
Mutational Characterization of Cutaneous Melanoma Supports Divergent Pathways Model for Melanoma Development

 , , , , ,
, , , , ,  and
and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Results
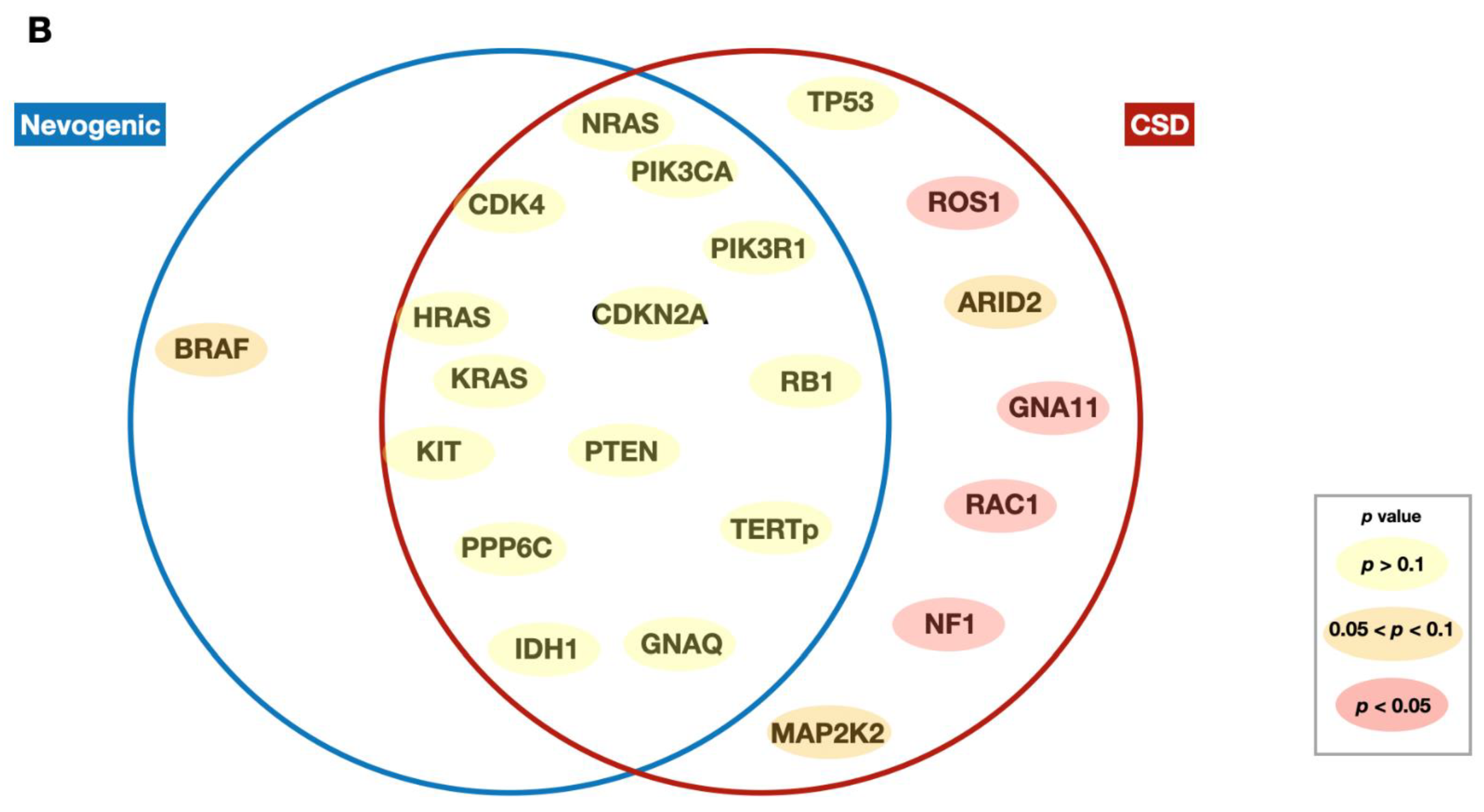
2.1. Mutational Distribution among Nevogenic and CSD Melanomas
2.2. Mechanistic Analysis of Pathways
3. Discussion
4. Materials and Methods
4.1. Patient Selection and Classification
4.2. Sample Preparation
4.3. Gene Panel and Library Construction
4.4. Next-Generation Sequencing
4.5. Mechanistic Analysis of Pathways
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Whiteman, D.C.; Parsons, P.; Green, A. p53 expression and risk factors for cutaneous melanoma: A case-control study. Int. J. Cancer 1998, 77, 843–848. [Google Scholar] [CrossRef]
- Lee, E.Y.; Williamson, R.; Watt, P.; Hughes, M.C.; Green, A.C.; Whiteman, D.C. Sun exposure and host phenotype as predictors of cutaneous melanoma associated with neval remnants or dermal elastosis. Int. J. Cancer 2006, 119, 636–642. [Google Scholar] [CrossRef]
- Gibbs, D.C.; Orlow, I.; Bramson, J.I.; Kanetsky, P.A.; Luo, L.; Kricker, A.; Armstrong, B.K.; Anton-Culver, H.; Gruber, S.B.; Marrett, L.D.; et al. Association of Interferon Regulatory Factor-4 Polymorphism rs12203592 With Divergent Melanoma Pathways. J. Natl. Cancer Inst. 2016, 108, 108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whiteman, D.C.; Watt, P.; Purdie, D.M.; Hughes, M.C.; Hayward, N.K.; Green, A.C. Melanocytic nevi, solar keratoses, and divergent pathways to cutaneous mela-noma. J. Natl. Cancer Inst. 2003, 95, 806–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carli, P.; Palli, D. Re: Melanocytic nevi, solar keratoses, and divergent pathways to cutaneous mela-noma. J. Natl. Cancer Inst. 2003, 95, 1801–1802. [Google Scholar] [CrossRef] [Green Version]
- Ghiasvand, R.; Robsahm, T.E.; Green, A.C.; Rueegg, C.S.; Weiderpass, E.; Lund, E.; Veierød, M.B. Association of Phenotypic Characteristics and UV Radiation Exposure With Risk of Melanoma on Different Body Sites. JAMA Dermatol. 2019, 155, 39–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Gorgojo, A.; Llinares, M.; Virós, A.; Requena, C.; Garcia-Casado, Z.; Traves, V.; Kumar, R.; Nagore, E. Cutaneous melanoma primary site is linked to nevus density. Oncotarget 2017, 8, 98876–98886. [Google Scholar] [CrossRef]
- Elder, D.E.; Bastian, B.C.; Cree, I.A.; Massi, D.; Scolyer, R.A. The 2018 World Health Organization Classification of Cutaneous, Mucosal, and Uveal Melanoma: Detailed Analysis of 9 Distinct Subtypes Defined by Their Evolutionary Pathway. Arch. Pathol. Lab. Med. 2020, 144, 500–522. [Google Scholar] [CrossRef] [Green Version]
