
Inhibition of Aryl Hydrocarbon Receptor (AhR) Expression Disrupts Cell Proliferation and Alters Energy Metabolism and Fatty Acid Synthesis in Colon Cancer Cells
 , ,
, ,  , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Generation of AhR Knock-Out HCT116 Cells
2.3. Cell Painting and Morphology Analysis
2.4. Immunohistochemistry
2.5. Cell Proliferation and Cell Cycle Analyses
2.6. Dual Luciferase Assay (TOP/FOP)
2.7. Real-Time Quantitative RT-PCR (RT-qPCR)
2.8. Western Blotting
2.9. Detection of ATP Production with SeaHorse Analyzer and Detection of Glucose Levels in Cell Culture Media
2.10. Analysis of Lipid and FA Content
2.11. Statistical Analysis
3. Results
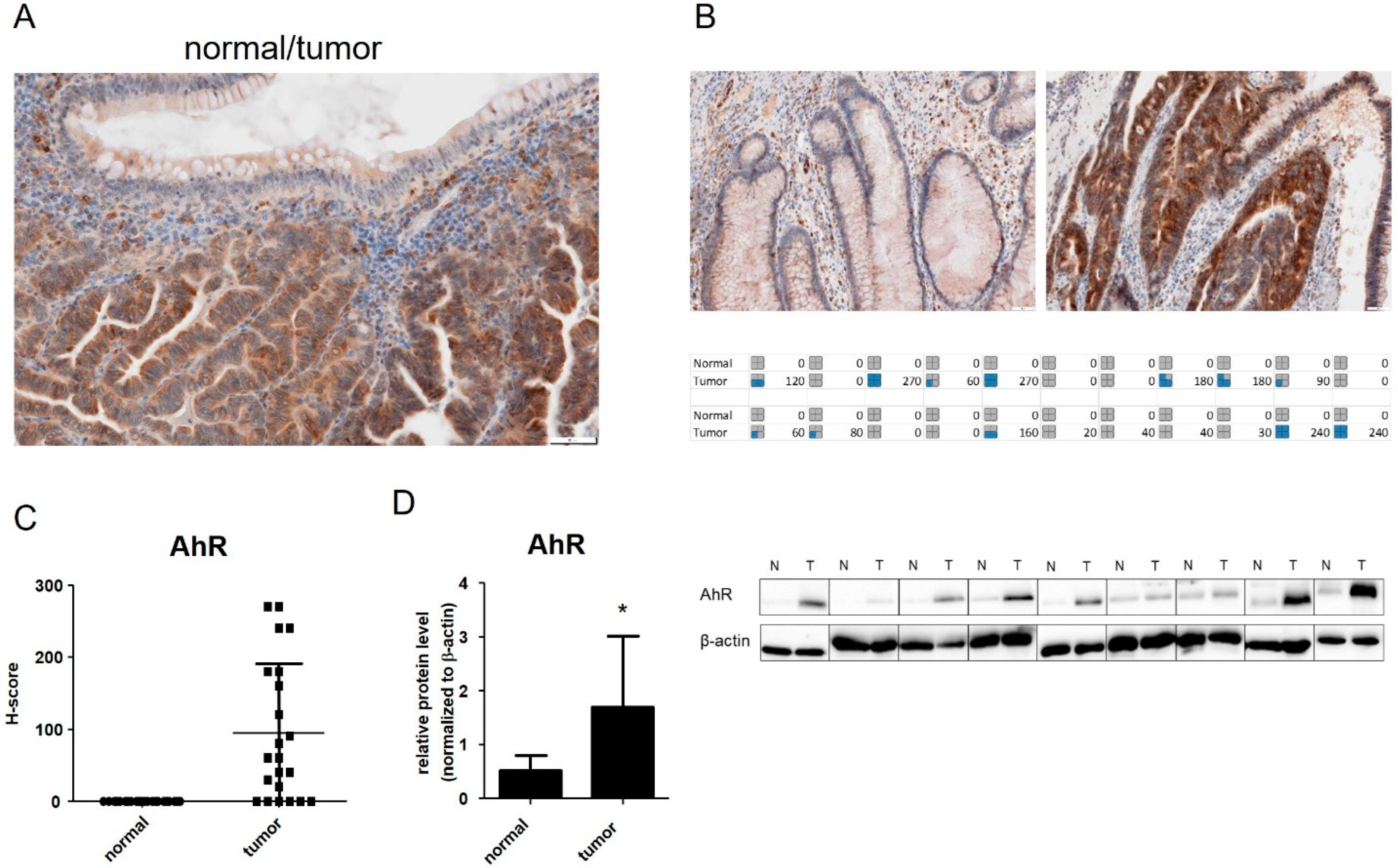
3.1. AhR Is Overexpressed in CRC Tumor Samples and in Isolated Colon Cancer Epithelial Cells
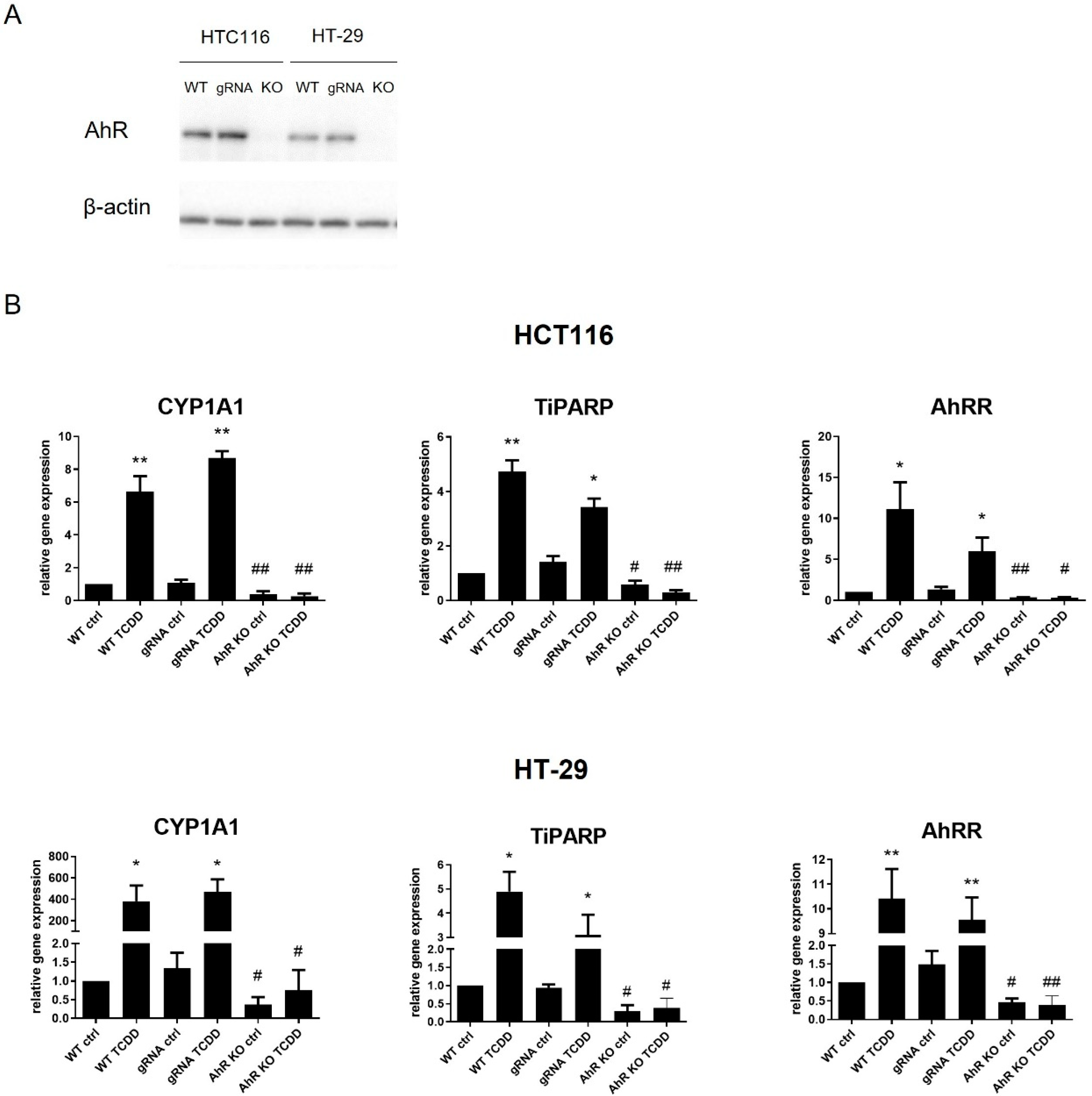
3.2. Loss of the AhR Reduces Proliferative Rate of Colon Cancer HCT116 and HT-29 Cells
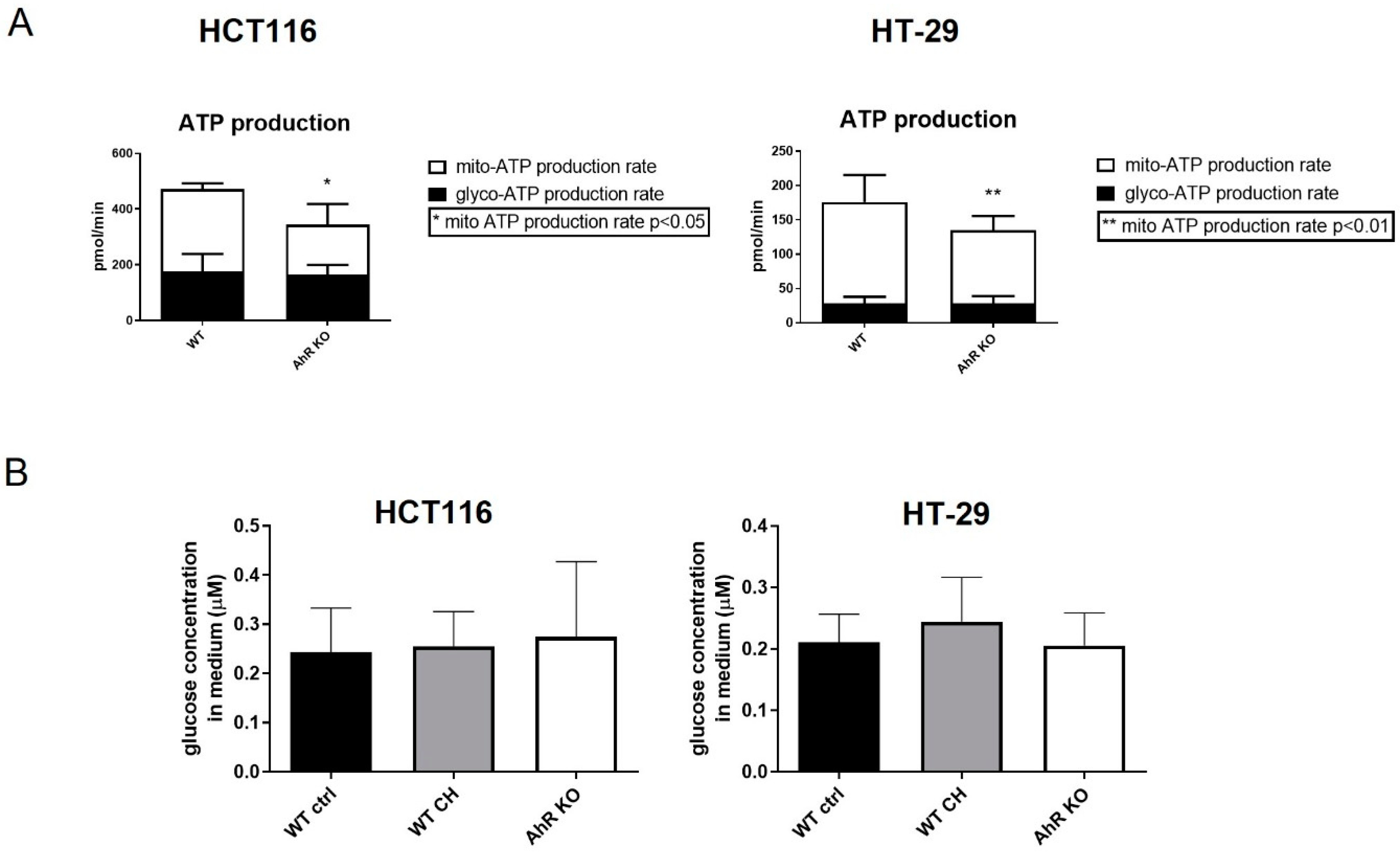
3.3. AhR Deficiency Alters Cellular Energy Metabolism in HCT116 and HT-29 Cells
3.4. Loss of the AhR Affects Expression of FA Synthesis Genes and Limits Akt Signaling
3.5. AhR Antagonist CH-223191 Inhibits Cell Proliferation and Expression of FA Synthesis-Related Genes and Their Regulators in Colon Cancer Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Goding Sauer, A.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef] [PubMed]
- Peck, B.; Schulze, A. Lipid metabolism at the nexus of diet and tumor microenvironment. Trends Cancer 2019, 5, 693–703. [Google Scholar] [CrossRef]
- Vander Heiden, M.G. Targeting cancer metabolism: A therapeutic window opens. Nat. Reviews Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the intersections between metabolism and cancer biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Zaytseva, Y. Lipid metabolism as a targetable metabolic vulnerability in colorectal cancer. Cancers 2021, 13, 301. [Google Scholar] [CrossRef]
- Murray, I.A.; Patterson, A.D.; Perdew, G.H. Aryl hydrocarbon receptor ligands in cancer: Friend and foe. Nat. Rev. Cancer 2014, 14, 801–814. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Safe, S.; Jayaraman, A.; Chapkin, R.S. Ah receptor ligands and their impacts on gut resilience: Structure–activity effects. Crit. Rev. Toxicol. 2020, 50, 463–473. [Google Scholar] [CrossRef]
- Andrysík, Z.; Vondráček, J.; Machala, M.; Krčmář, P.; Švihálková-Šindlerová, L.; Kranz, A.; Weiss, C.; Faust, D.; Kozubík, A.; Dietrich, C. The aryl hydrocarbon receptor-dependent deregulation of cell cycle control induced by polycyclic aromatic hydrocarbons in rat liver epithelial cells. Mutat. Res. 2007, 615, 87–97. [Google Scholar] [CrossRef]
- Dietrich, C. The AHR in the control of cell cycle and apoptosis. In The AH Receptor in Biology and Toxicology; Pohjanvirta, R., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; pp. 467–483. [Google Scholar]
