A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation?



Abstract
:Simple Summary
Abstract
1. Global Versus Local DSB Repair Pathway Selection and Regulation
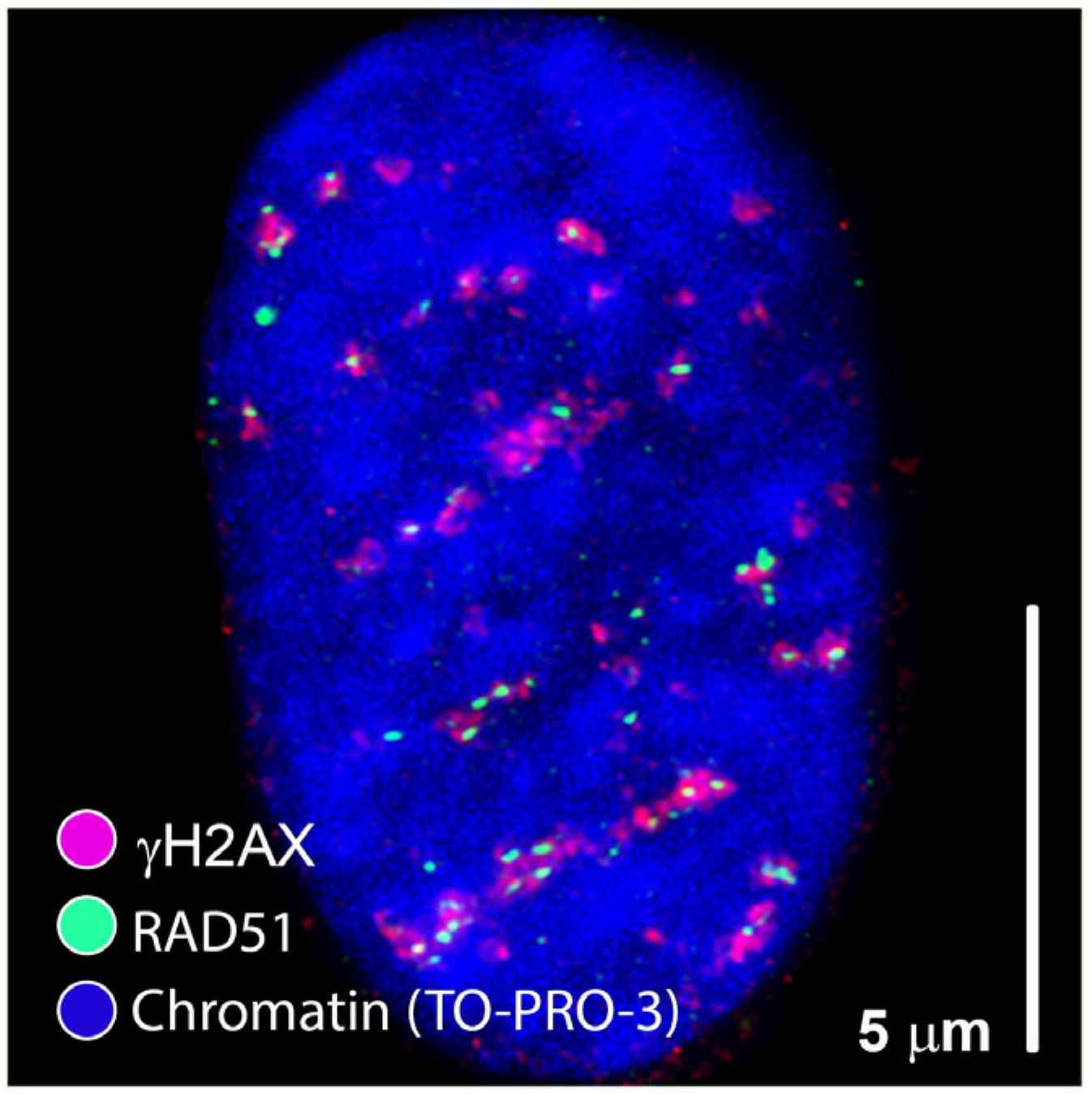
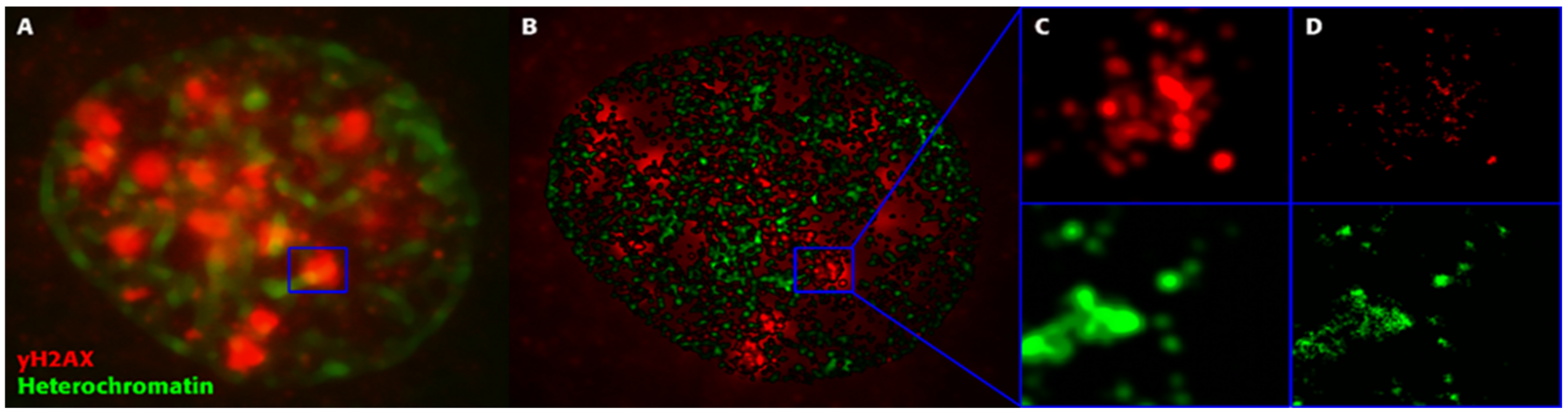
2. Is Regulation of DSB Repair Physically Controlled Through Chromatin and IRIF Architecture?
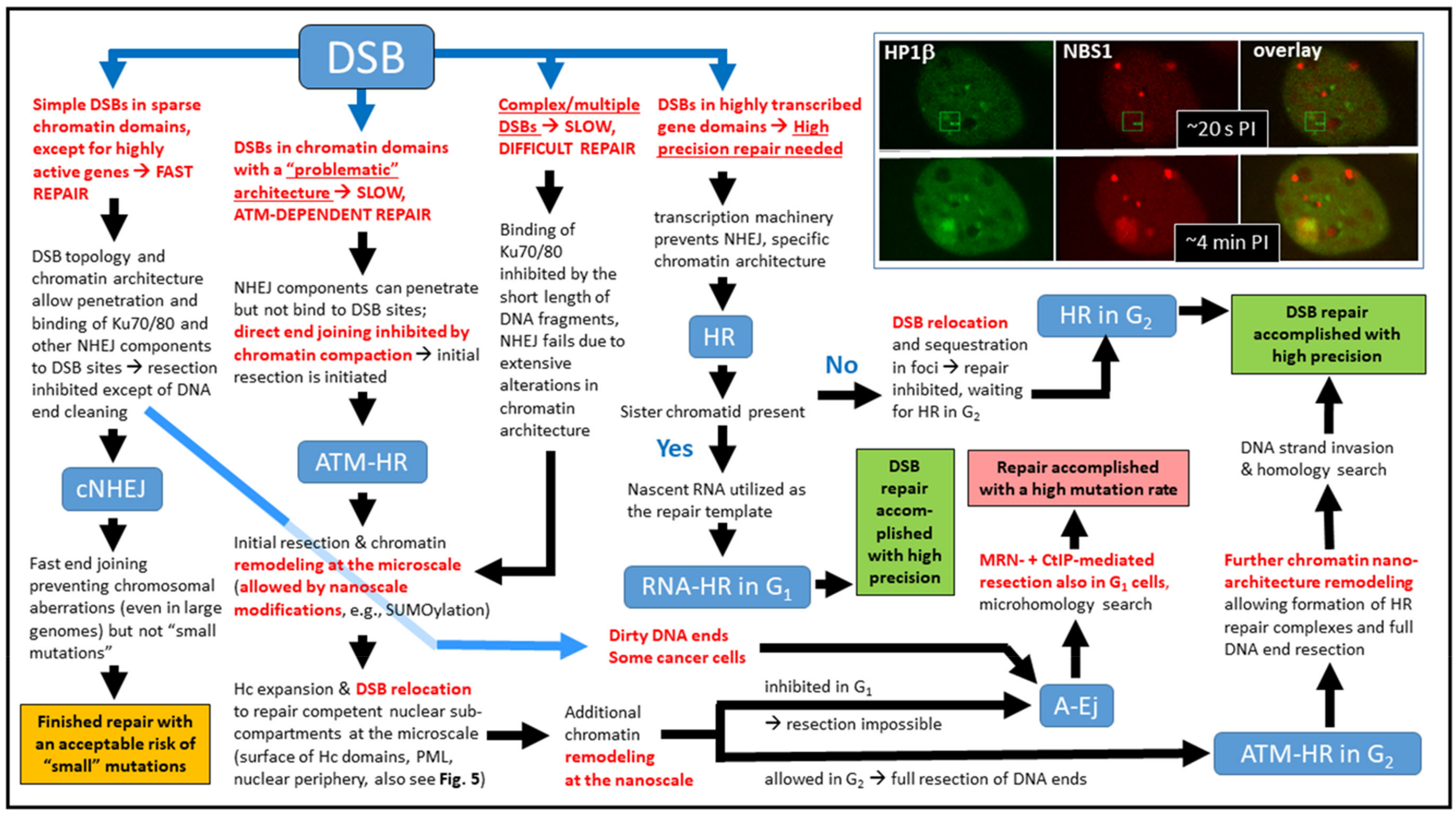
3. First Insights into DSB Repair and its Regulation at the Nanoscale
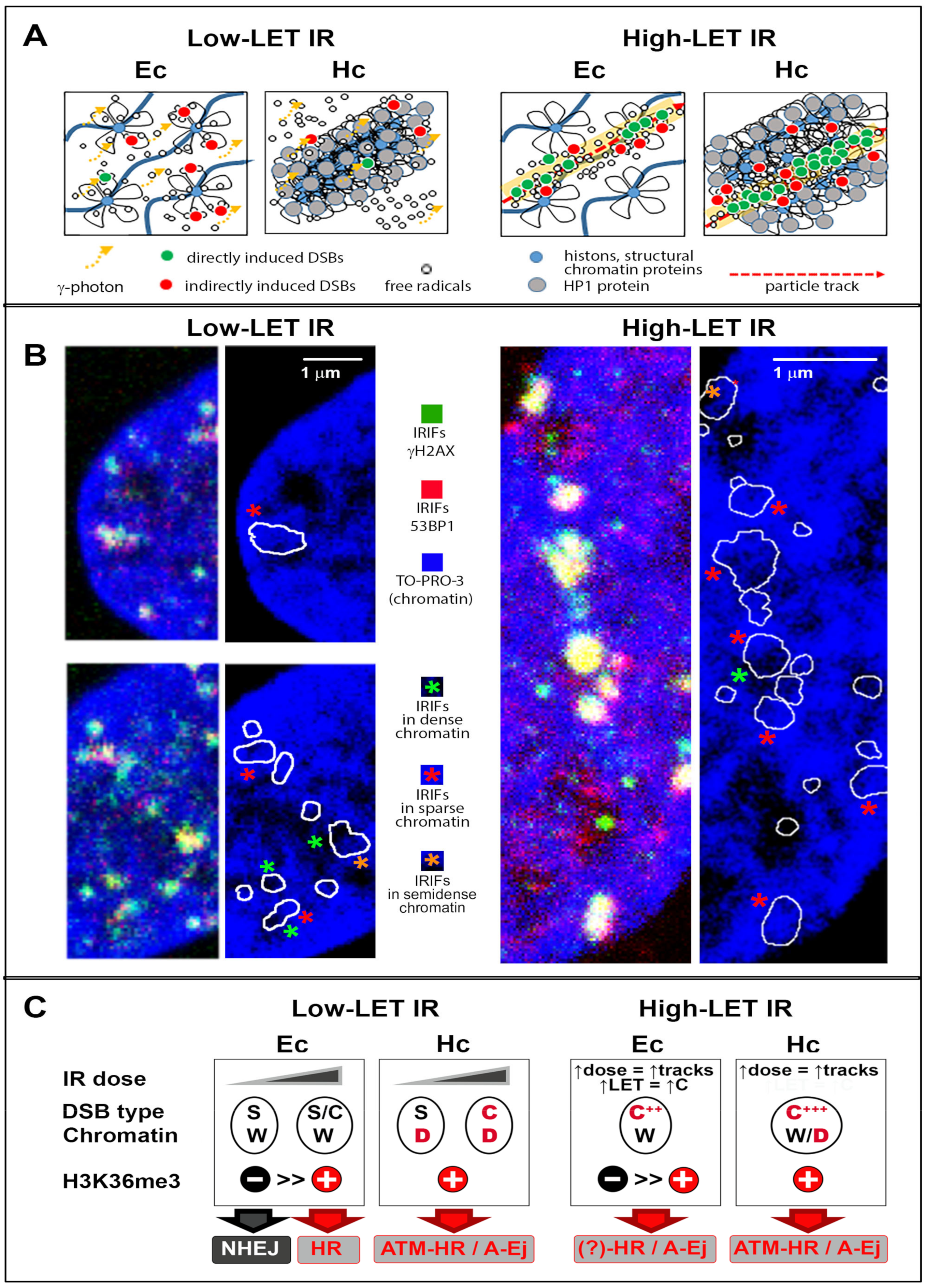
4. Specificities for High-LET Particle Radiation
5. Incorrect DSB Repair, Formation of Chromosomal Aberrations and Cancer
6. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A-Ej | alternative end joining mechanisms (alternative repair pathways) |
| cNHEJ | classical nonhomologous end joining (dependent on Ku70/80) |
| DSB | double-strand break |
| High-LET | high linear energy transfer |
| HR | homologous recombination |
| H3K9me3 | histone H3 trimethylated at lysine 9 |
| H3K36me3 | histone H3 trimethylated at lysine 39 |
| H4K20me3 | histone H4 trimethylated at lysine 20 |
| IRIF | ionizing radiation-induced focus/DSB repair complexes |
| IR | ionizing radiation |
| Low-LET | low linear energy transfer |
| MMEJ | microhomology-mediated end joining |
| NHEJ | nonhomologous end joining |
| SMLM | single-molecule localization microscopy |
| γH2AX | histone H2AX phosphorylated on serine 139 |
References
- Alhmoud, J.F.; Woolley, J.F.; Al Moustafa, A.-E.; Malki, M.I. DNA Damage/Repair Management in Cancers. Cancers 2020, 12, 1050. [Google Scholar] [CrossRef]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis: DNA Damage and Repair. Environ. Mol. Mutagen. 2017, 58, 235–263. [Google Scholar] [CrossRef] [Green Version]
- Rittich, B.; Spanová, A.; Falk, M.; Benes, M.J.; Hrubý, M. Cleavage of double stranded plasmid DNA by lanthanide complexes. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2004, 800, 169–173. [Google Scholar] [CrossRef]
- Bennett, C.B.; Lewis, A.L.; Baldwin, K.K.; Resnick, M.A. Lethality induced by a single site-specific double-strand break in a dispensable yeast plasmid. Proc. Natl. Acad. Sci. USA 1993, 90, 5613–5617. [Google Scholar] [CrossRef] [Green Version]
- Sharda, A.; Rashid, M.; Shah, S.G.; Sharma, A.K.; Singh, S.R.; Gera, P.; Chilkapati, M.K.; Gupta, S. Elevated HDAC activity and altered histone phospho-acetylation confer acquired radio-resistant phenotype to breast cancer cells. Clin. Epigenetics 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Ensminger, M.; Löbrich, M. One end to rule them all: Non-homologous end-joining and homologous recombination at DNA double-strand breaks. Br. J. Radiol. 2020, 2019, 1054. [Google Scholar] [CrossRef]
- Scully, R.; Panday, A.; Elango, R.; Willis, N.A. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat. Rev. Mol. Cell Biol. 2019, 20, 698–714. [Google Scholar] [CrossRef]
- Sallmyr, A.; Tomkinson, A.E. Repair of DNA double-strand breaks by mammalian alternative end-joining pathways. J. Biol. Chem. 2018, 293, 10536–10546. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Xiong, X.; Du, Z.; Wang, Y.; Feng, Z.; Fan, P.; Yan, C.; Willers, H.; Zhang, J. 53BP1 promotes microhomology-mediated end-joining in G1-phase cells. Nucleic Acids Res. 2015, 43, 1659–1670. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G. Backup pathways of NHEJ in cells of higher eukaryotes: Cell cycle dependence. Radiother. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.; Greenberg, R.A. Noncanonical views of homology-directed DNA repair. Genes Dev. 2016, 30, 1138–1154. [Google Scholar] [CrossRef] [Green Version]