- Rabbie, R.; Ferguson, P.M.; Aguilar, C.M.; Adams, D.J.; Robles-Espinoza, C.D. Melanoma subtypes: Genomic profiles, prognostic molecular markers and therapeutic possibilities. J. Pathol. 2018, 247, 539–551. [Google Scholar] [CrossRef]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The cutaneous melanoma genome: A high mutation rate and a signature of UVR exposure. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef]
- De Unamuno Bustos, B.; Murria Estal, R.; Pérez Simó, G.; de Juan Jimenez, I.; Escutia Muñoz, B.; Rodríguez Serna, M.; Alegre de Miquel, V.; Llavador Ros, M.; Ballester Sánchez, R.; Nagore Enguídanos, E.; et al. Towards Personalized Medicine in Melanoma: Implementation of a Clinical Next-Generation Sequencing Panel. Sci. Rep. 2017, 7, 495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Ayala, B.; et al. Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salavert, F.; Hidago, M.R.; Amadoz, A.; Çubuk, C.; Medina, I.; Crespo, D.; Carbonell-Caballero, J.; Dopazo, J. Actionable pathways: Interactive discovery of therapeutic targets using signal-ing pathway models. Nucleic Acids Res. 2016, 44, W212–W216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cubuk, C.; Hidalgo, M.R.; Amadoz, A.; Pujana, M.A.; Mateo, F.; Herranz, C.; Carbonell-Caballero, J.; Dopazo, J. Gene expression integration into pathway modules reveals a pan-cancer meta-bolic landscape. Cancer Res. 2018, 78, 6059–6072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peña-Chilet, M.; Esteban-Medina, M.; Falco, M.M.; Rian, K.; Hidalgo, M.R.; Loucera, C.; Dopazo, J. Using mechanistic models for the clinical interpretation of complex genomic variation. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Cirenajwis, H.; Lauss, M.; Ekedahl, H.; Törngren, T.; Kvist, A.; Saal, L.H.; Olsson, H.; Staaf, J.; Carneiro, A.; Ingvar, C.; et al. NF1-mutated melanoma tumors harbor distinct clinical and biological charac-teristics. Mol. Oncol. 2017, 11, 438–451. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Schoenfeld, A.J.; Siau, E.; Lu, Y.C.; Tai, H.; Suzawa, K.; Kubota, D.; Lui, A.J.; Qeriqi, B.; Mattar, M.; et al. MAPK Pathway Alterations Correlate with Poor Survival and Drive Resistance to Therapy in Patients with Lung Cancers Driven by ROS1 Fusions. Clin. Cancer Res. 2020, 26, 2932–2945. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.J.; Ha, B.H.; Holman, E.C.; Halaban, R.; Schlessinger, J.; Boggon, T.J. RAC1P29S is a spontaneously activating cancer-associated GTPase. Proc. Natl. Acad. Sci. USA 2013, 110, 912–917. [Google Scholar] [CrossRef] [Green Version]