- Dietrich, C.; Kaina, B. The aryl hydrocarbon receptor (AhR) in the regulation of cell-cell contact and tumor growth. Carcinogenesis 2010, 31, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Goode, G.; Pratap, S.; Eltom, S.E. Depletion of the aryl hydrocarbon receptor in MDA-MB-231 human breast cancer cells altered the expression of genes in key regulatory pathways of cancer. PLoS ONE 2014, 9, e100103. [Google Scholar] [CrossRef] [PubMed]
- Goode, G.D.; Ballard, B.R.; Manning, H.C.; Freeman, M.L.; Kang, Y.; Eltom, S.E. Knockdown of aberrantly upregulated aryl hydrocarbon receptor reduces tumor growth and metastasis of MDA-MB-231 human breast cancer cell line: AhR knockdown and breast cancer growth and metastasis. Int. J. Cancer 2013, 133, 2769–2780. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the immune response by the aryl hydrocarbon receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Fernandez-Salguero, P. The aryl hydrocarbon receptor: Studies using the AHR-null mice. Drug Metab. Dispos. 1998, 26, 1194–1198. [Google Scholar]
- Schmidt, J.V.; Su, G.H.; Reddy, J.K.; Simon, M.C.; Bradfield, C.A. Characterization of a murine Ahr null allele: Involvement of the Ah receptor in hepatic growth and development. Proc. Natl. Acad. Sci. USA 1996, 93, 6731–6736. [Google Scholar] [CrossRef]
- Safe, S.; Lee, S.O.; Jin, U.H. Role of the aryl hydrocarbon receptor in carcinogenesis and potential as a drug target. Toxicol. Sci. 2013, 135, 1–16. [Google Scholar] [CrossRef]
- Kawajiri, K.; Kobayashi, Y.; Ohtake, F.; Ikuta, T.; Matsushima, Y.; Mimura, J.; Pettersson, S.; Pollenz, R.S.; Sakaki, T.; Hirokawa, T.; et al. Aryl hydrocarbon receptor suppresses intestinal carcinogenesis in ApcMin/+ mice with natural ligands. Proc. Natl. Acad. Sci. USA 2009, 106, 13481–13486. [Google Scholar] [CrossRef]
- Díaz-Díaz, C.J.; Ronnekleiv-Kelly, S.M.; Nukaya, M.; Geiger, P.G.; Balbo, S.; Dator, R.; Megna, B.W.; Carney, P.R.; Bradfield, C.A.; Kennedy, G.D. The aryl hydrocarbon receptor is a repressor of inflammation-associated colorectal tumorigenesis in mouse. Ann. Surg. 2016, 264, 429–436. [Google Scholar] [CrossRef]
- Han, H.; Davidson, L.A.; Hensel, M.; Yoon, G.; Landrock, K.; Allred, C.; Jayaraman, A.; Ivanov, I.; Safe, S.H.; Chapkin, R.S. Loss of aryl hydrocarbon receptor promotes colon tumorigenesis in Apc(S580/+); Kras(G12D/+) mice. Mol. Cancer Res. 2021, 19, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Kurosumi, M.; Yatsuoka, T.; Nishimura, Y. Tissue distribution of aryl hydrocarbon receptor in the intestine: Implication of putative roles in tumor suppression. Exp. Cell Res. 2016, 343, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.-H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Sheng, B.; Pu, A.; Han, B.; Yang, K.; Wang, Q.; Sun, L.; Yang, H. Keratinocyte growth factor regulation of aryl hydrocarbon receptor activation in colorectal cancer cells. Dig. Dis. Sci. 2016, 61, 444–452. [Google Scholar] [CrossRef]
