. 2019, 7, 1980–1988. [Google Scholar] [CrossRef]
- Coupillaud, P.; Pinaud, J.; Guidolin, N.; Vignolle, J.; Fèvre, M.; Veaudecrenne, E.; Mecerreyes, D.; Taton, D. Poly(Ionic Liquid)s Based on Imidazolium Hydrogen Carbonate Monomer Units as Recyclable Polymer-Supported N-Heterocyclic Carbenes: Use in Organocatalysis. J. Polym. Sci. Part. A Polym. Chem. 2013, 51, 4530–4540. [Google Scholar] [CrossRef]
- Barrett, A.G.M.; Love, A.C.; Tedeschi, L. ROMPgel-Supported Thiazolium Iodide: An Efficient Supported Organic Catalyst for Parallel Stetter Reactions. Org. Lett. 2004, 6, 3377–3380. [Google Scholar] [CrossRef]
- Strašák, T.; Št’astná, L.Č.; Bílková, V.; Skoupá, V.; Karban, J.; Cuřínová, P.; Čermák, J. Synthesis and Fluorophilicity of Compounds with Tris(3,3,4,4,5,5,6,6,7,7,8,8,8-Tridecafluorooctyl)Silyl Substituent. J. Fluor. Chem. 2015, 178, 23–29. [Google Scholar] [CrossRef]
- Strašák, T.; Čermák, J.; Červenková Šťastná, L.; Sýkora, J.; Fajgar, R. Cobalt(I) and Cobalt(III) Cyclopentadienyl Complexes with New Silicon-Branched Fluorous Tags. J. Fluor. Chem. 2014, 159, 15–20. [Google Scholar] [CrossRef]
- Červenková Šťastná, L.; Bílková, V.; Cézová, T.; Cuřínová, P.; Karban, J.; Čermák, J.; Krupková, A.; Strašák, T. Imidazolium Based Fluorous N-Heterocyclic Carbenes as Effective and Recyclable Organocatalysts for Redox Esterification. Eur. J. Org. Chem. 2020, 2020, 3591–3598. [Google Scholar] [CrossRef]
- Liu, Y.; Cheng, W.; Zhang, Y.; Sun, J.; Zhang, S. Controllable Preparation of Phosphonium-Based Polymeric Ionic Liquids as Highly Selective Nanocatalysts for the Chemical Conversion of CO2 with Epoxides. Green Chem. 2017, 19, 2184–2193. [Google Scholar] [CrossRef]
- Müllerová, M.; Šabata, S.; Matoušek, J.; Kormunda, M.; Holubová, J.; Bálková, R.; Petričkovič, R.; Koštejn, M.; Kupčík, J.; Fajgar, R.; et al. Organoclays with Carbosilane Dendrimers Containing Ammonium or Phosphonium Groups. New J. Chem. 2018, 42, 1187–1196. [Google Scholar] [CrossRef]
- Ding, M.; Jiang, H.L. Incorporation of Imidazolium-Based Poly(Ionic Liquid)s into a Metal-Organic Framework for CO2 Capture and Conversion. ACS Catal. 2018, 8, 3194–3201. [Google Scholar] [CrossRef]
- Poli, R. Effects of Nanoconfinement on Catalysis; Poli, R., Ed.; Springer International Publishing: Cham, The Netherlands, 2017. [Google Scholar] [CrossRef]
- Tomalia, D.A. Dendrimers and Other Dendritic Polymers; Fréchet, J.M.J., Tomalia, D.A., Eds.; John Wiley & Sons, Ltd.: Chichester, UK, 2001; Volume 1. [Google Scholar] [CrossRef]
- Perrier, A.; Keller, M.; Caminade, A.M.; Majoral, J.P.; Ouali, A. Efficient and Recyclable Rare Earth-Based Catalysts for Friedel-Crafts Acylations under Microwave Heating: Dendrimers Show the Way. Green Chem. 2013, 15, 2075–2080. [Google Scholar] [CrossRef]