- Halaban, R. RAC1 and Melanoma. Clin. Ther. 2015, 37, 682–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, S.P.; Kim, D.W.; Lacey, C.L.; Hwu, P. GNA11 mutation in a patient with cutaneous origin melanoma a case report. Medicine 2016, 95, e2336. [Google Scholar] [CrossRef]
- Cadley, J.; Simpson, D.; Ferguson, R.; Pandya, A.; Hekal, T.; Richards, T.; Ibrahim, K.; Weber, J.S.; Osman, I.; Kirchhoff, T. Mutation burden in conjunction with MAPK-pathway mutation status as a prognostic biomarker of overall melanoma survival. J. Clin. Oncol. 2018, 36, 9584. [Google Scholar] [CrossRef]
- Simpson, D.; Ferguson, R.; Martinez, C.N.; Kazlow, E.; Moran, U.; Heguy, A.; Hanniford, D.; Hernando, E.; Osman, I.; Kirchhoff, T. Mutation burden as a potential prognostic marker of melanoma progression and survival. J. Clin. Oncol. 2017, 35, 9567. [Google Scholar] [CrossRef]
- Kang, K.; Xie, F.; Mao, J.; Bai, Y.; Wang, X. Significance of Tumor Mutation Burden in Immune Infiltration and Prognosis in Cutaneous Melanoma. Front. Oncol. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Byrne, E.H.; Fisher, D.E. Immune and molecular correlates in melanoma treated with immune checkpoint blockade. Cancer 2017, 123, 2143–2153. [Google Scholar] [CrossRef] [Green Version]
- Snyder, A.; Makarov, V.; Merghoub, T.; Yuan, J.; Zaretsky, J.M.; Desrichard, A.; Walsh, L.A.; Postow, M.A.; Wong, P.; Ho, T.S.; et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. N. Engl. J. Med. 2014, 371, 2189–2199. [Google Scholar] [CrossRef] [Green Version]
- Shtivelman, E.; A Davies, M.Q.; Hwu, P.; Yang, J.; Lotem, M.; Oren, M.; Flaherty, K.T.; Fisher, D.E. Pathways and therapeutic targets in melanoma. Oncotarget 2014, 5, 1701–1752. [Google Scholar] [CrossRef] [Green Version]
- Tsao, H.; Chin, L.; Garraway, L.A.; Fisher, D.E. Melanoma: From mutations to medicine. Genes Dev. 2012, 26, 1131–1155. [Google Scholar] [CrossRef] [Green Version]
- Davis, E.J.; Johnson, D.B.; Sosman, J.A.; Chandra, S. Melanoma: What do all the mutations mean? Cancer 2018, 124, 3490–3499. [Google Scholar] [CrossRef]
- Maldonado, J.L.; Fridlyand, J.; Patel, H.; Jain, A.N.; Busam, K.; Kageshita, T.; Ono, T.; Albertson, D.G.; Pinkel, D.; Bastian, B.C. Determinants of BRAF Mutations in Primary Melanomas. J. Natl. Cancer Inst. 2003, 95, 1878–1890. [Google Scholar] [CrossRef] [Green Version]
- Damsky, W.E.; Bosenberg, M. Melanocytic nevi and melanoma: Unraveling a complex relationship. Oncogene 2017, 36, 5771–5792. [Google Scholar] [CrossRef] [Green Version]