- Megna, B.W.; Carney, P.R.; Depke, M.G.; Nukaya, M.; McNally, J.; Larsen, L.; Rosengren, R.J.; Kennedy, G.D. The aryl hydrocarbon receptor as an antitumor target of synthetic curcuminoids in colorectal cancer. J. Surg. Res. 2017, 213, 16–24. [Google Scholar] [CrossRef]
- Ronnekleiv-Kelly, S.M.; Nukaya, M.; Díaz-Díaz, C.J.; Megna, B.W.; Carney, P.R.; Geiger, P.G.; Kennedy, G.D. Aryl hydrocarbon receptor-dependent apoptotic cell death induced by the flavonoid chrysin in human colorectal cancer cells. Cancer Lett. 2016, 370, 91–99. [Google Scholar] [CrossRef]
- Garcia-Villatoro, E.L.; DeLuca, J.A.A.; Callaway, E.S.; Allred, K.F.; Davidson, L.A.; Hensel, M.E.; Menon, R.; Ivanov, I.; Safe, S.H.; Jayaraman, A.; et al. Effects of high-fat diet and intestinal aryl hydrocarbon receptor deletion on colon carcinogenesis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 318, G451–G463. [Google Scholar] [CrossRef]
- Litzenburger, U.M.; Opitz, C.A.; Sahm, F.; Rauschenbach, K.J.; Trump, S.; Winter, M.; Ott, M.; Ochs, K.; Lutz, C.; Liu, X.; et al. Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 2014, 5, 1038–1051. [Google Scholar] [CrossRef]
- Xie, G.; Peng, Z.; Raufman, J.-P. Src-mediated aryl hydrocarbon and epidermal growth factor receptor cross talk stimulates colon cancer cell proliferation. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1006–G1015. [Google Scholar] [CrossRef]
- Ge, N.-L.; Elferink, C.J. A direct interaction between the aryl hydrocarbon receptor and retinoblastoma protein. J. Biol. Chem. 1998, 273, 22708–22713. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Weiss, C.; Koff, A.; Gottlicher, M. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev. 1999, 13, 1742–1753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlowe, J.L.; Knudsen, E.S.; Schwemberger, S.; Puga, A. The aryl hydrocarbon receptor displaces p300 from E2F-dependent promoters and represses S phase-specific gene expression. J. Biol. Chem. 2004, 279, 29013–29022. [Google Scholar] [CrossRef] [PubMed]
- Marlowe, J.L.; Puga, A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J. Cell. Biochem. 2005, 96, 1174–1184. [Google Scholar] [CrossRef]
- John, K.; Lahoti, T.S.; Wagner, K.; Hughes, J.M.; Perdew, G.H. The Ah receptor regulates growth factor expression in head and neck squamous cell carcinoma cell lines: Growth factor targets of AhR. Mol. Carcinog. 2014, 53, 765–776. [Google Scholar] [CrossRef]