- Keller, M.; Perrier, A.; Linhardt, R.; Travers, L.; Wittmann, S.; Caminade, A.M.; Majoral, J.P.; Reiser, O.; Ouali, A. Dendrimers or Nanoparticles as Supports for the Design of Efficient and Recoverable Organocatalysts? Adv. Synth. Catal. 2013, 355, 1748–1754. [Google Scholar] [CrossRef]
- Deng, G.-J.; Fan, Q.-H.; Chen, X.-M.; Liu, D.-S.; Chan, A.S.C. A Novel System Consisting of Easily Recyclable Dendritic Ru-BINAP Catalyst for Asymmetric Hydrogenation. Chem. Commun. 2002, 15, 1570–1571. [Google Scholar] [CrossRef]
- Huang, Y.-Y.; He, Y.-M.; Zhou, H.-F.; Wu, L.; Li, B.-L.; Fan, Q.-H. Thermomorphic System with Non-Fluorous Phase-Tagged Ru(BINAP) Catalyst: Facile Liquid/Solid Catalyst Separation and Application in Asymmetric Hydrogenation. J. Org. Chem. 2006, 71, 2874–2877. [Google Scholar] [CrossRef]
- Liu, J.; Ma, B.; Feng, Y.; He, Y.; Fan, Q.H. Dendritic [RuCl2(BINAP)(DPEN)] Catalysts with “Sandwich” Multi-Layer Structure for Asymmetric Hydrogenation of Simple Aryl Ketones. Inorg. Chim. Acta 2014, 409, 106–111. [Google Scholar] [CrossRef]
- Keller, M.; Collière, V.; Reiser, O.; Caminade, A.M.; Majoral, J.P.; Ouali, A. Pyrene-Tagged Dendritic Catalysts Noncovalently Grafted onto Magnetic Co/C Nanoparticles: An Efficient and Recyclable System for Drug Synthesis. Angew. Chem. Int. Ed. 2013, 52, 3626–3629. [Google Scholar] [CrossRef]
- Bronstein, L.M. Magnetically Recoverable Catalysts with Dendritic Ligands for Enhanced Catalysis and Easy Separation. ChemCatChem 2015, 7, 1058–1060. [Google Scholar] [CrossRef]
- Gaab, M.; Bellemin-Laponnaz, S.; Gade, L.H. “Catalysis in a Tea Bag”: Synthesis, Catalytic Performance and Recycling of Dendrimer-Immobilised Bis- and Trisoxazoline Copper Catalysts. Chemistry 2009, 15, 5450–5462. [Google Scholar] [CrossRef] [PubMed]
- Nemanashi, M.; Meijboom, R. “Cat in a Bag” Recycling of Dendrimer Encapsulated Au Nanoparticles by Use of Dialysis Membrane Bag in the Reduction of 4-Nitrophenol: Proof of Heterogeneous Catalysis. Catal. Commun. 2016, 83, 53–57. [Google Scholar] [CrossRef]
- Hovestad, N.J.; Eggeling, E.B.; Heidbüchel, H.J.; Jastrzebski, J.T.B.H.; Kragl, U.; Keim, W.; Vogt, D.; Van Koten, G. Selective Hydrovinylation of Styrene in a Membrane Reactor: Use of Carbosilane Dendrimers with Hemilabile P,O Ligands. Angew. Chem. Int. Ed. 1999, 38, 1655–1658. [Google Scholar] [CrossRef]
- Albrecht, M.; Hovestad, N.J.; Boersma, J.; Van Koten, G. Multiple Use of Soluble Metallodendritic Materials as Catalysts and Dyes. Chemistry 2001, 7, 1289–1294. [Google Scholar] [CrossRef]
- Guerra, J.; Cantillo, D.; Kappe, C.O. Visible-Light Photoredox Catalysis Using a Macromolecular Ruthenium Complex: Reactivity and Recovery by Size-Exclusion Nanofiltration in Continuous Flow. Catal. Sci. Technol. 2016, 6, 4695–4699. [Google Scholar] [CrossRef] [Green Version]
