
Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma
by
 , , and
, , and
Katerina Hadrava Vanova
1,2 ,
,
Chunzhang Yang
3,
Leah Meuter
1,
Jiri Neuzil
2,4 and
Karel Pacak
1,* 1
Section of Medical Neuroendocrinology, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA
2
Institute of Biotechnology, Czech Academy of Sciences, BIOCEV, Vestec, 252 50 Prague West, Czech Republic
3
Neuro-Oncology Branch, Center for Cancer Research, National Cancer Institute, National Institutes of Health, Bethesda, MD 20892, USA
4
School of Pharmacy and Medical Science, Griffith University, Southport, QLD 4222, Australia
*
Author to whom correspondence should be addressed.
Cancers 2021, 13(15), 3769; https://doi.org/10.3390/cancers13153769
Submission received: 28 June 2021
/
Revised: 21 July 2021
/
Accepted: 24 July 2021
/
Published: 27 July 2021
(This article belongs to the Special Issue Malignant Adrenal Tumors – from Bench to Bedside)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Pheochromocytoma and paraganglioma are rare neuroendocrine tumors that arise from chromaffin cells of the adrenal medulla or their neural crest progenitors located outside the adrenal gland, respectively. About 10–15% of patients develop metastatic disease for whom treatment options and availability are extremely limited. The risk of developing metastatic disease is increased for patients with mutations in succinate dehydrogenase subunit B, which leads to metabolic reprogramming and redox imbalance. From this perspective, we focus on redox imbalance caused by this mutation and explore potential opportunities to therapeutically target reactive oxygen species production in these rare tumors.
Abstract
Pheochromocytoma (PHEO) and paraganglioma (PGL) are rare neuroendocrine tumors derived from neural crest cells. Germline variants in approximately 20 PHEO/PGL susceptibility genes are found in about 40% of patients, half of which are found in the genes that encode succinate dehydrogenase (SDH). Patients with SDH subunit B (SDHB)-mutated PHEO/PGL exhibit a higher likelihood of developing metastatic disease, which can be partially explained by the metabolic cell reprogramming and redox imbalance caused by the mutation. Reactive oxygen species (ROS) are highly reactive molecules involved in a multitude of important signaling pathways. A moderate level of ROS production can help regulate cellular physiology; however, an excessive level of oxidative stress can lead to tumorigenic processes including stimulation of growth factor-dependent pathways and the induction of genetic instability. Tumor cells effectively exploit antioxidant enzymes in order to protect themselves against harmful intracellular ROS accumulation, which highlights the essential balance between ROS production and scavenging. Exploiting ROS accumulation can be used as a possible therapeutic strategy in ROS-scavenging tumor cells. Here, we focus on the role of ROS production in PHEO and PGL, predominantly in SDHB-mutated cases. We discuss potential strategies and approaches to anticancer therapies by enhancing ROS production in these difficult-to-treat tumors.
1. Introduction
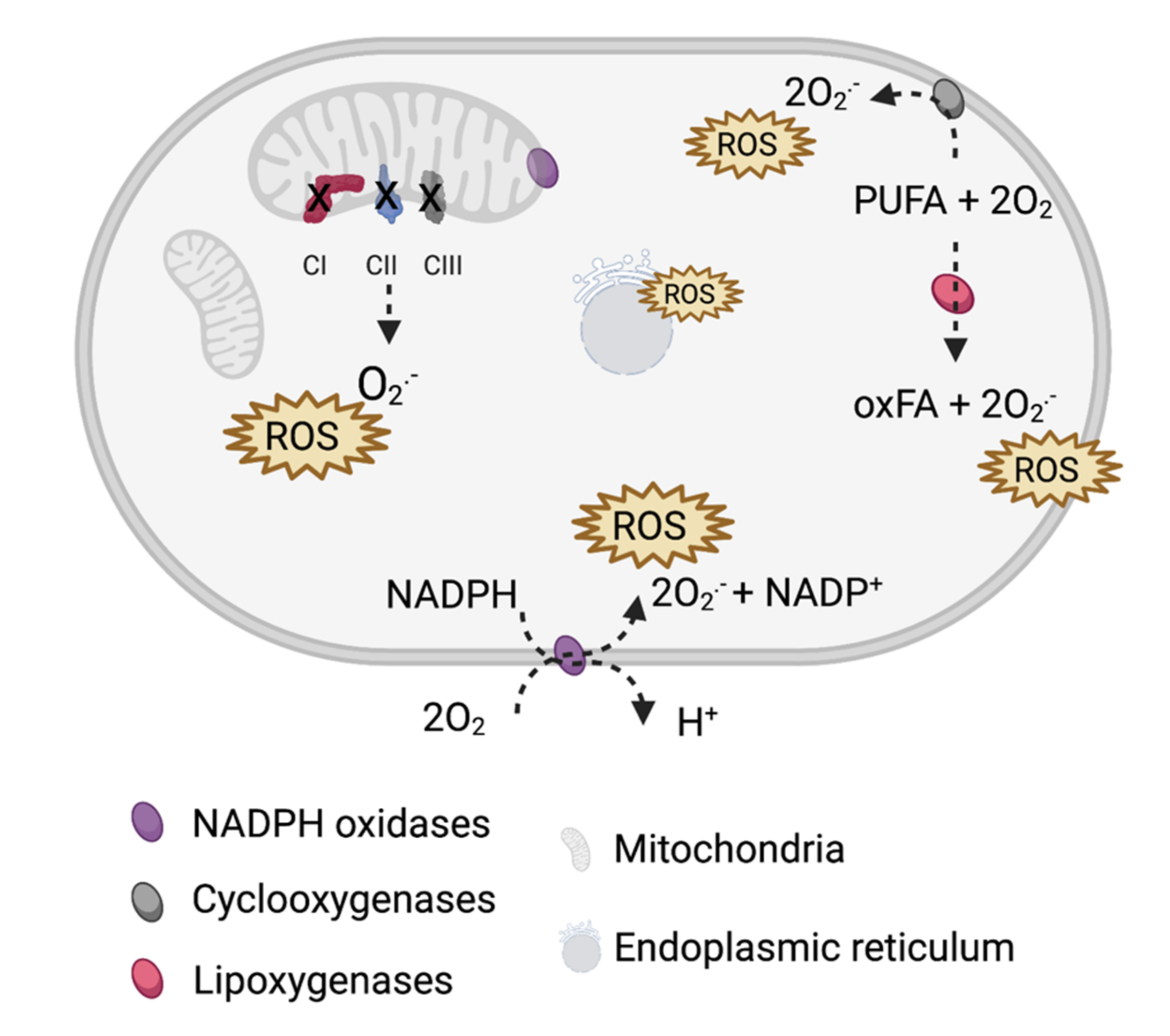
Reactive oxygen species (ROS) are oxygen-containing molecules with high reactivity including hydroxyl and superoxide radicals as well as superoxide non-radical molecules such as hydrogen peroxide. In eukaryotic cells, ROS are typically produced aerobically in mitochondria. Alternatively, ROS can be generated in peroxisomes via ß-oxidation of fatty acids and in the endoplasmic reticulum via protein oxidation (Figure 1).