- Kim, D.W.; Gazourian, L.; Quadri, S.A.; Romieu-Mourez, R.; Sherr, D.H.; Sonenshein, G.E. The RelA NF-kappaB subunit and the aryl hydrocarbon receptor (AhR) cooperate to transactivate the c-myc promoter in mammary cells. Oncogene 2000, 19, 5498–5506. [Google Scholar] [CrossRef] [PubMed]
- Al-Dhfyan, A.; Alhoshani, A.; Korashy, H.M. Aryl hydrocarbon receptor/cytochrome P450 1A1 pathway mediates breast cancer stem cells expansion through PTEN inhibition and β-Catenin and Akt activation. Mol. Cancer 2017, 16, 14. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Marín, N.; Merino, J.M.; Alvarez-Barrientos, A.; Patel, D.P.; Takahashi, S.; González-Sancho, J.M.; Gandolfo, P.; Rios, R.M.; Muñoz, A.; Gonzalez, F.J.; et al. Aryl hydrocarbon receptor promotes liver polyploidization and inhibits PI3K, ERK, and Wnt/β-catenin signaling. iScience 2018, 4, 44–63. [Google Scholar] [CrossRef]
- Wang, C.; Xu, C.-X.; Bu, Y.; Bottum, K.M.; Tischkau, S.A. Beta-naphthoflavone (DB06732) mediates estrogen receptor-positive breast cancer cell cycle arrest through AhR-dependent regulation of PI3K/AKT and MAPK/ERK signaling. Carcinogenesis 2014, 35, 703–713. [Google Scholar] [CrossRef]
- Lafita-Navarro, M.C.; Perez-Castro, L.; Zacharias, L.G.; Barnes, S.; DeBerardinis, R.J.; Conacci-Sorrell, M. The transcription factors aryl hydrocarbon receptor and MYC cooperate in the regulation of cellular metabolism. J. Biol. Chem. 2020, 295, 12398–12407. [Google Scholar] [CrossRef]
- Nault, R.; Fader, K.A.; Ammendolia, D.A.; Dornbos, P.; Potter, D.; Sharratt, B.; Kumagai, K.; Harkema, J.R.; Lunt, S.Y.; Matthews, J.; et al. Dose-dependent metabolic reprogramming and differential gene expression in TCDD-elicited hepatic fibrosis. Toxicol. Sci. 2016, 154, 253–266. [Google Scholar] [CrossRef]
- Nault, R.; Fader, K.A.; Lydic, T.A.; Zacharewski, T.R. Lipidomic evaluation of aryl hydrocarbon receptor-mediated hepatic steatosis in male and female mice elicited by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Chem. Res. Toxicol. 2017, 30, 1060–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, S.; Shirakawa, H.; Tomita, S.; Ohsaki, Y.; Haketa, K.; Tooi, O.; Santo, N.; Tohkin, M.; Furukawa, Y.; Gonzalez, F.J.; et al. Low-dose dioxins alter gene expression related to cholesterol biosynthesis, lipogenesis, and glucose metabolism through the aryl hydrocarbon receptor-mediated pathway in mouse liver. Toxicol. Appl. Pharmacol. 2008, 229, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Angrish, M.M.; Jones, A.D.; Harkema, J.R.; Zacharewski, T.R. Aryl hydrocarbon receptor–mediated induction of stearoyl-CoA desaturase 1 alters hepatic fatty acid composition in TCDD-elicited steatosis. Toxicol. Sci. 2011, 124, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Wada, T.; Febbraio, M.; He, J.; Matsubara, T.; Lee, M.J.; Gonzalez, F.J.; Xie, W. A novel role for the dioxin receptor in fatty acid metabolism and hepatic steatosis. Gastroenterology 2010, 139, 653–663. [Google Scholar] [CrossRef]