- Strašák, T.; Jaroschik, F.; Malý, M.; Čermák, J.; Sýkora, J.; Fajgar, R.; Karban, J.; Harakat, D. Titanocene Dichloride Complexes Bonded to Carbosilane Dendrimers via a Spacer of Variable Length—Molecular Dynamics Calculations and Catalysis of Allylic Coupling Reactions. Inorg. Chim. Acta 2014, 409, 137–146. [Google Scholar] [CrossRef]
- Marchetti, P.; Jimenez Solomon, M.F.; Szekely, G.; Livingston, A.G. Molecular Separation with Organic Solvent Nanofiltration: A Critical Review. Chem. Rev. 2014, 114, 10735–10805. [Google Scholar] [CrossRef]
- Červenková Šťastná, L.; Krupková, A.; Petrickovic, R.; Müllerová, M.; Matoušek, J.; Koštejn, M.; Cuřínová, P.; Jandová, V.; Šabata, S.; Strašák, T. Multivalent Bifunctional Carbosilane Dendrimer-Supported Ammonium and Phosphonium Organocatalysts for the Coupling of CO2 and Epoxides. ACS Sustain. Chem. Eng. 2020, 8, 11692–11703. [Google Scholar] [CrossRef]
- Strašák, T.; Malý, J.; Wróbel, D.; Malý, M.; Herma, R.; Čermák, J.; Müllerová, M.; Št′astná, L.Č.; Cuřínová, P. Phosphonium Carbosilane Dendrimers for Biomedical Applications-Synthesis, Characterization and Cytotoxicity Evaluation. RSC Adv. 2017, 7, 18724–18744. [Google Scholar] [CrossRef] [Green Version]
- Cuřínová, P.; Krupková, A.; Červenková Šťastná, L.; Müllerová, M.; Čermák, J.; Strašák, T. ESI-TOF Mass Spectrometry of Cationic Carbosilane Dendrimers: A Potent Tool for Characterization of Structural Defects. J. Mass Spectrom. 2018, 53, 986–996. [Google Scholar] [CrossRef]
- Chan, A.; Scheidt, K.A. Conversion of α,β-Unsaturated Aldehydes into Saturated Esters: An Umpolung Reaction Catalyzed by Nucleophilic Carbenes. Org. Lett. 2005, 7, 905–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.H.; Barsoum, D.; Schwamb, C.B.; Cohen, D.T.; Goess, B.C.; Riedrich, M.; Chan, A.; Maki, B.E.; Mishra, R.K.; Scheidt, K.A. Catalytic, Enantioselective β-Protonation through a Cooperative Activation Strategy. J. Org. Chem. 2017, 82, 4689–4702. [Google Scholar] [CrossRef] [PubMed]
- Mahatthananchai, J.; Bode, J.W. The Effect of the N-Mesityl Group in NHC-Catalyzed Reactions. Chem. Sci. 2012, 3, 192–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, B.E.; Patterson, E.V.; Cramer, C.J.; Scheidt, K.A. Impact of Solvent Polarity on N-Heterocyclic Carbene-Catalyzed β-Protonations of Homoenolate Equivalents. Org. Lett. 2009, 11, 3942–3945. [Google Scholar] [CrossRef] [Green Version]
- Maki, B.E.; Chan, A.; Scheidt, K.A. Protonation of Homoenolate Equivalents Generated by N-Heterocyclic Carbenes. Synthesis 2008, 8, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Sohn, S.S.; Bode, J.W. Catalytic Generation of Activated Carboxylates from Enals: A Product-Determining Role for the Base. Org. Lett. 2005, 7, 3873–3876. [Google Scholar] [CrossRef]
- Yatham, V.R.; Harnying, W.; Kootz, D.; Neudörfl, J.M.; Schlörer, N.E.; Berkessel, A. 1,4-Bis-Dipp/Mes-1,2,4-Triazolylidenes: Carbene Catalysts That Efficiently Overcome Steric Hindrance in the Redox Esterification of α- and β-Substituted α,β-Enals. J. Am. Chem. Soc. 2016, 138, 2670–2677. [Google Scholar] [CrossRef]