- Roh, M.R.; Eliades, P.; Gupta, S.; Tsao, H. Genetics of melanocytic nevi. Pigment Cell Melanoma Res. 2015, 28, 661–672. [Google Scholar] [CrossRef]
- Kumar, R.; Angelini, S.; Snellman, E.; Hemminki, K. BRAF Mutations Are Common Somatic Events in Melanocytic Nevi. J. Investig. Dermatol. 2004, 122, 342–348. [Google Scholar] [CrossRef] [Green Version]
- Mitra, D.; Luo, X.; Morgan, A.; Wang, J.; Hoang, M.P.; Lo, J.; Guerrero, C.R.; Lennerz, J.K.; Mihm, M.C.; Wargo, J.A.; et al. An ultraviolet-radiation-independent pathway to melanoma carcinogenesis in the red hair/fair skin background. Nature 2012, 491, 449–453. [Google Scholar] [CrossRef] [PubMed]
- Roider, E.; Fisher, D.E. Red Hair, Light Skin, and UV-Independent Risk for Melanoma Development in Humans. JAMA Dermatol. 2016, 152, 751–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, R.L.; Hennessey, R.C.; Weiss, T.J.; A Tallman, D.; Crawford, E.R.; Murphy, B.M.; Webb, A.; Zhang, S.; La Perle, K.M.; Burd, C.J.; et al. UVB mutagenesis differs in Nras- and Braf-mutant mouse models of melanoma. Life Sci. Alliance 2021, 4, e202101135. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV Signature Mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [Green Version]
- García-Nieto, P.E.; Schwartz, E.K.; A King, D.; Paulsen, J.; Collas, P.; E Herrera, R.; Morrison, A.J. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J. 2017, 36, 2829–2843. [Google Scholar] [CrossRef]
- Ferguson, B.; Handoko, H.Y.; Mukhopadhyay, P.; Chitsazan, A.; Balmer, L.; Morahan, G.; Walker, G.J. Different genetic mechanisms mediate spontaneous versus UVR-induced malignant melanoma. eLife 2019, 8, 8–10. [Google Scholar] [CrossRef]
- Heidenreich, B.; Kumar, R. TERT promoter mutations in telomere biology. Mutat. Res. Rev. Mutat. Res. 2017, 771, 15–31. [Google Scholar] [CrossRef]
- Rachakonda, S.; Hoheisel, J.D.; Kumar, R. Occurrence, functionality and abundance of the TERT promoter mutations. Int. J. Cancer 2021, 149, 1852–1862. [Google Scholar] [CrossRef]
- Heidenreich, B.; Nagore, E.; Rachakonda, P.S.; Garcia-Casado, Z.; Requena, C.; Traves, V.; Becker, J.; Soufir, N.; Hemminki, K.; Kumar, R. Telomerase reverse transcriptase promoter mutations in primary cuta-neous melanoma. Nat. Commun. 2014, 5, 3401. [Google Scholar] [CrossRef]
- Motaparthi, K.; Kim, J.; Andea, A.A.; Missall, T.A.; Novoa, R.A.; Vidal, C.I.; Fung, M.A.; Emanuel, P.O. TERT and TERT promoter in melanocytic neoplasms: Current concepts in patho-genesis, diagnosis, and prognosis. J. Cutan. Pathol. 2020, 47, 710–719. [Google Scholar] [CrossRef] [Green Version]