Growing evidence suggests that ROS (usually considered a metabolic waste) could serve as regulators of essential signaling and metabolic pathways. Continuous ROS production helps maintain control of cell proliferation and differentiation. Antioxidant enzymes closely regulate ROS production based on a scavenging system that is crucial for protecting eukaryotic cells from oxidative damage and other pathological processes including tumorigenesis [1,2,3]. Although cancer cells thrive on slightly higher levels of ROS compared to normal cells [3,4,5,6], cancer cells are more sensitive to external stimuli, which leads to significant redox adaptation and cell death induction [5,7]. Currently, anticancer therapy agents used to induce levels of ROS have been introduced in pancreatic, breast, colon, rectal, bladder, lung, and prostate cancers as well as melanoma, glioblastoma, and lymphoma (reviewed in [8]). From this perspective, we will focus on ROS production in pheochromocytoma (PHEO) and paraganglioma (PGL) and explore potential opportunities to therapeutically target ROS production in these rare tumors.
2. ROS in PHEO and PGL
PHEO and PGL are rare neuroendocrine neoplasms derived from chromaffin cells [9]. These tumors have a strong genetic disposition including variants in many Krebs cycle enzymes such as succinate dehydrogenase (SDH), fumarate hydratase, and malate dehydrogenase, which affect energy homeostasis and hypoxia signaling in cells [9,10]. Such defects in the Krebs cycle can result in accumulation of succinate and fumarate, which contribute to cancer development [11]. SDH mutations are found in approximately 17–28% of PHEO/PGL cases [12,13,14,15,16,17,18]. In particular, SDH subunit B (SDHB)-mutated PHEO/PGL is associated with a more aggressive tumor behavior and higher rates of metastatic disease when compared to all other PHEO/PGL cases (reviewed in [19]). These mutations alter the function of mitochondrial complex II and result in succinate accumulation, thus directly modifying oxidative phosphorylation and increasing ROS production in the SDHx-mutated tumor [20]. Currently, treatments for metastatic SDHB-related PHEO/PGL are palliative with very limited clinical benefit. There is an urgent need for more transformative therapies in order to improve disease outcomes in patients with SDHB-related PHEO/PGL. Recently, Moog et al. suggested angiogenesis, pseudohypoxia, epigenetics, metabolic reprogramming, and redox imbalance as therapeutic target options for the treatment of SDH-deficient PGLs [21].
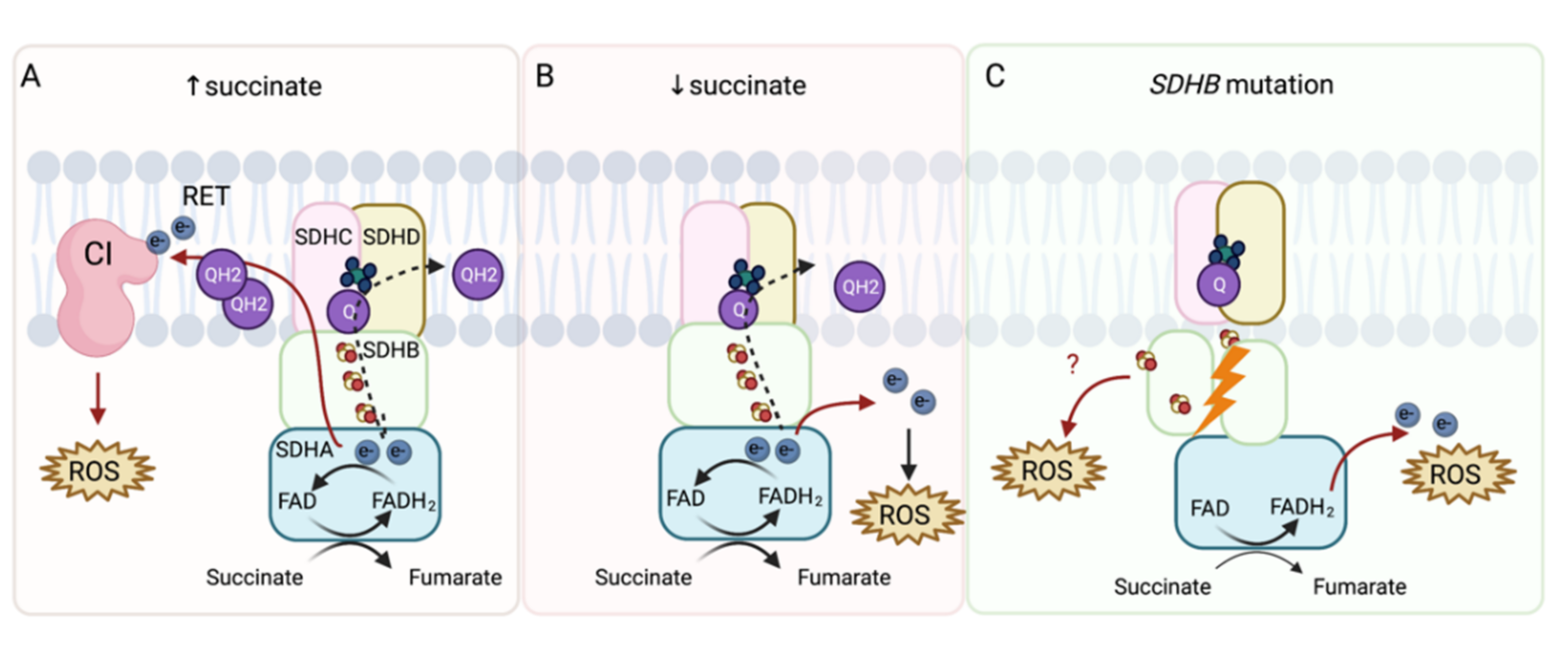
Mitochondrial complex II significantly contributes to ROS production directly and indirectly via reverse electron transfer in CI [20,22,23,24,25], suggesting that this pathway could be used as a potential target in tumors with compromised SDH (Figure 2). Indeed, pharmacologic inhibition of SDHB in rat PHEO cells (PC12 cells) led to increased ROS production [26], which is consistent with findings from transgenic mouse cell line (NIH3T3 cells) with SDHC mutations [27] and Chinese hamster models of fibroblasts expressing truncated SDHC protein [28]. These findings resulted in the discovery of mitochondrial complex II as a target for cancer therapy [29,30]. Additionally, SDHD-mutated cDNA introduced into Chinese hamster