- de Saint Laumer, J.-Y.; Cicchetti, E.; Merle, P.; Egger, J.; Chaintreau, A. Quantification in Gas Chromatography: Prediction of Flame Ionization Detector Response Factors from Combustion Enthalpies and Molecular Structures. Anal. Chem. 2010, 82, 6457–6462. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Precatalyst | Loading [mol%] | Base [mol%] | Conversion [%] a | Yield 3a/4/5a [%] a | ||
| 1 | G0MeImI | 10 | 10 | 96 | 87 | 7 | 2 |
| 2 | G1(MeImI)4 | 10 | 10 | 25 | 20 | 3 | 1 |
| 3 | G2(MeImI)8 | 10 | 10 | 20 | 15 | 1 | 1 |
| 4 | G3(MeImI)16 | 10 | 10 | 21 | 16 | 4 | 1 |
| 5 | G0iPrImI | 10 | 10 | 38 | 31 | 6 | 1 |
| 6 | G1(iPrImI)4 | 10 | 10 | 10 | 2 | 7 | 1 |
| 7 | G2(iPrImI)8 | 10 | 10 | 9 | 2 | 5 | - |
| 8 b | G0DippImI | 10 | 10 | 100 | 94 | 5 | 1 |
| 9 c | G0DippImI | 1 | 10 | 100 | 94 | 6 | - |
| 10 | G0DippImI | 1 | 2 | 73 | 72 | - | 1 |
| 11 d | G0DippImI | 10 | 10 | 100 | 93 | 3 | 3 |
| 12 e | G0DippImI | 10 | 10 | 100 | 65 | 9 | 8 |
| 13 f | G0DippImI | 10 | 10 | 52 | 15 | 8 | 12 |
| 14 | G1(DippImI)4 | 1 | 10 | 72 | 69 | 1 | 2 |
| 15 | G1(DippImI)4 | 1 | 2 | 51 | 47 | - | 1 |
| 16 | G2(DippImI)8 | 1 | 10 | 63 | 61 | 0.5 | - |
| 17 | G2(DippImI)8 | 1 | 2 | 21 | 20 | - | 0.4 |
| 18 | G3(DippImI)16 | 1 | 10 | 26 | 22 | 0.4 | 2 |
| 19 | G3(DippImI)16 | 1 | 2 | 7 | 4 | - | - |
| Loading c | Substrate (Product) | Benzyl alcohol 2a (3a) | 1-Butanol 2b (3b) | Allyl alcohol 2c (3c) | Propargyl alcohol 2d (3d) | ||||
| Time [h] | 5 | 24 | 5 | 24 | 5 | 24 | 5 | 24 | |
| 2 | G0DippImI | 91 (93) | 98 (100) | 91 (100) | - | 86 (89) | 96 (100) | 80 (83) | 99 (100) |
| G1(DippImI)4 | 57 (58) | 96 (97) | 36 (45) | 75 (86) | 15 (20) | 44 (52) | 6 (6) | 43 (48) | |
| G2(DippImI)8 | 31 (31) | 74 (76) | 26 (32) | 66 (76) | 6 (7) | 32 (35) | 4 (5) | 28 (36) | |
| 10 | G1(DippImI)4 | - | - | - | - | 72 (85) | 83 (100) | 73 (86) | 84 (100) |
| G2(DippImI)8 | - | - | - | - | 69 (78) | 83 (98) | 57 (72) | 78 (98) | |
| Loading c | Substrate (Product) | Cyclopentanol 2e (3e) | Cyclohexanol 2f (3f) | Cycloheptanol 2g (3g) | Borneol 2h (3h) | ||||
| Time [h] | 5 | 100 | 5 | 100 | 5 | 100 | 5 | 100 | |
| 2 | G0DippImI | 31 (46) | 82 (100) | 20 (39) | 74 (100) | 8 (21) | 50 (88) | 3 (26) | 13 (67) |
| G1(DippImI)4 | 11 (22) | 50 (71) | 11 (23) | 45 (83) | 4 (13) | 10 (28) | 1 (13) | 4 (30) | |
| G2(DippImI)8 | 6 (12) | 38 (57) | 2 (13) | 20 (43) | 3 (10) | 11 (32) | 1 (8) | 3 (18) | |
| 10 | G1(DippImI)4 | 29 (57) | 65 (98) | 18 (53) | 55 (100) | 17 (58) | 34 (92) | - | - |
| G2(DippImI)8 | 23 (48) | 68 (99) | 24 (58) | 57 (100) | 9 (45) | 34 (92) | - | - | |
| G0MeImI | 66 (82) | 81 (100) d | 52 (73) | 71 (93)d | 53 (82) | 66 (100) d | 58 (83) | 71 (100) d | |
| G1(MeImI)4 | 13 (20) | 69 (86) | 10 (19) | 64 (87) | 9 (22) | 48 (78) | 9 (21) | 42 (74) | |