- Nagore, E.; Rachakonda, S.; Kumar, R. TERT promoter mutations in melanoma survival. Oncotarget 2019, 10, 1546–1548. [Google Scholar] [CrossRef] [PubMed]
- Andres-Lencina, J.J.; Rachakonda, S.; García-Casado, Z.; Srinivas, N.; Skorokhod, A.; Requena, C.; Soriano, V.; Kumar, R.; Nagore, E. TERT promoter mutation subtypes and survival in stage I and II melanoma patients. Int. J. Cancer 2019, 144, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Nagore, E.; Heidenreich, B.; Requena, C.; García-Casado, Z.; Martorell-Calatayud, A.; Pont-Sanjuan, V.; Jimenez-Sanchez, A.I.; Kumar, R. TERT promoter mutations associate with fast-growing melanoma. Pigment Cell Melanoma Res. 2016, 29, 236–238. [Google Scholar] [CrossRef] [PubMed]
- Calomarde-Rees, L.; García-Calatayud, R.; Requena Caballero, C.; Manrique-Silva, E.; Traves, V.; García-Casado, Z.; Soriano, V.; Kumar, R.; Nagore, E. Risk Factors for Lymphatic and Hematogenous Dis-semination in Patients With Stages I to II Cutaneous Melanoma. JAMA Dermatol. 2019, 155, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.A.; Montesion, M.; Shah, N.; Sharaf, R.; Pavlick, D.C.; Sokol, E.S.; Alexander, B.; Venstrom, J.; Elvin, J.A.; Ross, J.S.; et al. Melanoma with in-frame deletion of MAP2K1: A distinct molecular subtype of cutaneous melanoma mutually exclusive from BRAF, NRAS, and NF1 mutations. Mod. Pathol. 2020, 33, 2397–2406. [Google Scholar] [CrossRef]
- Cho, J.; Kim, S.Y.; Kim, Y.J.; Sim, M.H.; Kim, S.T.; Kim, N.K.D.; Kim, K.; Park, W.; Kim, J.H.; Jang, K.T.; et al. Emergence of CTNNB1 mutation at acquired resistance to KIT inhibitor in metastatic mela-noma. Clin. Transl. Oncol. 2017, 19, 1247–1252. [Google Scholar] [CrossRef]
- Curtin, J.A.; Fridlyand, J.; Kageshita, T.; Patel, H.N.; Busam, K.J.; Kutzner, H.; Cho, K.-H.; Aiba, S.; Bröcker, E.-B.; LeBoit, P.E.; et al. Distinct Sets of Genetic Alterations in Melanoma. N. Engl. J. Med. 2005, 353, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Peña-Vilabelda, M.M.; García-Casado, Z.; Requena, C.; Traves, V.; López-Guerrero, J.A.; Guillén, C.; Kumar, R.; Nagorea, E. Características clínicas de los pacientes con melanoma cutáneo en función de las variaciones en el gen del receptor 1 de la melanocortina (MC1R). Actas Dermo-Sifiliogr. 2014, 105, 159–171. [Google Scholar] [CrossRef] [PubMed]