- Vanoli, F.; Prakash, R.; White, T.; Jasin, M. Interhomolog Homologous Recombination in Mouse Embryonic Stem Cells. In Homologous Recombination; Aguilera, A., Carreira, A., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; Volume 2153, pp. 127–143. ISBN 978-1-07-160643-8. [Google Scholar]
- Fernandez, J.; Bloomer, H.; Kellam, N.; LaRocque, J.R. Chromosome Preference During Homologous Recombination Repair of DNA Double-Strand Breaks in Drosophila melanogaster. Genes Genomes Genet. 2019, 9, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Campos, A.; Clemente-Blanco, A. Cell Cycle and DNA Repair Regulation in the Damage Response: Protein Phosphatases Take Over the Reins. IJMS 2020, 21, 446. [Google Scholar] [CrossRef] [Green Version]
- Hustedt, N.; Durocher, D. The control of DNA repair by the cell cycle. Nat. Cell Biol. 2017, 19, 1–9. [Google Scholar] [CrossRef]
- You, Z.; Bailis, J.M. DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol. 2010, 20, 402–409. [Google Scholar] [CrossRef] [Green Version]
- Bordelet, H.; Dubrana, K. Keep moving and stay in a good shape to find your homologous recombination partner. Curr. Genet. 2019, 65, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Katsuki, Y.; Jeggo, P.A.; Uchihara, Y.; Takata, M.; Shibata, A. DNA double-strand break end resection: A critical relay point for determining the pathway of repair and signaling. Genome Instab. Dis. 2020, 1, 155–171. [Google Scholar] [CrossRef]
- Choi, E.-H.; Yoon, S.; Koh, Y.E.; Seo, Y.-J.; Kim, K.P. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp. Mol. Med. 2020, 52, 1220–1229. [Google Scholar] [CrossRef]
- Kohutova, A.; Raška, J.; Kruta, M.; Seneklova, M.; Barta, T.; Fojtik, P.; Jurakova, T.; Walter, C.A.; Hampl, A.; Dvorak, P.; et al. Ligase 3–mediated end-joining maintains genome stability of human embryonic stem cells. FASEB J. 2019, 33, 6778–6788. [Google Scholar] [CrossRef]
- Shrivastav, M.; De Haro, L.P.; Nickoloff, J.A. Regulation of DNA double-strand break repair pathway choice. Cell Res. 2008, 18, 134–147. [Google Scholar] [CrossRef] [Green Version]
- Serrano, L.; Liang, L.; Chang, Y.; Deng, L.; Maulion, C.; Nguyen, S.; Tischfield, J.A. Homologous Recombination Conserves DNA Sequence Integrity Throughout the Cell Cycle in Embryonic Stem Cells. Stem Cells Dev. 2011, 20, 363–374. [Google Scholar] [CrossRef] [Green Version]
- Mujoo, K.; Pandita, R.K.; Tiwari, A.; Charaka, V.; Chakraborty, S.; Singh, D.K.; Hambarde, S.; Hittelman, W.N.; Horikoshi, N.; Hunt, C.R.; et al. Differentiation of Human Induced Pluripotent or Embryonic Stem Cells Decreases the DNA Damage Repair by Homologous Recombination. Stem Cell Rep. 2017, 9, 1660–1674. [Google Scholar] [CrossRef] [Green Version]
- Lukášová, E.; Kořistek, Z.; Klabusay, M.; Ondřej, V.; Grigoryev, S.; Bačíková, A.; Řezáčová, M.; Falk, M.; Vávrová, J.; Kohútová, V.; et al. Granulocyte maturation determines ability to release chromatin NETs and loss of DNA damage response; these properties are absent in immature AML granulocytes. Biochim. Biophys. Acta 2013, 1833, 767–779. [Google Scholar] [CrossRef] [Green Version]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Dietlein, F.; Thelen, L.; Reinhardt, H.C. Cancer-specific defects in DNA repair pathways as targets for personalized therapeutic approaches. Trends Genet. 2014, 30, 326–339. [Google Scholar] [CrossRef]
- Ma, J.; Setton, J.; Lee, N.Y.; Riaz, N.; Powell, S.N. The therapeutic significance of mutational signatures from DNA repair deficiency in cancer. Nat. Commun. 2018, 9, 1–12. [Google Scholar] [CrossRef]
- Delabaere, L.; Ertl, H.A.; Massey, D.J.; Hofley, C.M.; Sohail, F.; Bienenstock, E.J.; Sebastian, H.; Chiolo, I.; LaRocque, J.R. Aging impairs double-strand break repair by homologous recombination in Drosophila germ cells. Aging Cell 2017, 16, 320–328. [Google Scholar] [CrossRef]