- Xu, C.-X.; Wang, C.; Zhang, Z.-M.; Jaeger, C.D.; Krager, S.L.; Bottum, K.M.; Liu, J.; Liao, D.-F.; Tischkau, S.A. Aryl hydrocarbon receptor deficiency protects mice from diet-induced adiposity and metabolic disorders through increased energy expenditure. Int. J. Obes. 2015, 39, 1300–1309. [Google Scholar] [CrossRef]
- Hofmanová, J.; Slavík, J.; Ciganek, M.; Ovesná, P.; Tylichová, Z.; Karasová, M.; Zapletal, O.; Straková, N.; Procházková, J.; Bouchal, J.; et al. Complex aterations of fatty acid metabolism and phospholipidome uncovered in isolated colon cancer epithelial cells. Int. J. Mol. Sci. 2021, 22, 6650. [Google Scholar] [CrossRef]
- Procházková, J.; Slavík, J.; Bouchal, J.; Levková, M.; Hušková, Z.; Ehrmann, J.; Ovesná, P.; Kolář, Z.; Skalický, P.; Straková, N.; et al. Specific alterations of sphingolipid metabolism identified in EpCAM-positive cells isolated from human colon tumors. Biochim. Biophys. Acta 2020, 1865, 158742. [Google Scholar] [CrossRef]
- Vyhlídalová, B.; Krasulová, K.; Pečínková, P.; Marcalíková, A.; Vrzal, R.; Zemánková, L.; Vančo, J.; Trávníček, Z.; Vondráček, J.; Karasová, M.; et al. Gut microbial catabolites of tryptophan are ligands and agonists of the aryl hydrocarbon receptor: A detailed characterization. Int. J. Mol. Sci. 2020, 21, 2614. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.S.; Waldman, T. PTEN gene targeting reveals a radiation-induced size checkpoint in human cancer cells. Cancer Res. 2004, 64, 6906–6914. [Google Scholar] [CrossRef]
- Bray, M.-A.; Singh, S.; Han, H.; Davis, C.T.; Borgeson, B.; Hartland, C.; Kost-Alimova, M.; Gustafsdottir, S.M.; Gibson, C.C.; Carpenter, A.E. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat. Protoc. 2016, 11, 1757–1774. [Google Scholar] [CrossRef]
- Veeman, M.T.; Slusarski, D.C.; Kaykas, A.; Louie, S.H.; Moon, R.T. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr. Biol. 2003, 13, 680–685. [Google Scholar] [CrossRef] [Green Version]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Tylichová, Z.; Slavík, J.; Ciganek, M.; Ovesná, P.; Krčmář, P.; Straková, N.; Machala, M.; Kozubík, A.; Hofmanová, J.; Vondráček, J. Butyrate and docosahexaenoic acid interact in alterations of specific lipid classes in differentiating colon cancer cells. J. Cell Biochem. 2018, 119, 4664–4679. [Google Scholar] [CrossRef] [PubMed]
- Kosti, I.; Jain, N.; Aran, D.; Butte, A.J.; Sirota, M. Cross-tissue Analysis of Gene and Protein Expression in Normal and Cancer Tissues. Sci. Rep. 2016, 6, 24799. [Google Scholar] [CrossRef] [PubMed]
- Koundouros, N.; Poulogiannis, G. Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 2020, 122, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Cross, D.A.; Alessi, D.R.; Cohen, P.; Andjelkovich, M.; Hemmings, B.A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995, 378, 785–789. [Google Scholar] [CrossRef]