- Landi, M.T.; Bauer, J.; Pfeiffer, R.M.; Elder, D.E.; Hulley, B.; Minghetti, P.; Calista, D.; Kanetsky, P.A.; Pinkel, D.; Bastian, B.C. MC1R Germline Variants Confer Risk for BRAF -Mutant Melanoma. Science 2006, 313, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Aguet, F.; Anand, S.; Ardlie, K.G.; Gabriel, S.; Getz, G.A.; Graubert, A.; Hadley, K.; Handsaker, R.E.; Huang, K.H.; Kashin, S.; et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020, 369, 1318–1330. [Google Scholar]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Hidalgo, M.R.; Cubuk, C.; Amadoz, A.; Salavert, F.; Carbonell-Caballero, J.; Dopazo, J. High throughput estimation of functional cell activities reveals disease mecha-nisms and predicts relevant clinical outcomes. Oncotarget 2017, 8, 5160–5178. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear Models and Empirical Bayes Methods for Assessing Differential Expression in Microarray Experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Baker, S.; Ali, I.; Silins, I.; Pyysalo, S.; Guo, Y.; Högberg, J.; Stenius, U.; Korhonen, A. Cancer Hallmarks Analytics Tool (CHAT): A text mining approach to organize and evaluate scientific literature on cancer. Bioinformatics 2017, 33, 3973–3981. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Variables | Total | Nevogenic | CSD | ||||
|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | ||
| Sex | Male | 65 | 54.6 | 42 | 51.2 | 23 | 62.2 |
| Female | 54 | 45.4 | 40 | 48.8 | 14 | 37.8 | |
| Phototype | 1 | 2 | 1.7 | 2 | 2.4 | 0 | 0 |
| 2 | 32 | 26.9 | 23 | 28.0 | 9 | 24.3 | |
| 3 | 68 | 57.1 | 46 | 56.1 | 22 | 59.5 | |
| 4 | 15 | 12.6 | 10 | 12.2 | 5 | 13.5 | |
| 5 | 1 | 0.8 | 0 | 0 | 1 | 2.7 | |
| Unknown | 1 | 0.8 | 1 | 1.2 | 0 | 0 | |
| Sunburns at the area of melanoma | None | 12 | 26.7 | 8 | 27.6 | 4 | 25.0 |
| Mild | 15 | 33.3 | 9 | 31.0 | 6 | 37.5 | |
| Severe | 16 | 35.6 | 10 | 34.5 | 6 | 37.5 | |
| N/A | 1 | 2.2 | 1 | 3.4 | 0 | 0 | |
| Unknown | 1 | 2.2 | 1 | 3.4 | 0 | 0 | |
| Basal Cell Carcinoma | No | 96 | 81.4 | 72 | 88.9 | 24 | 64.9 |
| Yes | 22 | 18.6 | 9 | 11.1 | 13 | 35.1 | |
| Multiple Melanoma | No | 109 | 93.2 | 76 | 93.8 | 33 | 91.7 |
| Yes | 8 | 6.8 | 5 | 6.2 | 3 | 8.3 | |
| Familial Melanoma | No | 102 | 87.2 | 67 | 82.7 | 35 | 94.6 |
| Yes | 15 | 12.8 | 13 | 16.0 | 2 | 5.4 | |
| Anatomical location | Head/Neck | 30 | 25.2 | 2 | 2.4 | 28 | 75.7 |
| Limb | 25 | 21.0 | 18 | 22.0 | 7 | 18.9 | |
| Trunk | 59 | 49.6 | 57 | 69.5 | 2 | 5.4 | |
| Acral | 4 | 3.4 | 4 | 4.9 | 0 | 0 | |
| Other | 1 | 0.8 | 1 | 1.2 | 0 | 0 | |
| Histological type | LMM | 18 | 15.1 | 1 | 1.2 | 17 | 45.9 |
| SSM | 73 | 61.3 | 60 | 73.2 | 13 | 35.1 | |
| NM | 15 | 12.6 | 11 | 13.4 | 4 | 10.8 | |
| ALM | 3 | 2.5 | 3 | 3.7 | 0 | 0 | |