- Bristow, R.G.; Hill, R.P. Hypoxia, DNA repair and genetic instability. Nat. Rev. Cancer 2008, 8, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Carreau, A.; Hafny-Rahbi, B.E.; Matejuk, A.; Grillon, C.; Kieda, C. Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 2011, 15, 1239–1253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagiwara, Y.; Oike, T.; Niimi, A.; Yamauchi, M.; Sato, H.; Limsirichaikul, S.; Held, K.D.; Nakano, T.; Shibata, A. Clustered DNA double-strand break formation and the repair pathway following heavy-ion irradiation. J. Radiat. Res. 2019, 60, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogueira, A.; Fernandes, M.; Catarino, R.; Medeiros, R. RAD52 Functions in Homologous Recombination and Its Importance on Genomic Integrity Maintenance and Cancer Therapy. Cancers 2019, 11, 1622. [Google Scholar] [CrossRef] [Green Version]
- Löbrich, M.; Jeggo, P. A Process of Resection-Dependent Nonhomologous End Joining Involving the Goddess Artemis. Trends Biochem. Sci. 2017, 42, 690–701. [Google Scholar] [CrossRef] [Green Version]
- Kakarougkas, A.; Jeggo, P.A. DNA DSB repair pathway choice: An orchestrated handover mechanism. Br. J. Radiol. 2014, 87, 20130685. [Google Scholar] [CrossRef]
- Bader, A.S.; Hawley, B.R.; Wilczynska, A.; Bushell, M. The roles of RNA in DNA double-strand break repair. Br. J. Cancer 2020, 122, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Ingram, S.P.; Warmenhoven, J.W.; Henthorn, N.T.; Smith, E.A.K.; Chadwick, A.L.; Burnet, N.G.; Mackay, R.I.; Kirkby, N.F.; Kirkby, K.J.; Merchant, M.J. Mechanistic modelling supports entwined rather than exclusively competitive DNA double-strand break repair pathway. Sci. Rep. 2019, 9, 6359. [Google Scholar] [CrossRef]
- Jezkova, L.; Zadneprianetc, M.; Kulikova, E.; Smirnova, E.; Bulanova, T.; Depes, D.; Falkova, I.; Boreyko, A.; Krasavin, E.; Davidkova, M.; et al. Particles with similar LET values generate DNA breaks of different complexity and reparability: A high-resolution microscopy analysis of γH2AX/53BP1 foci. Nanoscale 2018, 10, 1162–1179. [Google Scholar] [CrossRef] [Green Version]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [Green Version]
- Cruz, G.A.S. Microdosimetry: Principles and applications. Rep. Pract. Oncol. Radiother. 2016, 21, 135–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, W.; Li, W.B.; Friedland, W.; Miller, B.W.; Madas, B.; Bardiès, M.; Balásházy, I. Internal microdosimetry of alpha-emitting radionuclides. Radiat. Environ. Biophys. 2020, 59, 29–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozubek, S.; Lukásová, E.; Jirsová, P.; Koutná, I.; Kozubek, M.; Ganová, A.; Bártová, E.; Falk, M.; Paseková, R. 3D Structure of the human genome: Order in randomness. Chromosoma 2002, 111, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Cremer, T.; Cremer, M.; Hübner, B.; Strickfaden, H.; Smeets, D.; Popken, J.; Sterr, M.; Markaki, Y.; Rippe, K.; Cremer, C. The 4D nucleome: Evidence for a dynamic nuclear landscape based on co-aligned active and inactive nuclear compartments. FEBS Lett. 2015, 589, 2931–2943. [Google Scholar] [CrossRef] [Green Version]
- Lukásová, E.; Kozubek, S.; Kozubek, M.; Falk, M.; Amrichová, J. The 3D structure of human chromosomes in cell nuclei. Chromosome Res. 2002, 10, 535–548. [Google Scholar] [CrossRef]
- Crosetto, N.; Bienko, M. Radial Organization in the Mammalian Nucleus. Front. Genet. 2020, 11, 33. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukasova, E.; Kozubek, S. Higher-order chromatin structure in DSB induction, repair and misrepair. Mutat. Res. 2010, 704, 88–100. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falková, I.; Ježková, L.; et al. Determining Omics spatiotemporal dimensions using exciting new nanoscopy techniques to assess complex cell responses to DNA damage: Part A—Radiomics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 205–223. [Google Scholar] [CrossRef]