fibroblasts resulted in increases of superoxide production with subsequent increases in mutation frequency and rate. This demonstrates the role of SDH-produced ROS in genomic instability within mammalian cells [31]. Furthermore, Guzy et al. found that SDHB inhibition (Hep3B human hepatoma cells, A549 human alveolar epithelium-derived tumor cells, and 143B human osteosarcoma cells) led to the accumulation of ROS [32], which was later confirmed by SDHB silencing in rat PHEO cells (PC12 cells) [33], mouse PHEO cells (MPC cells) [34,35], immortalized mouse chromaffin cells [36], and human hPheo1 cells [34,35]. Moreover, an increase in mitochondrial ROS production was observed in our model of the SDHB knock-out in human PHEO cell line (hPheo1, unpublished data).
3. Targeting of ROS Production in PHEO and PGL
Recently, several studies have uncovered ways to suppress PHEO/PGL cell growth by ROS manipulation. Inhibition of pyruvate dehydrogenase kinase, either alone or in combination with an inhibitor of mitochondrial complex I (CI) metformin, resulted in oxidative metabolism promotion and intracellular ROS accumulation in immortalized head and neck PGL cell cultures from two patients carrying the SDHC and SDHD mutations [37]. This treatment promoted cell cycle arrest and apoptosis, which are the processes previously associated with ROS accumulation [37]. The increase in oxidative stress was confirmed in SDHB-silenced PHEO mouse models (MPC cells) and resulted in an increased demand for antioxidative defense [35]. In this study, the authors targeted glutathione (GSH) de novo synthesis with a nuclear factor erythroid-2 related factor 2 (NRF2) inhibitor. The targeted disruption of ROS homeostasis with the NRF2 inhibitor, brusatol, suppressed the growth of metastatic lesions from SDHB-silenced mouse PHEO cells (MPC) in vivo and prolonged overall survival of mice [35]. In addition to the significant ROS production, SDHB-silenced tumor cell lines (MPC and human pheochromocytoma precursor cells; hPheo1) accumulate iron, whereby treatment with ascorbic acid disrupts redox hemostasis, leading to ROS overload. This ultimately results in cell death in vitro and delays tumor growth in vivo [34]. Recently, dysregulation of iron homeostasis and ascorbate-induced accumulation of ROS resulting in cell death has been observed in SDHB-mutated immortalized chromaffin cells [36].
Interestingly, Pang et al. found that SDHB-mutated PHEO/PGL tumor tissue and SDHBKD PHEO cell culture (mouse metastatic tumor tissue; MTT) had increased mitochondrial complex I activity, which catalyzes the first step of the electron transport chain and oxidizes NADH to NAD+ [38]. NAD+ activates poly (ADP-ribose) polymerase (PARP), which stimulates DNA repair mechanisms and protects against DNA damage-associated cell death [39]. Inhibition and depletion of PARP activity leads to DNA damage and ROS induction [40]. Indeed, in SDHB-silenced PHEO models (MTT cells), PARP inhibition with olaparib in combination with a chemotherapeutic agent effectively reduced tumor proliferation, metastasis, and aggressive phenotypes in vivo [38]. These studies suggest that both increasing ROS accumulation to override the tolerated ROS pool and/or targeting the protective antioxidant mechanisms can suppress PHEO/PGL progression.