- Feng, S.; Cao, Z.; Wang, X. Role of aryl hydrocarbon receptor in cancer. Biochim. Biophys. Acta 2013, 1836, 197–210. [Google Scholar] [CrossRef]
- Huang, Y.H.; Cao, Y.F.; Jiang, Z.Y.; Zhang, S.; Gao, F. Th22 cell accumulation is associated with colorectal cancer development. World J. Gastroenterol. 2015, 21, 4216–4224. [Google Scholar] [CrossRef]
- Céspedes, M.V.; Espina, C.; García-Cabezas, M.A.; Trias, M.; Boluda, A.; Gómez del Pulgar, M.T.; Sancho, F.J.; Nistal, M.; Lacal, J.C.; Mangues, R. Orthotopic microinjection of human colon cancer cells in nude mice induces tumor foci in all clinically relevant metastatic sites. Am. J. Pathol. 2007, 170, 1077–1085. [Google Scholar] [CrossRef]
- El-Brolosy, M.A.; Stainier, D.Y.R. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017, 13, e1006780. [Google Scholar] [CrossRef]
- Oberg, M.; Bergander, L.; Hakansson, H.; Rannug, U.; Rannug, A. Identification of the tryptophan photoproduct 6-formylindolo[3,2-b]carbazole, in cell culture medium, as a factor that controls the background aryl hydrocarbon receptor activity. Toxicol. Sci. 2005, 85, 935–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rannug, A. How the AHR Became Important in Intestinal Homeostasis—A Diurnal FICZ/AHR/CYP1A1 Feedback Controls Both Immunity and Immunopathology. Int. J. Mol. Sci. 2020, 21, 5681. [Google Scholar] [CrossRef] [PubMed]
- Abdelrahim, M.; Smith, R., 3rd; Safe, S. Aryl hydrocarbon receptor gene silencing with small inhibitory RNA differentially modulates Ah-responsiveness in MCF-7 and HepG2 cancer cells. Mol. Pharmacol. 2003, 63, 1373–1381. [Google Scholar] [CrossRef]
- Ma, Q.; Whitlock, J.P., Jr. The aromatic hydrocarbon receptor modulates the Hepa 1c1c7 cell cycle and differentiated state independently of dioxin. Mol. Cell Biol. 1996, 16, 2144–2150. [Google Scholar] [CrossRef]
- Elferink, C.J. Aryl hydrocarbon receptor-mediated cell cycle control. Prog. Cell Cycle Res. 2003, 5, 261–267. [Google Scholar] [PubMed]
- Elferink, C.J.; Ge, N.L.; Levine, A. Maximal aryl hydrocarbon receptor activity depends on an interaction with the retinoblastoma protein. Mol. Pharmacol. 2001, 59, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Elson, D.J.; Nguyen, B.D.; Wood, R.; Zhang, Y.; Puig-Sanvicens, V.; Kolluri, S.K. The cyclin-dependent kinase inhibitor p27(Kip1) interacts with the aryl hydrocarbon receptor and negatively regulates its transcriptional activity. FEBS Lett. 2022, 596, 2056–2071. [Google Scholar] [CrossRef]