- Falk, M.; Hausmann, M.; Lukášová, E.; Biswas, A.; Hildenbrand, G.; Davídková, M.; Krasavin, E.; Kleibl, Z.; Falková, I.; Ježková, L.; et al. Determining Omics spatiotemporal dimensions using exciting new nanoscopy techniques to assess complex cell responses to DNA damage: Part B—Structuromics. Crit. Rev. Eukaryot. Gene Expr. 2014, 24, 225–247. [Google Scholar] [CrossRef]
- Zhao, L.; Bao, C.; Shang, Y.; He, X.; Ma, C.; Lei, X.; Mi, D.; Sun, Y. The Determinant of DNA Repair Pathway Choices in Ionising Radiation-Induced DNA Double-Strand Breaks. Biomed Res. Int. 2020, 2020, 1–12. [Google Scholar] [CrossRef]
- Ruebe, C.E.; Lorat, Y.; Schanz, S.; Schuler, N.; Ruebe, C. DNA Double-strand Break Repair in the Context of Chromatin. Int. J. Radiat. Oncol. Biol. Phys. 2011, 81, 23. [Google Scholar] [CrossRef]
- Shibata, A. Regulation of repair pathway choice at two-ended DNA double-strand breaks. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2017, 803–805, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Clouaire, T.; Legube, G. DNA double strand break repair pathway choice: A chromatin based decision? Nucleus 2015, 6, 107–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodarzi, A.A.; Jeggo, P.A. The Heterochromatic Barrier to DNA Double Strand Break Repair: How to Get the Entry Visa. IJMS 2012, 13, 11844–11860. [Google Scholar] [CrossRef] [Green Version]
- Noon, A.T.; Shibata, A.; Rief, N.; Löbrich, M.; Stewart, G.S.; Jeggo, P.A.; Goodarzi, A.A. 53BP1-dependent robust localized KAP-1 phosphorylation is essential for heterochromatic DNA double-strand break repair. Nat. Cell Biol. 2010, 12, 177–184. [Google Scholar] [CrossRef]
- Ježková, L.; Falk, M.; Falková, I.; Davídková, M.; Bačíková, A.; Štefančíková, L.; Vachelová, J.; Michaelidesová, A.; Lukášová, E.; Boreyko, A.; et al. Function of chromatin structure and dynamics in DNA damage, repair and misrepair: γ-rays and protons in action. Appl. Radiat. Isot. 2014, 83 Pt B, 128–136. [Google Scholar] [CrossRef]
- Lemaître, C.; Grabarz, A.; Tsouroula, K.; Andronov, L.; Furst, A.; Pankotai, T.; Heyer, V.; Rogier, M.; Attwood, K.M.; Kessler, P.; et al. Nuclear position dictates DNA repair pathway choice. Genes Dev. 2014, 28, 2450–2463. [Google Scholar] [CrossRef] [Green Version]
- Her, J.; Bunting, S.F. How cells ensure correct repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10502–10511. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813.e6. [Google Scholar] [CrossRef] [Green Version]
- Cannan, W.J.; Pederson, D.S. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin: Double-Strand Dna Break Formation in Chromatin. J. Cell. Physiol. 2016, 231, 3–14. [Google Scholar] [CrossRef] [Green Version]
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavragani, I.V.; Nikitaki, Z.; Kalospyros, S.A.; Georgakilas, A.G. Ionizing Radiation and Complex DNA Damage: From Prediction to Detection Challenges and Biological Significance. Cancers 2019, 11, 1789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipov, A.N.; Grekhova, A.; Pustovalova, M.; Ozerov, I.V.; Eremin, P.; Vorobyeva, N.; Lazareva, N.; Pulin, A.; Zhavoronkov, A.; Roumiantsev, S.; et al. Activation of homologous recombination DNA repair in human skin fibroblasts continuously exposed to X-ray radiation. Oncotarget 2015, 6, 26876–26885. [Google Scholar] [CrossRef] [PubMed]