4. Future Directions
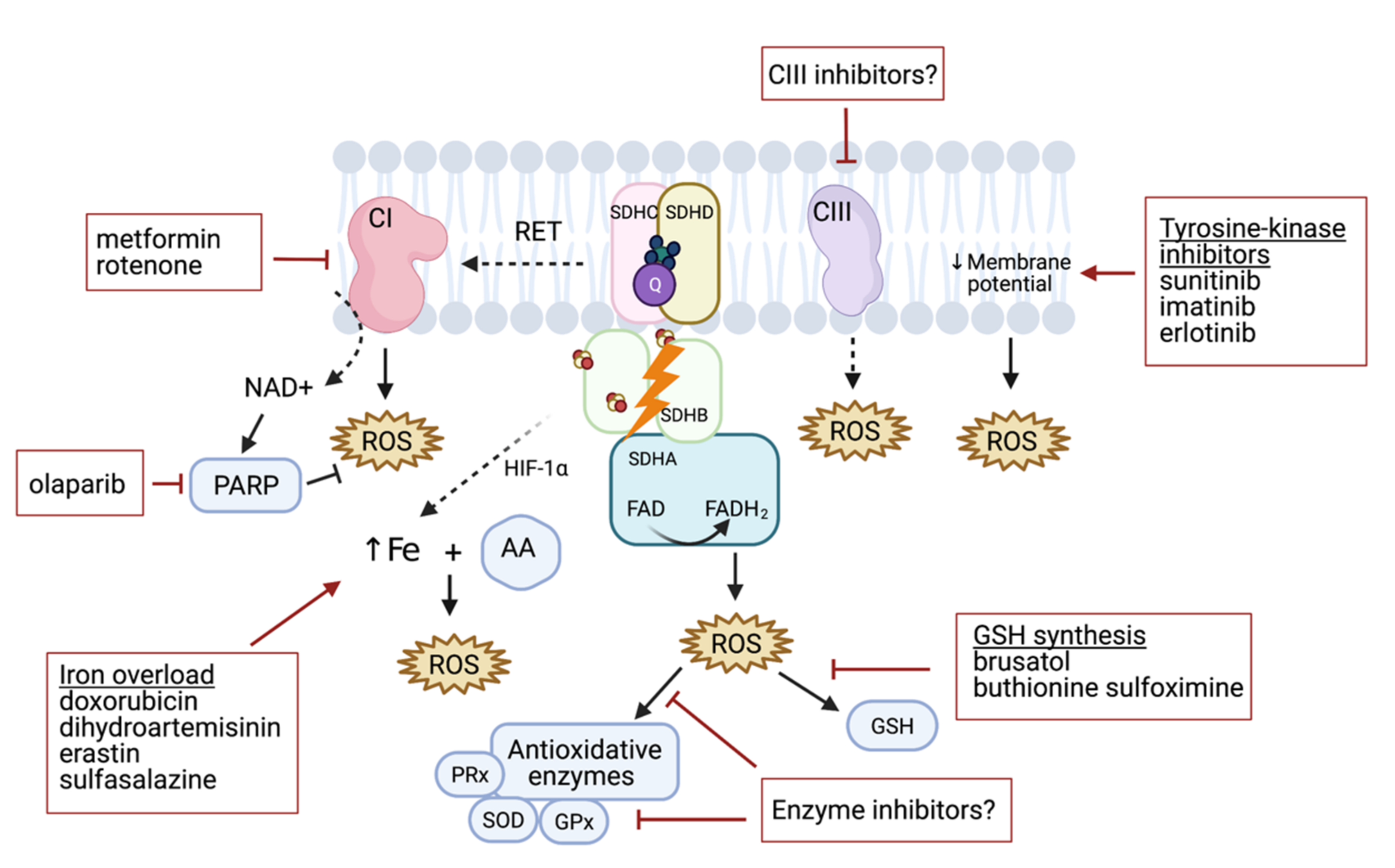
Elevated levels of ROS are common hallmarks of cancer progression and resistance to treatment [41,42]. High ROS burden in SDHB deficient solid tumors offers a promising target for treatment. Although there are several pathways that activate ROS production, at the present time, there is little evidence to prove its effectiveness as an antiproliferative approach in SDHB-silenced PHEOs/PGLs in vitro and in animal models [34,35,37]. The benefits of high-dose ascorbic acid treatment in SDHBKD models resulting in the Fenton reaction (due to iron accumulation and consequent ROS production [34]) is a potential way to disrupt redox imbalance. For example, doxorubicin can induce iron-mediated increases in ROS [43], and this drug was previously found to limit in vitro growth of MTT cells that are considered to be a more aggressive model [44]. Fliedner et al. suggested that this may be linked to increased ROS production if compared to MPC cells [45]. However, the authors focused on the hypoxia signaling pathway rather than ROS production in the doxorubicin study [46], and thus the effectiveness of the drug in SDHx-models has yet to be verified. Other iron-dependent ROS generators such as dihydroartemisinin, erastin, and sulfasalazine [47], and/or the combination of these drugs with a high-dose ascorbic acid treatment, could potentially serve as a future iron/ROS-targeted strategy for SDHx-mutated tumors (Figure 3).
Tyrosine kinase inhibitors (TKIs) have been extensively studied for their antitumoral properties. TKIs target functions of different cellular compartments and signaling pathways. Certain TKIs induce mitochondrial dysfunction (i.e., uncoupling components of the electron transport chain), resulting in a drop in mitochondrial membrane potential and increased ROS production [48,49,50,51,52]. Treatment with certain TKIs such as imatinib or erlotinib was not effective in PHEO/PGL [53]. However, the insignificant findings of this study may be partially explained by the limited number of PHEO/PGL cases and lack of cases with SDHB mutations associated with ROS-mediated anticancer effects as well as by the specificity of certain TKIs. Indeed, treatment with a different inhibitor, sunitinib, led to a reduction in tumor size, stabilization of disease, and improvement in hypertension among SDHB-mutated PHEO/PGL patients [54], warranting further evaluation of this compound. While mitochondrial complexes I and III are relevant producers of ROS themselves [3], targeted disruption of oxidative phosphorylation was shown to increase ROS levels in several cancer models [55,56,57,58,59,60,61]. Targeting CI with metformin in rat PHEO cells (PC12 cells) revealed the antiproliferative potential of this drug [62,63] in vitro, however, these results have yet to be confirmed in vivo. Similarly, Florio et al. showed that metformin treatment promoted oxidative metabolism and decreased proliferation in cells isolated from SDHC- and SDHD-mutated patients [37]. Furthermore, rotenone, an inhibitor of CI, induced rat PHEO cell (PC12 cells) apoptosis by ROS production [64]. Even though these drugs have not been extensively tested in SDHx-mutated models, further accumulation of ROS could lead to ROS overload in mutated cells, resulting in cell apoptosis. Other CI and CIII inhibitors have yet to be tested (Figure 3).
NRF2 was found to be a promising therapeutic target in other neoplasms [65]. Targeting NRF2-dependent GSH synthesis was effective in the metastatic model of SDHB-silenced PHEO/PGL [35], thus, more strategies to prevent the overall production and/or availability of GSH in tumor cells may be beneficial in future PHEO/PGL therapies. Buthionine sulfoximine, an inhibitor of the enzyme glutamate cysteine ligase (required for GSH synthesis), depletes GSH, and exhibits anticancer activity [6]. When considering the benefits of GSH synthesis disruption in SDHBKD cells, we hypothesize that targeting other antioxidant pathways could be a potential strategy to expose cells to endogenously produced ROS. For example, it has been shown that MTT cells, more aggressive mouse PHEO cells derived from MPC cells, produce higher amounts of ROS when compared to MPC cells. This is accompanied by an increase in superoxide dismutase 1 [45] and such an increase in antioxidant protection can thus be hypothesized as a consequence of ROS accumulation in SDHx-mutated PHEO/PGL models. Similarly, SDHB-mutated PHEO/PGL showed increased expression of superoxide dismutase 2 when compared to VHL-mutated PHEO/PGL [45], which may play a role in the malignancy rates of patients with these mutations [66]. Thus, enzyme inhibitors may enable overproduction of ROS with consequent tumor cell death (Figure 3).