| Desmoplastic | 2 | 1.7 | 2 | 2.4 | 0 | 0 | |
| Spitzoid | 2 | 1.7 | 2 | 2.4 | 0 | 0 | |
| Other | 6 | 5.0 | 3 | 3.7 | 3 | 8.1 | |
| Ulceration | No | 99 | 83.2 | 70 | 85.4 | 29 | 78.4 |
| Yes | 20 | 16.8 | 12 | 14.6 | 8 | 21.6 | |
| Sentinel node | Negative | 19 | 67.9 | 14 | 82.4 | 5 | 45.5 |
| Positive | 6 | 21.4 | 3 | 17.6 | 3 | 27.3 | |
| Unknown | 3 | 10.7 | 0 | 0 | 3 | 27.3 | |
| Age * | <=59 | 59 | 50.0 | 58 | 71.6 | 1 | 2.7 |
| >59 | 59 | 50.0 | 23 | 28.4 | 36 | 97.3 | |
| Breslow * | <=1.08 | 59 | 50.0 | 48 | 59.3 | 11 | 29.7 |
| >1.08 | 59 | 50.0 | 33 | 40.7 | 26 | 70.3 | |
| Gene | Mutation Prevalence | Gene | Mutation Prevalence |
|---|---|---|---|
| TERTp | 52.21 | RB1 | 4.20 |
| BRAF | 50.42 | PIK3R1 | 4.20 |
| NF1 | 16.81 | GNA11 | 4.20 |
| NRAS | 13.45 | CDK4 | 3.36 |
| ROS1 | 11.76 | PPP6C | 3.36 |
| TP53 | 10.92 | PTEN | 2.52 |
| ARID2 | 9.24 | HRAS | 1.68 |
| CDKN2A | 7.56 | MAP2K2 | 1.68 |
| RAC1 | 5.88 | GNAQ | 0.84 |
| IDH1 | 5.04 | KRAS | 0.84 |
| KIT | 4.20 | PIK3CA | 0.84 |
| Molecular subgroup | % within cohort | Molecular subgroup | % within cohort |
| “BRAF+” | 40.3 | “BRAF+RAS+” | 2.5 |
| “RAS+” | 12.6 | “BRAF+NF1+” | 7.6 |
| “NF1+” | 8.4 | “RAS+NF1+” | 0.8 |
| “3wt” | 27.7 |
| Gene | Status | Total | Nevogenic | CSD | p-Value | |||
|---|---|---|---|---|---|---|---|---|
| N | % | N | % | N | % | |||
| TP53 | WT | 106 | 89.1 | 76 | 92.7 | 30 | 81.1 | 0.108 |
| Mutated | 13 | 10.9 | 6 | 7.3 | 7 | 18.9 | ||
| NF1 | WT | 99 | 83.2 | 76 | 92.7 | 23 | 62.2 | <0.001 |
| Mutated | 20 | 16.8 | 6 | 7.3 | 14 | 37.8 | ||
| BRAF | WT | 59 | 49.6 | 36 | 43.9 | 23 | 62.2 | 0.077 |
| Mutated | 60 | 50.4 | 46 | 56.1 | 14 | 37.8 | ||
| ROS1 | WT | 105 | 88.2 | 78 | 95.1 | 27 | 73.0 | 0.001 |
| Mutated | 14 | 11.8 | 4 | 4.9 | 10 | 27.0 | ||
| NRAS | WT | 103 | 86.6 | 70 | 85.4 | 33 | 89.2 | 0.773 |
| Mutated | 16 | 13.4 | 12 | 14.6 | 4 | 10.8 | ||
| CDK4 | WT | 115 | 96.6 | 79 | 96.3 | 36 | 97.3 | 1 |
| Mutated | 4 | 3.4 | 3 | 3.7 | 1 | 2.7 | ||
| ARID2 | WT | 108 | 90.8 | 77 | 93.9 | 31 | 83.8 | 0.094 |
| Mutated | 11 | 9.2 | 5 | 6.1 | 6 | 16.2 | ||
| CDKN2A | WT | 110 | 92.4 | 76 | 92.7 | 34 | 91.9 | 1 |
| Mutated | 9 | 7.6 | 6 | 7.3 | 3 | 8.1 | ||
| KIT | WT | 114 | 95.8 | 80 | 97.6 | 34 | 91.9 | 0.173 |
| Mutated | 5 | 4.2 | 2 | 2.4 | 3 | 8.2 | ||
| RB1 | WT | 114 | 95.8 | 79 | 96.3 | 35 | 94.6 | 0.646 |
| Mutated | 5 | 4.2 | 3 | 3.7 | 2 | 5.4 | ||
| PPP6C | WT | 115 | 96.6 | 79 | 96.3 | 36 | 97.3 | 1 |
| Mutated | 4 | 3.4 | 3 | 3.7 | 1 | 2.7 | ||
| PTEN | WT | 116 | 97.5 | 80 | 97.6 | 36 | 97.3 | 1 |