- Somaiah, N.; Yarnold, J.; Lagerqvist, A.; Rothkamm, K.; Helleday, T. Homologous recombination mediates cellular resistance and fraction size sensitivity to radiation therapy. Radiother. Oncol. 2013, 108, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Salles, B. Inhibition of the non homologous end joining process in the context of hypoxic tumor cells. Transl. Cancer Res. 2013, 2, 215–226. [Google Scholar]
- Cowman, S.; Pizer, B.; Sée, V. Downregulation of both mismatch repair and non-homologous end-joining pathways in hypoxic brain tumour cell lines. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Redon, C.E.; Dickey, J.S.; Bonner, W.M.; Sedelnikova, O.A. γ-H2AX as a biomarker of DNA damage induced by ionizing radiation in human peripheral blood lymphocytes and artificial skin. Adv. Space Res. 2009, 43, 1171–1178. [Google Scholar] [CrossRef] [Green Version]
- Rogakou, E.P.; Boon, C.; Redon, C.; Bonner, W.M. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J. Cell Biol. 1999, 146, 905–916. [Google Scholar] [CrossRef] [Green Version]
- Paull, T.T. Mechanisms of ATM Activation. Annu. Rev. Biochem. 2015, 84, 711–738. [Google Scholar] [CrossRef]
- Podhorecka, M.; Skladanowski, A.; Bozko, P. H2AX Phosphorylation: Its Role in DNA Damage Response and Cancer Therapy. J. Nucleic Acids 2010, 2010, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bekker-Jensen, S.; Mailand, N. Assembly and function of DNA double-strand break repair foci in mammalian cells. DNA Repair 2010, 9, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Vítor, A.C.; Huertas, P.; Legube, G.; de Almeida, S.F. Studying DNA Double-Strand Break Repair: An Ever-Growing Toolbox. Front. Mol. Biosci. 2020, 7, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, M.; Falk, M.; Komůrková, D.; Falková, I.; Bačíková, A.; Klejdus, B.; Pagáčová, E.; Štefančíková, L.; Weiterová, L.; Angelis, K.J.; et al. Two New Faces of Amifostine: Protector from DNA Damage in Normal Cells and Inhibitor of DNA Repair in Cancer Cells. J. Med. Chem. 2016, 59, 3003–3017. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Lukasova, E.; Gabrielova, B.; Ondrej, V.; Kozubek, S. Chromatin dynamics during DSB repair. Biochim. Biophys. Acta 2007, 1773, 1534–1545. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukásová, E.; Kozubek, S. Chromatin structure influences the sensitivity of DNA to gamma-radiation. Biochim. Biophys. Acta 2008, 1783, 2398–2414. [Google Scholar] [CrossRef] [Green Version]
- Falk, M.; Lukášová, E.; Štefančíková, L.; Baranová, E.; Falková, I.; Ježková, L.; Davídková, M.; Bačíková, A.; Vachelová, J.; Michaelidesová, A.; et al. Heterochromatinization associated with cell differentiation as a model to study DNA double strand break induction and repair in the context of higher-order chromatin structure. Appl. Radiat. Isot. 2014, 83, 177–185. [Google Scholar] [CrossRef]
- Belyaev, I.Y. Radiation-induced DNA repair foci: Spatio-temporal aspects of formation, application for assessment of radiosensitivity and biological dosimetry. Mutat. Res. Rev. Mutat. Res. 2010, 704, 132–141. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.A. Irradiation induced foci (IRIF) as a biomarker for radiosensitivity. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2012, 736, 39–47. [Google Scholar] [CrossRef]
- Rothkamm, K.; Barnard, S.; Moquet, J.; Ellender, M.; Rana, Z.; Burdak-Rothkamm, S. DNA damage foci: Meaning and significance. Environ. Mol. Mutagen. 2015, 56, 491–504. [Google Scholar] [CrossRef]
- Tonnemacher, S.; Eltsov, M.; Jakob, B. Correlative Light and Electron Microscopy (CLEM) Analysis of Nuclear Reorganization Induced by Clustered DNA Damage Upon Charged Particle Irradiation. IJMS 2020, 21, 1911. [Google Scholar] [CrossRef] [Green Version]
- Entrz Pubmed Database. Available online: https://pubmed.ncbi.nlm.nih.gov (accessed on 7 November 2020).