5. Conclusions
In conclusion, patients with SDHB-mutated PHEO/PGL have a higher likelihood of metastatic disease with limited therapeutic options and poor prognosis. Data regarding anticancer ROS-related drugs as potential therapeutic candidates for SDHB-mutated PHEO/PGL are still very limited. More studies are needed to evaluate the effectiveness and safety of these drugs in PHEO/PGL patients. Given the accumulation of promising evidence, ROS targeting may become an effective anticancer therapy in PHEO/PGL as well as other tumor types in the near future.
Author Contributions
Conceptualization, K.H.V. and K.P.; Writing—original draft preparation, K.H.V.; Writing—review and editing, K.H.V., C.Y., L.M., J.N. and K.P., Visualization, K.H.V. and C.Y.; Supervision, J.N. and K.P. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Intramural Research Programs of the National Institutes of Health: Eunice Kennedy Shriver National Institute of Child Health and Human Development, and National Cancer Institute.
Acknowledgments
Figures created with BioRender.com (accessed on 19 June 2021).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, A.; Scholer-Dahirel, A.; Mechta-Grigoriou, F. The role of reactive oxygen species and metabolism on cancer cells and their microenvironment. Semin. Cancer Biol. 2014, 25, 23–32. [Google Scholar] [CrossRef]
- Nishikawa, M. Reactive oxygen species in tumor metastasis. Cancer Lett. 2008, 266, 53–59. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS Function in Redox Signaling and Oxidative Stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [Green Version]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liaison in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Fishbein, L.; Leshchiner, I.; Walter, V.; Danilova, L.; Robertson, A.G.; Johnson, A.R.; Lichtenberg, T.M.; Murray, B.A.; Ghayee, H.K.; Else, T.; et al. Comprehensive Molecular Characterization of Pheochromocytoma and Paraganglioma. Cancer Cell 2017, 31, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remacha, L.; Comino-Méndez, I.; Richter, S.; Contreras, L.; Currás-Freixes, M.; Pita, G.; Letón, R.; Galarreta, A.; Torres-Pérez, R.; Honrado, E.; et al. Targeted Exome Sequencing of Krebs Cycle Genes Reveals Candidate Cancer-Predisposing Mutations in Pheochromocytomas and Paragangliomas. Clin. Cancer Res. 2017, 23, 6315–6324. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yang, C. Oncometabolites in Cancer: Current Understanding and Challenges. Cancer Res. 2021, 81, 2820–2823. [Google Scholar] [CrossRef]
- Neumann, H.P.; Bausch, B.; McWhinney, S.R.; Bender, B.U.; Gimm, O.; Franke, G.; Schipper, J.; Klisch, J.; Altehoefer, C.; Zerres, K.; et al. Germ-line mutations in nonsyndromic pheochromocytoma. N. Engl. J. Med. 2002, 346, 1459–1466. [Google Scholar] [CrossRef] [Green Version]
- Eijkelenkamp, K.; Osinga, T.E.; Links, T.P.; Van der Horst-Schrivers, A.N.A. Clinical implications of the oncometabolite succinate in SDHx-mutation carriers. Clin. Genet. 2020, 97, 39–53. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.P.; Pawlu, C.; Peczkowska, M.; Bausch, B.; McWhinney, S.R.; Muresan, M.; Buchta, M.; Franke, G.; Klisch, J.; Bley, T.A.; et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA 2004, 292, 943–951. [Google Scholar] [CrossRef] [Green Version]
- Else, T.; Marvin, M.L.; Everett, J.N.; Gruber, S.B.; Arts, H.A.; Stoffel, E.M.; Auchus, R.J.; Raymond, V.M. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3). J. Clin. Endocrinol. Metab. 2014, 99, E1482–E1486. [Google Scholar] [CrossRef] [Green Version]
- Amar, L.; Bertherat, J.; Baudin, E.; Ajzenberg, C.; Bressac-de Paillerets, B.; Chabre, O.; Chamontin, B.; Delemer, B.; Giraud, S.; Murat, A.; et al. Genetic testing in pheochromocytoma or functional paraganglioma. J. Clin. Oncol. 2005, 23, 8812–8818. [Google Scholar] [CrossRef]
- Heesterman, B.L.; Bayley, J.P.; Tops, C.M.; Hes, F.J.; Van Brussel, B.T.; Corssmit, E.P.; Hamming, J.F.; Van der Mey, A.G.; Jansen, J.C. High prevalence of occult paragangliomas in asymptomatic carriers of SDHD and SDHB gene mutations. Eur. J. Hum. Genet. 2013, 21, 469–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Tuin, K.; Mensenkamp, A.R.; Tops, C.M.J.; Corssmit, E.P.M.; Dinjens, W.N.; Van de Horst-Schrivers, A.N.A.; Jansen, J.C.; De Jong, M.M.; Kunst, H.P.M.; Kusters, B.; et al. Clinical Aspects of SDHA-Related Pheochromocytoma and Paraganglioma: A Nationwide Study. J. Clin. Endocrinol. Metab. 2018, 103, 438–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilanchezhian, M.; Jha, A.; Pacak, K.; Del Rivero, J. Emerging Treatments for Advanced/Metastatic Pheochromocytoma and Paraganglioma. Curr. Treat. Options Oncol. 2020, 21, 85. [Google Scholar] [CrossRef] [PubMed]
- Hadrava Vanova, K.; Kraus, M.; Neuzil, J.; Rohlena, J. Mitochondrial complex II and reactive oxygen species in disease and therapy. Redox Rep. 2020, 25, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Moog, S.; Lussey-Lepoutre, C.; Favier, J. Epigenetic and metabolic reprogramming of SDH-deficient paragangliomas. Endocr. Relat. Cancer 2020, 27, R451–R463. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. 2009, 554, 165–181. [Google Scholar] [CrossRef] [PubMed]
- Chouchani, E.T.; Pell, V.R.; Gaude, E.; Aksentijević, D.; Sundier, S.Y.; Robb, E.L.; Logan, A.; Nadtochiy, S.M.; Ord, E.N.J.; Smith, A.C.; et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 2014, 515, 431–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, C.L.; Orr, A.L.; Perevoshchikova, I.V.; Treberg, J.R.; Ackrell, B.A.; Brand, M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012, 287, 27255–27264. [Google Scholar] [CrossRef] [Green Version]