| G2(MeImI)8 | 30 (35) | 81 (95) | 12 (20) | 68 (88) | 19 (32) | 64 (92) | 26 (42) | 63 (91) | |
| Loading c | Substrate (Product) | 2,2,3,3-Tetrafluoropropanol 2i (3i) | 2,2-Dichloroethanol 2j (3j) | Isobutanol 2k (3k) | 2-Butanol 2l (3l) | ||||
| Time [h] | 5 | 100 | 5 | 100 | 5 | 24 | 5 | 100 | |
| 2 | G0DippImI | 1 (3) | 99 (100) | 10 (13) | 90 (90) | 76 (85) | 95 (100) | 3 (20) | 5 (30) d |
| G1(DippImI)4 | 0.4 (1) | 8 (8) | 2 (5) | 20 (20) | 15 (21) | 32 (44) | 2 (19) | 3 (24) d | |
| G2(DippImI)8 | 0.4 (1) | 19 (20) | 1 (2) | 20 (21) | 10 (11) | 26 (34) | 2 (14) | 4 (23) d | |
| 10 | G1(DippImI)4 | 41 (54) | 83 (100) d | 48 (58) | 80 (91) | 67 (88) | 77 (100) | - | - |
| G2(DippImI)8 | 35 (48) | 94 (100) d | 35 (48) | 60 (83) | 57 (72) | 79 (100) | - | - | |
| G0MeImI | - | - | 17 (23) | 59 (70) | - | - | 41 (66) | 65 (100) | |
| G1(MeImI)4 | - | - | 1 (7) | 4 (9) | - | - | 4 (15) | 8 (45) | |
| G2(MeImI)8 | - | - | 5 (9) | 15 (23) | - | - | 8 (25) | 14 (50) | |
| Entry | Substrate | Conversion b | Product | Yield b |
|---|---|---|---|---|
| 1 | 2-methyl-2-butenal 1b | 7 | - | - |
| 2 | 3-methyl-2-butenal 1c | 12 | 5m | 3 |
| 3 | trans-2-hexenal 1d | 96 | 3n | 76 |
| 4 | trans,trans-2,4-decadienal 1e | 100 | 3o + 5o | 66 + 10 |
| 5 | α-methylcinnamaldehyde 1f | 2 | - | - |
| 6 | 3-(4-(dimethylamino)phenyl)-2-propenal 1g | 82 | 3p + 5p | 73 + 1 |
| 7 | 3-(2-nitrophenyl)-2-propenal 1h | 93 | 3q + 5q | 45 + 16 |
| Run | Reaction Time [h] | TOF [h−1] b | Isolated Yield [%] | GC Purity [%] | Catalyst Recovery [%] c |
|---|---|---|---|---|---|
| 1 d | 3 | 3.3 | 79 | 93 | 77 |
| 2 | 2 | 5.0 | 73 | 97 | 89 |
| 3 | 1.5 | 6.7 | 68 | 97 | 95 |
| 4 | 1 | 10 | 73 | 98 | 92 |
| 5 | 1 | 10 | 72 | 98 | 95 |
| 6 | 1 | 10 | 73 | 98 | 92 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krupková, A.; Kubátová, K.; Červenková Šťastná, L.; Cuřínová, P.; Müllerová, M.; Karban, J.; Čermák, J.; Strašák, T. Poly(imidazolium) Carbosilane Dendrimers: Synthesis, Catalytic Activity in Redox Esterification of α,β-Unsaturated Aldehydes and Recycling via Organic Solvent Nanofiltration. Catalysts 2021, 11, 1317. https://doi.org/10.3390/catal11111317
Krupková A, Kubátová K, Červenková Šťastná L, Cuřínová P, Müllerová M, Karban J, Čermák J, Strašák T. Poly(imidazolium) Carbosilane Dendrimers: Synthesis, Catalytic Activity in Redox Esterification of α,β-Unsaturated Aldehydes and Recycling via Organic Solvent Nanofiltration. Catalysts. 2021; 11(11):1317. https://doi.org/10.3390/catal11111317
Chicago/Turabian StyleKrupková, Alena, Klára Kubátová, Lucie Červenková Šťastná, Petra Cuřínová, Monika Müllerová, Jindřich Karban, Jan Čermák, and Tomáš Strašák. 2021. "Poly(imidazolium) Carbosilane Dendrimers: Synthesis, Catalytic Activity in Redox Esterification of α,β-Unsaturated Aldehydes and Recycling via Organic Solvent Nanofiltration" Catalysts 11, no. 11: 1317. https://doi.org/10.3390/catal11111317