- Shiizaki, K.; Kido, K.; Mizuta, Y. Insight into the relationship between aryl-hydrocarbon receptor and β-catenin in human colon cancer cells. PLoS ONE 2019, 14, e0224613. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Hazlett, H.F.; Nemani, K.V.; Trask, H.W.; West, R.J.; Lupien, L.E.; Collins, A.J.; Ringelberg, C.S.; et al. Inhibition of the aryl hydrocarbon receptor prevents Western diet-induced obesity. Model for AHR activation by kynurenine via oxidized-LDL, TLR2/4, TGFβ, and IDO1. Toxicol. Appl. Pharmacol. 2016, 300, 13–24. [Google Scholar] [CrossRef]
- Moyer, B.J.; Rojas, I.Y.; Kerley-Hamilton, J.S.; Nemani, K.V.; Trask, H.W.; Ringelberg, C.S.; Gimi, B.; Demidenko, E.; Tomlinson, C.R. Obesity and fatty liver are prevented by inhibition of the aryl hydrocarbon receptor in both female and male mice. Nutr. Res. 2017, 44, 38–50. [Google Scholar] [CrossRef]
- Hwang, H.J.; Dornbos, P.; Steidemann, M.; Dunivin, T.K.; Rizzo, M.; LaPres, J.J. Mitochondrial-targeted aryl hydrocarbon receptor and the impact of 2,3,7,8-tetrachlorodibenzo-p-dioxin on cellular respiration and the mitochondrial proteome. Toxicol. Appl. Pharmacol. 2016, 304, 121–132. [Google Scholar] [CrossRef] [Green Version]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer: Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Else, P.L. The highly unnatural fatty acid profile of cells in culture. Prog. Lipid. Res. 2020, 77, 101017. [Google Scholar] [CrossRef] [PubMed]
- Angrish, M.M.; Dominici, C.Y.; Zacharewski, T.R. TCDD-elicited effects on liver, serum, and adipose lipid composition in C57BL/6 Mice. Toxicol. Sci. 2013, 131, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Kania-Korwel, I.; Wu, X.; Wang, K.; Lehmler, H.-J. Identification of lipidomic markers of chronic 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) exposure in the male rat liver. Toxicology 2017, 390, 124–134. [Google Scholar] [CrossRef]
- Li, F.; Xiang, B.; Jin, Y.; Li, C.; Ren, S.; Wu, Y.; Li, J.; Luo, Q. Hepatotoxic effects of inhalation exposure to polycyclic aromatic hydrocarbons on lipid metabolism of C57BL/6 mice. Environ. Int. 2020, 134, 105000. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, L.; Allman, E.L.; Hubbard, T.D.; Murray, I.A.; Hao, F.; Tian, Y.; Gui, W.; Nichols, R.G.; Smith, P.B.; et al. The aryl hydrocarbon receptor activates ceramide biosynthesis in mice contributing to hepatic lipogenesis. Toxicology 2021, 458, 152831. [Google Scholar] [CrossRef]
- Zaytseva, Y.Y.; Rychahou, P.G.; Gulhati, P.; Elliott, V.A.; Mustain, W.C.; O’Connor, K.; Morris, A.J.; Sunkara, M.; Weiss, H.L.; Lee, E.Y.; et al. Inhibition of fatty acid synthase attenuates CD44-associated signaling and reduces metastasis in colorectal cancer. Cancer Res. 2012, 72, 1504–1517. [Google Scholar] [CrossRef]
- Mason, P.; Liang, B.; Li, L.; Fremgen, T.; Murphy, E.; Quinn, A.; Madden, S.L.; Biemann, H.-P.; Wang, B.; Cohen, A.; et al. SCD1 inhibition causes cancer cell death by depleting mono-unsaturated fatty acids. PLoS ONE 2012, 7, e33823. [Google Scholar] [CrossRef]