- Robb, E.L.; Hall, A.R.; Prime, T.A.; Eaton, S.; Szibor, M.; Viscomi, C.; James, A.M.; Murphy, M.P. Control of mitochondrial superoxide production by reverse electron transport at complex I. J. Biol. Chem. 2018, 293, 9869–9879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Nakamura, E.; Yang, H.; Wei, W.; Linggi, M.S.; Sajan, M.P.; Farese, R.V.; Freeman, R.S.; Carter, B.D.; Kaelin, W.G.; et al. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: Developmental culling and cancer. Cancer Cell 2005, 8, 155–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishii, T.; Yasuda, K.; Akatsuka, A.; Hino, O.; Hartman, P.S.; Ishii, N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005, 65, 203–209. [Google Scholar]
- Slane, B.G.; Aykin-Burns, N.; Smith, B.J.; Kalen, A.L.; Goswami, P.C.; Domann, F.E.; Spitz, D.R. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Res. 2006, 66, 7615–7620. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Low, P.; Dyason, J.C.; Wang, X.F.; Prochazka, L.; Witting, P.K.; Freeman, R.; Swettenham, E.; Valis, K.; Liu, J.; et al. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene 2008, 27, 4324–4335. [Google Scholar] [CrossRef] [Green Version]
- Dong, L.F.; Freeman, R.; Liu, J.; Zobalova, R.; Marin-Hernandez, A.; Stantic, M.; Rohlena, J.; Valis, K.; Rodriguez-Enriquez, S.; Butcher, B.; et al. Suppression of tumor growth in vivo by the mitocan alpha-tocopheryl succinate requires respiratory complex II. Clin. Cancer Res. 2009, 15, 1593–1600. [Google Scholar] [CrossRef] [Green Version]
- Owens, K.M.; Aykin-Burns, N.; Dayal, D.; Coleman, M.C.; Domann, F.E.; Spitz, D.R. Genomic instability induced by mutant succinate dehydrogenase subunit D (SDHD) is mediated by O2(-•) and H2O2. Free Radic. Biol. Med. 2012, 52, 160–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzy, R.D.; Sharma, B.; Bell, E.; Chandel, N.S.; Schumacker, P.T. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell Biol. 2008, 28, 718–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, Y.; Ishii, K.A.; Aita, Y.; Ikeda, T.; Kawakami, Y.; Shimano, H.; Hara, H.; Takekoshi, K. Loss of SDHB Elevates Catecholamine Synthesis and Secretion Depending on ROS Production and HIF Stabilization. Neurochem. Res. 2016, 41, 696–706. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Pang, Y.; Zhu, B.; Uher, O.; Caisova, V.; Huynh, T.T.; Taieb, D.; Hadrava Vanova, K.; Ghayee, H.K.; Neuzil, J.; et al. Therapeutic Targeting of SDHB-Mutated Pheochromocytoma/Paraganglioma with Pharmacologic Ascorbic Acid. Clin. Cancer Res. 2020, 26, 3868–3880. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Pang, Y.; Caisova, V.; Ding, J.; Yu, D.; Zhou, Y.; Huynh, T.T.; Ghayee, H.; Pacak, K.; Yang, C. Targeting NRF2-Governed Glutathione Synthesis for SDHB-Mutated Pheochromocytoma and Paraganglioma. Cancers 2020, 12. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, J.; Moog, S.; Morin, A.; Gentric, G.; Müller, S.; Morrell, A.P.; Kluckova, K.; Stewart, T.J.; Andoniadou, C.L.; Lussey-Lepoutre, C.; et al. Loss of SDHB Promotes Dysregulated Iron Homeostasis, Oxidative Stress, and Sensitivity to Ascorbate. Cancer Res. 2021, 81, 3480–3494. [Google Scholar] [CrossRef]
- Florio, R.; De Lellis, L.; Veschi, S.; Verginelli, F.; Di Giacomo, V.; Gallorini, M.; Perconti, S.; Sanna, M.; Mariani-Costantini, R.; Natale, A.; et al. Effects of dichloroacetate as single agent or in combination with GW6471 and metformin in paraganglioma cells. Sci. Rep. 2018, 8, 13610. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.; Lu, Y.; Caisova, V.; Liu, Y.; Bullova, P.; Huynh, T.T.; Zhou, Y.; Yu, D.; Frysak, Z.; Hartmann, I.; et al. Targeting NAD(+)/PARP DNA Repair Pathway as a Novel Therapeutic Approach to SDHB-Mutated Cluster I Pheochromocytoma and Paraganglioma. Clin. Cancer Res. 2018, 24, 3423–3432. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T.; Petermann, E.; Lundin, C.; Hodgson, B.; Sharma, R.A. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer 2008, 8, 193–204. [Google Scholar] [CrossRef]
- Hou, D.; Liu, Z.; Xu, X.; Liu, Q.; Zhang, X.; Kong, B.; Wei, J.-J.; Gong, Y.; Shao, C. Increased oxidative stress mediates the antitumor effect of PARP inhibition in ovarian cancer. Redox Biol. 2018, 17, 99–111. [Google Scholar] [CrossRef]
- Okon, I.S.; Zou, M.-H. Mitochondrial ROS and cancer drug resistance: Implications for therapy. Pharm. Res. 2015, 100, 170–174. [Google Scholar] [CrossRef] [Green Version]
- Bhardwaj, V.; He, J. Reactive Oxygen Species, Metabolic Plasticity, and Drug Resistance in Cancer. Int. J. Mol. Sci. 2020, 21, 3412. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, Y.; Ghanefar, M.; Bayeva, M.; Wu, R.; Khechaduri, A.; Naga Prasad, S.V.; Mutharasan, R.K.; Naik, T.J.; Ardehali, H. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J. Clin. Investig. 2014, 124, 617–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martiniova, L.; Lai, E.W.; Elkahloun, A.G.; Abu-Asab, M.; Wickremasinghe, A.; Solis, D.C.; Perera, S.M.; Huynh, T.T.; Lubensky, I.A.; Tischler, A.S.; et al. Characterization of an animal model of aggressive metastatic pheochromocytoma linked to a specific gene signature. Clin. Exp. Metastasis 2009, 26, 239–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fliedner, S.M.J.; Kaludercic, N.; Jiang, X.-S.; Hansikova, H.; Hajkova, Z.; Sladkova, J.; Limpuangthip, A.; Backlund, P.S.; Wesley, R.; Martiniova, L.; et al. Warburg Effect’s Manifestation in Aggressive Pheochromocytomas and Paragangliomas: Insights from a Mouse Cell Model Applied to Human Tumor Tissue. PLoS ONE 2012, 7, e40949. [Google Scholar] [CrossRef]