- Falk, M.; Hausmann, M. Advances in research of DNA damage and repair in cells exposed to various types of ionizing radiation in the era of super-resolution optical microscopy. Cas Lek Cesk 2020, 159, 7–8, (pages in press). [Google Scholar]
- Fabre, E.; Zimmer, C. From dynamic chromatin architecture to DNA damage repair and back. Nucleus 2018, 9, 161–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnould, C.; Legube, G. The Secret Life of Chromosome Loops upon DNA Double-Strand Break. J. Mol. Biol. 2020, 432, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Downs, J.A. Roles of chromatin remodellers in DNA double strand break repair. Exp. Cell Res. 2014, 329, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Watts, F. Repair of DNA Double-Strand Breaks in Heterochromatin. Biomolecules 2016, 6, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caron, H.; van Schaik, B.; van der Mee, M.; Baas, F.; Riggins, G.; van Sluis, P.; Hermus, M.C.; van Asperen, R.; Boon, K.; Voûte, P.A.; et al. The human transcriptome map: Clustering of highly expressed genes in chromosomal domains. Science 2001, 291, 1289–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Versteeg, R. The Human Transcriptome Map Reveals Extremes in Gene Density, Intron Length, GC Content, and Repeat Pattern for Domains of Highly and Weakly Expressed Genes. Genome Res. 2003, 13, 1998–2004. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, A.A.; Noon, A.T.; Jeggo, P.A. The impact of heterochromatin on DSB repair. Biochem. Soc. Trans. 2009, 37, 569–576. [Google Scholar] [CrossRef]
- Goodarzi, A.A.; Jeggo, P.; Lobrich, M. The influence of heterochromatin on DNA double strand break repair: Getting the strong, silent type to relax. DNA Repair 2010, 9, 1273–1282. [Google Scholar] [CrossRef]
- Baldock, R.A.; Day, M.; Wilkinson, O.J.; Cloney, R.; Jeggo, P.A.; Oliver, A.W.; Watts, F.Z.; Pearl, L.H. ATM Localization and Heterochromatin Repair Depend on Direct Interaction of the 53BP1-BRCT2 Domain with γH2AX. Cell Rep. 2015, 13, 2081–2089. [Google Scholar] [CrossRef] [Green Version]
- Goodarzi, A.A.; Noon, A.T.; Deckbar, D.; Ziv, Y.; Shiloh, Y.; Löbrich, M.; Jeggo, P.A. ATM signaling facilitates repair of DNA double-strand breaks associated with heterochromatin. Mol. Cell 2008, 31, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Fortuny, A.; Polo, S.E. The response to DNA damage in heterochromatin domains. Chromosoma 2018, 127, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Chapuis, C.; Huet, S.; Ellenberg, J. Crowded chromatin is not sufficient for heterochromatin formation and not required for its maintenance. J. Struct. Biol. 2013, 184, 445–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jakob, B.; Splinter, J.; Conrad, S.; Voss, K.-O.; Zink, D.; Durante, M.; Löbrich, M.; Taucher-Scholz, G. DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 2011, 39, 6489–6499. [Google Scholar] [CrossRef] [PubMed]
- Svetličič, M.; Bomhard, A.; Sterr, C.; Brückner, F.; Płódowska, M.; Lisowska, H.; Lundholm, L. Alpha Radiation as a Way to Target Heterochromatic and Gamma Radiation-Exposed Breast Cancer Cells. Cells 2020, 9, 1165. [Google Scholar] [CrossRef]
- Yu, S.; Yang, F.; Shen, W.H. Genome maintenance in the context of 4D chromatin condensation. Cell. Mol. Life Sci. 2016, 73, 3137–3150. [Google Scholar] [CrossRef] [Green Version]
- Chiolo, I.; Minoda, A.; Colmenares, S.U.; Polyzos, A.; Costes, S.V.; Karpen, G.H. Double-Strand Breaks in Heterochromatin Move Outside of a Dynamic HP1a Domain to Complete Recombinational Repair. Cell 2011, 144, 732–744. [Google Scholar] [CrossRef] [Green Version]
- Luijsterburg, M.S.; van Attikum, H. Close encounters of the RNF8th kind: When chromatin meets DNA repair. Curr. Opin. Cell Biol. 2012, 24, 439–447. [Google Scholar] [CrossRef]
- Amaral, N.; Ryu, T.; Li, X.; Chiolo, I. Nuclear Dynamics of Heterochromatin Repair. Trends Genet. 2017, 33, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Foltánková, V.; Legartová, S.; Kozubek, S.; Hofer, M.; Bártová, E. DNA-damage response in chromatin of ribosomal genes and the surrounding genome. Gene 2013, 522, 156–167. [Google Scholar] [CrossRef]
- Lorat, Y.; Timm, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered double-strand breaks in heterochromatin perturb DNA repair after high linear energy transfer irradiation. Radiother. Oncol. 2016, 121, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Timm, S.; Lorat, Y.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Clustered DNA damage concentrated in particle trajectories causes persistent large-scale rearrangements in chromatin architecture. Radiother. Oncol. 2018, 129, 600–610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Heermann, D.W. DNA double-strand breaks: Linking gene expression to chromosome morphology and mobility. Chromosoma 2014, 123, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Hausmann, M.; Wagner, E.; Lee, J.-H.; Schrock, G.; Schaufler, W.; Krufczik, M.; Papenfuß, F.; Port, M.; Bestvater, F.; Scherthan, H. Super-resolution localization microscopy of radiation-induced histone H2AX-phosphorylation in relation to H3K9-trimethylation in HeLa cells. Nanoscale 2018, 10, 4320–4331. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Máté, G.; Müller, P.; Hillebrandt, S.; Krufczik, M.; Bach, M.; Kaufmann, R.; Hausmann, M.; Heermann, D.W. Radiation induced chromatin conformation changes analysed by fluorescent localization microscopy, statistical physics, and graph theory. PLoS ONE 2015, 10, e0128555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aymard, F.; Bugler, B.; Schmidt, C.K.; Guillou, E.; Caron, P.; Briois, S.; Iacovoni, J.S.; Daburon, V.; Miller, K.M.; Jackson, S.P.; et al. Transcriptionally active chromatin recruits homologous recombination at DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 366–374. [Google Scholar] [CrossRef] [Green Version]
- Aymard, F.; Aguirrebengoa, M.; Guillou, E.; Javierre, B.M.; Bugler, B.; Arnould, C.; Rocher, V.; Iacovoni, J.S.; Biernacka, A.; Skrzypczak, M.; et al. Genome-wide mapping of long-range contacts unveils clustering of DNA double-strand breaks at damaged active genes. Nat. Struct. Mol. Biol. 2017, 24, 353–361. [Google Scholar] [CrossRef]
- Deckbar, D.; Jeggo, P.A.; Löbrich, M. Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 271–283. [Google Scholar] [CrossRef] [Green Version]