| Mutated | 3 | 2.5 | 2 | 2.4 | 1 | 2.7 | ||
| IDH1 | WT | 113 | 95.0 | 78 | 95.1 | 35 | 94.6 | 1 |
| Mutated | 6 | 5.0 | 4 | 4.9 | 2 | 5.4 | ||
| GNA11 | WT | 114 | 95.8 | 81 | 98.8 | 33 | 89.2 | 0.032 |
| Mutated | 5 | 4.2 | 1 | 1.2 | 4 | 10.8 | ||
| GNAQ | WT | 118 | 99.2 | 82 | 100.0 | 36 | 97.3 | 0.311 |
| Mutated | 1 | 0.8 | 0 | 0 | 1 | 2.7 | ||
| RAC1 | WT | 112 | 94.1 | 81 | 98.8 | 31 | 83.8 | 0.004 |
| Mutated | 7 | 5.9 | 1 | 1.2 | 6 | 16.2 | ||
| KRAS | WT | 118 | 99.2 | 81 | 98.8 | 37 | 100.0 | 1 |
| Mutated | 1 | 0.8 | 1 | 1.2 | 0 | 0 | ||
| HRAS | WT | 117 | 98.3 | 81 | 98.8 | 36 | 97.3 | 0.527 |
| Mutated | 2 | 1.7 | 1 | 1.2 | 1 | 2.7 | ||
| MAP2K2 | WT | 117 | 98.3 | 82 | 100.0 | 35 | 94.6 | 0.095 |
| Mutated | 2 | 1.7 | 0 | 0 | 2 | 5.4 | ||
| PIK3CA | WT | 118 | 99.2 | 82 | 100.0 | 36 | 97.3 | 0.311 |
| Mutated | 1 | 0.8 | 0 | 0 | 1 | 2.7 | ||
| PIK3R1 | WT | 114 | 95.8 | 80 | 97.6 | 34 | 91.9 | 0.173 |
| Mutated | 5 | 4.2 | 2 | 2.4 | 3 | 8.1 | ||
| TERTp | WT | 54 | 47.8 | 41 | 51.9 | 13 | 38.2 | 0.220 |
| Mutated | 59 | 52.2 | 38 | 48.1 | 21 | 61.8 | ||
| Pathogenic mutations | <=2 | 74 | 62.2 | 55 | 67.1 | 19 | 51.4 | 0.108 |
| >2 | 45 | 37.8 | 27 | 32.9 | 18 | 48.6 | ||
| Mutational subtype * | “BRAF+” | 48 | 45.3 | 39 | 52.0 | 9 | 29.0 | <0.001 |
| “RAS+” | 15 | 14.2 | 12 | 16.0 | 3 | 9.7 | ||
| “NF1+” | 10 | 9.4 | 1 | 1.3 | 9 | 29.0 | ||
| “3wt” | 33 | 32.4 | 23 | 30.7 | 10 | 32.3 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Millán-Esteban, D.; Peña-Chilet, M.; García-Casado, Z.; Manrique-Silva, E.; Requena, C.; Bañuls, J.; López-Guerrero, J.A.; Rodríguez-Hernández, A.; Traves, V.; Dopazo, J.; et al. Mutational Characterization of Cutaneous Melanoma Supports Divergent Pathways Model for Melanoma Development. Cancers 2021, 13, 5219. https://doi.org/10.3390/cancers13205219
Millán-Esteban D, Peña-Chilet M, García-Casado Z, Manrique-Silva E, Requena C, Bañuls J, López-Guerrero JA, Rodríguez-Hernández A, Traves V, Dopazo J, et al. Mutational Characterization of Cutaneous Melanoma Supports Divergent Pathways Model for Melanoma Development. Cancers. 2021; 13(20):5219. https://doi.org/10.3390/cancers13205219
Chicago/Turabian StyleMillán-Esteban, David, María Peña-Chilet, Zaida García-Casado, Esperanza Manrique-Silva, Celia Requena, José Bañuls, Jose Antonio López-Guerrero, Aranzazu Rodríguez-Hernández, Víctor Traves, Joaquín Dopazo, and et al. 2021. "Mutational Characterization of Cutaneous Melanoma Supports Divergent Pathways Model for Melanoma Development" Cancers 13, no. 20: 5219. https://doi.org/10.3390/cancers13205219