- Pang, Y.; Yang, C.; Schovanek, J.; Wang, H.; Bullova, P.; Caisova, V.; Gupta, G.; Wolf, K.I.; Semenza, G.L.; Zhuang, Z.; et al. Anthracyclines suppress pheochromocytoma cell characteristics, including metastasis, through inhibition of the hypoxia signaling pathway. Oncotarget 2017, 8, 22313–22324. [Google Scholar] [CrossRef] [Green Version]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Paech, F.; Bouitbir, J.; Krähenbühl, S. Hepatocellular Toxicity Associated with Tyrosine Kinase Inhibitors: Mitochondrial Damage and Inhibition of Glycolysis. Front. Pharm. 2017, 8, 367. [Google Scholar] [CrossRef] [Green Version]
- Paech, F.; Mingard, C.; Grünig, D.; Abegg, V.F.; Bouitbir, J.; Krähenbühl, S. Mechanisms of mitochondrial toxicity of the kinase inhibitors ponatinib, regorafenib and sorafenib in human hepatic HepG2 cells. Toxicology 2018, 395, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Park, Y.J.; Shin, J.H.; Kim, E.S.; Hwang, J.J.; Jin, D.H.; Kim, J.C.; Cho, D.H. A receptor tyrosine kinase inhibitor, Tyrphostin A9 induces cancer cell death through Drp1 dependent mitochondria fragmentation. Biochem. Biophys. Res. Commun. 2011, 408, 465–470. [Google Scholar] [CrossRef]
- Will, Y.; Dykens, J.A.; Nadanaciva, S.; Hirakawa, B.; Jamieson, J.; Marroquin, L.D.; Hynes, J.; Patyna, S.; Jessen, B.A. Effect of the Multitargeted Tyrosine Kinase Inhibitors Imatinib, Dasatinib, Sunitinib, and Sorafenib on Mitochondrial Function in Isolated Rat Heart Mitochondria and H9c2 Cells. Toxicol. Sci. 2008, 106, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Hernández, M.A.; De la Cruz-Ojeda, P.; López-Grueso, M.J.; Navarro-Villarán, E.; Requejo-Aguilar, R.; Castejón-Vega, B.; Negrete, M.; Gallego, P.; Vega-Ochoa, Á.; Victor, V.M.; et al. Integrated molecular signaling involving mitochondrial dysfunction and alteration of cell metabolism induced by tyrosine kinase inhibitors in cancer. Redox Biol. 2020, 36, 101510. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.Q.; Bezerra-Neto, J.E.; Mendonça, B.B.; Latronico, A.C.; Fragoso, M.C.B.V. Primary malignant tumors of the adrenal glands. Clinics 2018, 73, e756s. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Ramirez, M.; Chougnet, C.N.; Habra, M.A.; Palmer, J.L.; Leboulleux, S.; Cabanillas, M.E.; Caramella, C.; Anderson, P.; Al Ghuzlan, A.; Waguespack, S.G.; et al. Treatment with sunitinib for patients with progressive metastatic pheochromocytomas and sympathetic paragangliomas. J. Clin. Endocrinol. Metab. 2012, 97, 4040–4050. [Google Scholar] [CrossRef] [PubMed]
- Warkad, M.S.; Kim, C.H.; Kang, B.G.; Park, S.H.; Jung, J.S.; Feng, J.H.; Inci, G.; Kim, S.C.; Suh, H.W.; Lim, S.S.; et al. Metformin-induced ROS upregulation as amplified by apigenin causes profound anticancer activity while sparing normal cells. Sci. Rep. 2021, 11, 14002. [Google Scholar] [CrossRef]
- Li, B.; Zhou, P.; Xu, K.; Chen, T.; Jiao, J.; Wei, H.; Yang, X.; Xu, W.; Wan, W.; Xiao, J. Metformin induces cell cycle arrest, apoptosis and autophagy through ROS/JNK signaling pathway in human osteosarcoma. Int. J. Biol. Sci. 2020, 16, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Duan, Q.; Wang, T.; Ahmed, M.; Zhang, N.; Li, Y.; Li, L.; Yao, X. Mitochondrial Respiratory Chain Inhibitors Involved in ROS Production Induced by Acute High Concentrations of Iodide and the Effects of SOD as a Protective Factor. Oxid. Med. Cell Longev. 2015, 2015, 217670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin transiently inhibits colorectal cancer cell proliferation as a result of either AMPK activation or increased ROS production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Park, W.H.; Han, Y.W.; Kim, S.H.; Kim, S.Z. An ROS generator, antimycin A, inhibits the growth of HeLa cells via apoptosis. J. Cell Biochem. 2007, 102, 98–109. [Google Scholar] [CrossRef]
- Park, D. Metformin Induces Oxidative Stress-Mediated Apoptosis without the Blockade of Glycolysis in H4IIE Hepatocellular Carcinoma Cells. Biol. Pharm. Bull. 2019, 42, 2002–2008. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Jiang, X.; Su, T.; Jiang, L.; Zhou, W.; Wang, W. Metformin Suppresses Proliferation and Viability of Rat Pheochromocytoma Cells. Med. Sci. Monit. 2017, 23, 3253–3260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, S.; Daley, B.; Klubo-Gwiezdzinska, J. The role of an anti-diabetic drug metformin in the treatment of endocrine tumors. J. Mol. Endocrinol. 2019, 63, R17–R35. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Liu, C.; Liu, W.; Zhang, H.; Zhang, R.; Liu, J.; Zhang, J.; Xu, C.; Liu, L.; Huang, S.; et al. Rotenone Induction of Hydrogen Peroxide Inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E Pathways, Leading to Neuronal Apoptosis. Toxicol. Sci. 2015, 143, 81–96. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lang, F.; Yang, C. NRF2 in human neoplasm: Cancer biology and potential therapeutic target. Pharm. Ther. 2021, 217, 107664. [Google Scholar] [CrossRef]
- Hempel, N.; Carrico, P.M.; Melendez, J.A. Manganese superoxide dismutase (Sod2) and redox-control of signaling events that drive metastasis. Anticancer. Agents Med. Chem. 2011, 11, 191–201. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Main sources of reactive oxygen species (ROS) in cells. ROS are primarily generated by electron leaks from mitochondrial complexes I–III (CI–CIII) and ROS-producing enzymes: NADPH oxidases, cyclooxygenases, and lipoxygenases that catalyze the oxygenation of polyunsaturated fatty acids (PUFA). Additionally, protein oxidation on the endoplasmic reticulum (ER) and ER-related stress add to the ROS pool.