- Chao, H.X.; Poovey, C.E.; Privette, A.A.; Grant, G.D.; Chao, H.Y.; Cook, J.G.; Purvis, J.E. Orchestration of DNA Damage Checkpoint Dynamics across the Human Cell Cycle. Cell Syst. 2017, 5, 445–459.e5. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.; Tapryal, N.; Venkova, T.; Horikoshi, N.; Pandita, R.K.; Sarker, A.H.; Sarkar, P.S.; Pandita, T.K.; Hazra, T.K. Classical non-homologous end-joining pathway utilizes nascent RNA for error-free double-strand break repair of transcribed genes. Nat. Commun. 2016, 7, 13049. [Google Scholar] [CrossRef] [Green Version]
- Domingo-Prim, J.; Bonath, F.; Visa, N. RNA at DNA Double-Strand Breaks: The Challenge of Dealing with DNA: RNA Hybrids. BioEssays 2020, 42, 1900225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monajembashi, S.; Rapp, A.; Schmitt, E.; Dittmar, H.; Greulich, K.-O.; Hausmann, M. Spatial Association of Homologous Pericentric Regions in Human Lymphocyte Nuclei during Repair. Biophys. J. 2005, 88, 2309–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen, A.; Colmenares, S.U.; Lee, T.; Karpen, G.H. Timely double-strand break repair and pathway choice in pericentromeric heterochromatin depend on the histone demethylase dKDM4A. Genes Dev. 2019, 33, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyodorov, D.V.; Zhou, B.-R.; Skoultchi, A.I.; Bai, Y. Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Falk, M.; Falková, I.; Kopečná, O.; Bačíková, A.; Pagáčová, E.; Šimek, D.; Golan, M.; Kozubek, S.; Pekarová, M.; Follett, S.E.; et al. Chromatin architecture changes and DNA replication fork collapse are critical features in cryopreserved cells that are differentially controlled by cryoprotectants. Sci. Rep. 2018, 8, 14694. [Google Scholar] [CrossRef] [Green Version]
- Caron, P.; van der Linden, J.; van Attikum, H. Bon voyage: A transcriptional journey around DNA breaks. DNA Repair 2019, 82, 102686. [Google Scholar] [CrossRef]
- Kalousi, A.; Soutoglou, E. Nuclear compartmentalization of DNA repair. Curr. Opin. Genet. Dev. 2016, 37, 148–157. [Google Scholar] [CrossRef]
- Bobkova, E.; Depes, D.; Lee, J.-H.; Jezkova, L.; Falkova, I.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; Kulikova, E.; et al. Recruitment of 53BP1 Proteins for DNA Repair and Persistence of Repair Clusters Differ for Cell Types as Detected by Single Molecule Localization Microscopy. Int. J. Mol. Sci. 2018, 19, 3713. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, B.; Friedl, A.A.; Girst, S.; Dollinger, G.; Reindl, J. Nanoscopic analysis of 53BP1, BRCA1 and Rad51 reveals new insights in temporal progression of DNA-repair and pathway choice. Mutat. Res. 2019, 816–818, 111675. [Google Scholar] [CrossRef]
- Jakob, B.; Splinter, J.; Durante, M.; Taucher-Scholz, G. Live cell microscopy analysis of radiation-induced DNA double-strand break motion. Proc. Natl. Acad. Sci. USA 2009, 106, 3172–3177. [Google Scholar] [CrossRef] [Green Version]
- Costes, S.V.; Ponomarev, A.; Chen, J.L.; Nguyen, D.; Cucinotta, F.A.; Barcellos-Hoff, M.H. Image-based modeling reveals dynamic redistribution of DNA damage into nuclear sub-domains. PLoS Comput. Biol. 2007, 3, e155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sundaravinayagam, D.; Rahjouei, A.; Andreani, M.; Tupiņa, D.; Balasubramanian, S.; Saha, T.; Delgado-Benito, V.; Coralluzzo, V.; Daumke, O.; Di Virgilio, M. 53BP1 Supports Immunoglobulin Class Switch Recombination Independently of Its DNA Double-Strand Break End Protection Function. Cell Rep. 2019, 28, 1389–1399.e6. [Google Scholar] [CrossRef] [PubMed]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhász, S.; Künzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell 2017, 65, 671–684.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kallimasioti-Pazi, E.M.; Chathoth, K.T.; Taylor, G.C.; Meynert, A.; Ballinger, T.; Kelder, M.J.E.; Lalevée, S.; Sanli, I.; Feil, R.; Wood, A.J. Heterochromatin delays CRISPR-Cas9 mutagenesis but does not influence the outcome of mutagenic DNA repair. PLoS Biol. 2018, 16, e2005595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stadler, J.; Richly, H. Regulation of DNA Repair Mechanisms: How the Chromatin Environment Regulates the DNA Damage Response. Int. J. Mol. Sci. 2017, 18, 1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Lan, L.; Hong, Z.; Yasui, A.; Ishioka, C.; Chiba, N. Rapid recruitment of BRCA1 to DNA double-strand breaks is dependent on its association with Ku80. Mol. Cell. Biol. 2008, 28, 7380–7393. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T.; Soutoglou, E. The emerging role of nuclear architecture in DNA repair and genome maintenance. Nat. Rev. Mol. Cell Biol. 2009, 10, 243–254. [Google Scholar] [CrossRef]
- Groth, A.; Rocha, W.; Verreault, A.; Almouzni, G. Chromatin Challenges during DNA Replication and Repair. Cell 2007, 128, 721–733. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.; Oberdoerffer, P. Chromatin dynamics in DNA double-strand break repair. Biochim. Biophys. Acta Gene Regul. Mech. 2012, 1819, 811–819. [Google Scholar] [CrossRef] [Green Version]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Cremer, C.; Birk, U. Perspectives in Super-Resolved Fluorescence Microscopy: What Comes Next? Front. Phys. 2016, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Cremer, C.; Masters, B.R. Resolution enhancement techniques in microscopy. Eur. Phys. J. H 2013, 38, 281–344. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, R.; Lemmer, P.; Gunkel, M.; Weiland, Y.; Müller, P.; Hausmann, M.; Baddeley, D.; Amberger, R.; Cremer, C. SPDM: Single Molecule Superresolution of Cellular Nanostructures. In Single Molecule Spectroscopy and Imaging II, Proceedings of SPIE BiOS, San Jose, CA, USA, 24 February 2009; Enderlein, J., Gryczynski, Z.K., Erdmann, R., Eds.; Proc. SPIE: San Jose, CA, USA, 2009; p. 7185. [Google Scholar] [CrossRef] [Green Version]
- Lemmer, P.; Gunkel, M.; Weiland, Y.; Müller, P.; Baddeley, D.; Kaufmann, R.; Urich, A.; Eipel, H.; Amberger, R.; Hausmann, M.; et al. Using conventional fluorescent markers for far-field fluorescence localization nanoscopy allows resolution in the 10-nm range. J. Microsc. 2009, 235, 163–171. [Google Scholar] [CrossRef]
- Depes, D.; Lee, J.-H.; Bobkova, E.; Jezkova, L.; Falkova, I.; Bestvater, F.; Pagacova, E.; Kopecna, O.; Zadneprianetc, M.; Bacikova, A.; et al. Single-molecule localization microscopy as a promising tool for γH2AX/53BP1 foci exploration. Eur. Phys. J. 2018, 72, e2018–e90148. [Google Scholar] [CrossRef]