- Sanchez-Martinez, R.; Cruz-Gil, S.; Gomez de Cedron, M.; Alvarez-Fernandez, M.; Vargas, T.; Molina, S.; Garcia, B.; Herranz, J.; Moreno-Rubio, J.; Reglero, G.; et al. A link between lipid metabolism and epithelial-mesenchymal transition provides a target for colon cancer therapy. Oncotarget 2015, 6, 38719–38736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruz-Gil, S.; Sanchez-Martinez, R.; Gomez de Cedron, M.; Martin-Hernandez, R.; Vargas, T.; Molina, S.; Herranz, J.; Davalos, A.; Reglero, G.; Ramirez de Molina, A. Targeting the lipid metabolic axis ACSL/SCD in colorectal cancer progression by therapeutic miRNAs: miR-19b-1 role. J. Lipid Res. 2018, 59, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Taniue, K.; Kurimoto, A.; Sugimasa, H.; Nasu, E.; Takeda, Y.; Iwasaki, K.; Nagashima, T.; Okada-Hatakeyama, M.; Oyama, M.; Kozuka-Hata, H.; et al. Long noncoding RNA UPAT promotes colon tumorigenesis by inhibiting degradation of UHRF1. Proc. Natl. Acad. Sci. USA 2016, 113, 1273–1278. [Google Scholar] [CrossRef] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K–AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.-L.; Schulze, A. SREBP Activity Is Regulated by mTORC1 and Contributes to Akt-Dependent Cell Growth. Cell Metabol. 2008, 8, 224–236. [Google Scholar] [CrossRef]
- Wu, R.; Zhang, L.; Hoagland, M.S.; Swanson, H.I. Lack of the aryl hydrocarbon receptor leads to impaired activation of AKT/Protein Kinase B and enhanced sensitivity to apoptosis induced via the intrinsic pathway. J. Pharmacol. Exp. Therap. 2006, 320, 448–457. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karasová, M.; Procházková, J.; Tylichová, Z.; Fedr, R.; Ciganek, M.; Machala, M.; Dvořák, Z.; Vyhlídalová, B.; Zůvalová, I.; Ehrmann, J.; et al. Inhibition of Aryl Hydrocarbon Receptor (AhR) Expression Disrupts Cell Proliferation and Alters Energy Metabolism and Fatty Acid Synthesis in Colon Cancer Cells. Cancers 2022, 14, 4245. https://doi.org/10.3390/cancers14174245
Karasová M, Procházková J, Tylichová Z, Fedr R, Ciganek M, Machala M, Dvořák Z, Vyhlídalová B, Zůvalová I, Ehrmann J, et al. Inhibition of Aryl Hydrocarbon Receptor (AhR) Expression Disrupts Cell Proliferation and Alters Energy Metabolism and Fatty Acid Synthesis in Colon Cancer Cells. Cancers. 2022; 14(17):4245. https://doi.org/10.3390/cancers14174245
Chicago/Turabian StyleKarasová, Martina, Jiřina Procházková, Zuzana Tylichová, Radek Fedr, Miroslav Ciganek, Miroslav Machala, Zdeněk Dvořák, Barbora Vyhlídalová, Iveta Zůvalová, Jiří Ehrmann, and et al. 2022. "Inhibition of Aryl Hydrocarbon Receptor (AhR) Expression Disrupts Cell Proliferation and Alters Energy Metabolism and Fatty Acid Synthesis in Colon Cancer Cells" Cancers 14, no. 17: 4245. https://doi.org/10.3390/cancers14174245