Figure 1.
Main sources of reactive oxygen species (ROS) in cells. ROS are primarily generated by electron leaks from mitochondrial complexes I–III (CI–CIII) and ROS-producing enzymes: NADPH oxidases, cyclooxygenases, and lipoxygenases that catalyze the oxygenation of polyunsaturated fatty acids (PUFA). Additionally, protein oxidation on the endoplasmic reticulum (ER) and ER-related stress add to the ROS pool.

Figure 2.
ROS production from SDH under physiological and pathophysiological conditions. The SDHA subunit covalently binds flavin adenine dinucleotide (FAD), which removes electrons from succinate to form fumarate. SDHB then transfers electrons via three iron–sulfur (FeS) clusters to the ubiquinone molecule (Q) located in the ubiquinone-binding site, formed by the SDHB, SDHC, and SDHD subunit. Ubiquinone is further reduced to ubiquinol, fueling CIII and CIV (dashed arrow). (A) At high succinate concentrations (≥5 mM), succinate-derived electrons reduce the ubiquinone pool and electrons are forced backward to CI in a process called reverse electron transfer (RET), which leads to indirect ROS production. (B) At physiological succinate concentrations, ROS can be produced directly. FAD is reduced to FADH2, which then reacts with oxygen within an unoccupied binding site to form ROS. (C) In cases of SDHB mutation, incorrect assembly of SDH can result in ROS production directly from reactions between oxygen and FADH2 or exposed FeS clusters.
Figure 2.
ROS production from SDH under physiological and pathophysiological conditions. The SDHA subunit covalently binds flavin adenine dinucleotide (FAD), which removes electrons from succinate to form fumarate. SDHB then transfers electrons via three iron–sulfur (FeS) clusters to the ubiquinone molecule (Q) located in the ubiquinone-binding site, formed by the SDHB, SDHC, and SDHD subunit. Ubiquinone is further reduced to ubiquinol, fueling CIII and CIV (dashed arrow). (A) At high succinate concentrations (≥5 mM), succinate-derived electrons reduce the ubiquinone pool and electrons are forced backward to CI in a process called reverse electron transfer (RET), which leads to indirect ROS production. (B) At physiological succinate concentrations, ROS can be produced directly. FAD is reduced to FADH2, which then reacts with oxygen within an unoccupied binding site to form ROS. (C) In cases of SDHB mutation, incorrect assembly of SDH can result in ROS production directly from reactions between oxygen and FADH2 or exposed FeS clusters.

Figure 3.
Targeting different mechanisms to induce and enhance ROS production in PHEO/PGL. ROS are generated by mitochondrial complex CI–CIII dysfunction, which may be further supported by CI, CII, and CIII inhibitors. Similarly, decreasing the membrane potential can lead to ROS accumulation. CII-related ROS production can be enhanced by increasing the iron pool and consequently increasing ROS production under ascorbate (AA) treatment. Downregulating glutathione (GSH) synthesis can decrease antioxidant protection with other antioxidative enzyme inhibitors including peroxidases (PRx), superoxide dismutase (SOD), and glutathione peroxidase (GPx).
Figure 3.
Targeting different mechanisms to induce and enhance ROS production in PHEO/PGL. ROS are generated by mitochondrial complex CI–CIII dysfunction, which may be further supported by CI, CII, and CIII inhibitors. Similarly, decreasing the membrane potential can lead to ROS accumulation. CII-related ROS production can be enhanced by increasing the iron pool and consequently increasing ROS production under ascorbate (AA) treatment. Downregulating glutathione (GSH) synthesis can decrease antioxidant protection with other antioxidative enzyme inhibitors including peroxidases (PRx), superoxide dismutase (SOD), and glutathione peroxidase (GPx).

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Hadrava Vanova, K.; Yang, C.; Meuter, L.; Neuzil, J.; Pacak, K. Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma. Cancers 2021, 13, 3769. https://doi.org/10.3390/cancers13153769
AMA Style
Hadrava Vanova K, Yang C, Meuter L, Neuzil J, Pacak K. Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma. Cancers. 2021; 13(15):3769. https://doi.org/10.3390/cancers13153769
Chicago/Turabian StyleHadrava Vanova, Katerina, Chunzhang Yang, Leah Meuter, Jiri Neuzil, and Karel Pacak. 2021. "Reactive Oxygen Species: A Promising Therapeutic Target for SDHx-Mutated Pheochromocytoma and Paraganglioma" Cancers 13, no. 15: 3769. https://doi.org/10.3390/cancers13153769
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.