- Hausmann, M.; Ilić, N.; Pilarczyk, G.; Lee, J.-H.; Logeswaran, A.; Borroni, A.P.; Krufczik, M.; Theda, F.; Waltrich, N.; Bestvater, F.; et al. Challenges for super-resolution localization microscopy and biomolecular fluorescent nano-probing in cancer research. Int. J. Mol. Sci. 2017, 18, 2066. [Google Scholar] [CrossRef] [Green Version]
- Cremer, C.; Kaufmann, R.; Gunkel, M.; Pres, S.; Weiland, Y.; Müller, P.; Ruckelshausen, T.; Lemmer, P.; Geiger, F.; Degenhard, S.; et al. Superresolution imaging of biological nanostructures by spectral precision distance microscopy. Biotechnol. J. 2011, 6, 1037–1051. [Google Scholar] [CrossRef]
- Scherthan, H.; Lee, J.-H.; Maus, E.; Schumann, S.; Muhtadi, R.; Chojowski, R.; Port, M.; Lassmann, M.; Bestvater, F.; Hausmann, M. Nanostructure of Clustered DNA Damage in Leukocytes after In-Solution Irradiation with the Alpha Emitter Ra-223. Cancers 2019, 11, 1877. [Google Scholar] [CrossRef] [Green Version]
- Natale, F.; Rapp, A.; Yu, W.; Maiser, A.; Harz, H.; Scholl, A.; Grulich, S.; Anton, T.; Hörl, D.; Chen, W.; et al. Identification of the elementary structural units of the DNA damage response. Nat. Commun. 2017, 8, 15760. [Google Scholar] [CrossRef]
- Ripley, B.D. Modelling Spatial Patterns. J. R. Stat. Soc. Ser. B 1977, 39, 172–192. [Google Scholar] [CrossRef]
- Hausmann, M.; Neitzel, C.; Bobkova, E.; Nagel, D.; Hofmann, A.; Chramko, T.; Smirnova, E.; Kopečná, O.; Pagáčová, E.; Boreyko, A.; et al. Single Molecule Localization Microscopy Analyses of DNA-Repair Foci and Clusters Detected along Particle Damage Tracks. Front. Phys. Sect. Med. Phys. Imaging 2020, 8, 473. [Google Scholar]
- Hofmann, A.; Krufczik, M.; Heermann, D.W.; Hausmann, M. Using Persistent Homology as a New Approach for Super-Resolution Localization Microscopy Data Analysis and Classification of γH2AX Foci/Clusters. Int. J. Mol. Sci. 2018, 19, 2263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iacovoni, J.S.; Caron, P.; Lassadi, I.; Nicolas, E.; Massip, L.; Trouche, D.; Legube, G. High-resolution profiling of γH2AX around DNA double strand breaks in the mammalian genome. EMBO J. 2010, 29, 1446–1457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnould, C.; Rocher, V.; Clouaire, T.; Caron, P.; Philippe, E.M.; Emiliano, P.R.; Mourad, R.; Noordermeer, D.; Legube, G. Loop extrusion as a mechanism for DNA Double-Strand Breaks repair foci formation. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, M.; de Lange, T. 53BP1: Prochoice in DNA repair. Trends Cell Biol. 2014, 24, 108–117. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Eckelmann, B.; Adhikari, S.; Ahmed, K.M.; Sengupta, S.; Pandey, A.; Hegde, P.M.; Tsai, M.-S.; Tainer, J.A.; Weinfeld, M.; et al. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017, 45, 1262. [Google Scholar] [CrossRef] [Green Version]
- Ochs, F.; Karemore, G.; Miron, E.; Brown, J.; Sedlackova, H.; Rask, M.-B.; Lampe, M.; Buckle, V.; Schermelleh, L.; Lukas, J.; et al. Stabilization of chromatin topology safeguards genome integrity. Nature 2019, 574, 571–574. [Google Scholar] [CrossRef]
- Bártová, E.; Legartová, S.; Dundr, M.; Suchánková, J. A role of the 53BP1 protein in genome protection: Structural and functional characteristics of 53BP1-dependent DNA repair. Aging 2019, 11, 2488–2511. [Google Scholar] [CrossRef]
- Reindl, J.; Drexler, G.A.; Girst, S.; Greubel, C.; Siebenwirth, C.; Drexler, S.E.; Dollinger, G.; Friedl, A.A. Nanoscopic exclusion between Rad51 and 53BP1 after ion irradiation in human HeLa cells. Phys. Biol. 2015, 12, 066005. [Google Scholar] [CrossRef]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780. [Google Scholar] [CrossRef]
- Varga, D.; Majoros, H.; Ujfaludi, Z.; Erdélyi, M.; Pankotai, T. Quantification of DNA damage induced repair focus formation via super-resolution dSTORM localization microscopy. Nanoscale 2019, 11, 14226–14236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Wang, Y. Heavier Ions with a Different Linear Energy Transfer Spectrum Kill More Cells Due to Similar Interference with the Ku-Dependent DNA Repair Pathway. Radiat. Res. 2014, 182, 458–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, M.; Park, D.; You, S.; Li, R.; Owonikoko, T.K.; Wang, Y.; Doetsch, P.W.; Deng, X. Bcl2 inhibits recruitment of Mre11 complex to DNA double-strand breaks in response to high-linear energy transfer radiation. Nucleic Acids Res. 2015, 43, 960–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roobol, S.J.; van den Bent, I.; van Cappellen, W.A.; Abraham, T.E.; Paul, M.W.; Kanaar, R.; Houtsmuller, A.B.; van Gent, D.C.; Essers, J. Comparison of High- and Low-LET Radiation-Induced DNA Double-Strand Break Processing in Living Cells. Int. J. Mol. Sci. 2020, 21, 6602. [Google Scholar] [CrossRef]
- Scuric, Z.; Chan, C.Y.; Hafer, K.; Schiestl, R.H. Ionizing Radiation Induces Microhomology-Mediated End Joining in trans in Yeast and Mammalian Cells. Radiat. Res. 2009, 171, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Löbrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase: DSB repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Jeggo, P.A.; Löbrich, M. DNA non-homologous end-joining enters the resection arena. Oncotarget 2017, 8, 93317–93318. [Google Scholar] [CrossRef]
- Advances in Radiation Therapy; Guckenberger, M.; Combs, S.E.; Zips, D. (Eds.) Progress in Tumor Research; Karger: Basel, Switzerland; New York, NY, USA, 2018; ISBN 978-3-318-06361-5. [Google Scholar]
- Lorat, Y.; Brunner, C.U.; Schanz, S.; Jakob, B.; Taucher-Scholz, G.; Rübe, C.E. Nanoscale analysis of clustered DNA damage after high-LET irradiation by quantitative electron microscopy–The heavy burden to repair. DNA Repair 2015, 28, 93–106. [Google Scholar] [CrossRef]
- Tang, N.; Bueno, M.; Meylan, S.; Incerti, S.; Tran, H.N.; Vaurijoux, A.; Gruel, G.; Villagrasa, C. Influence of chromatin compaction on simulated early radiation-induced DNA damage using Geant4-DNA. Med. Phys. 2019, 46, 1501–1511. [Google Scholar] [CrossRef] [Green Version]
- Shibata, A.; Jeggo, P.A. Canonical DNA non-homologous end-joining; capacity versus fidelity. Br. J. Radiol. 2020, 2019, 966. [Google Scholar] [CrossRef]
- Iliakis, G. The Biological Foundations of Risks from Ionizing Radiation Exposures: How an Understanding of Associated Effects Will Help Their Quantification and Mitigation. In Sustainable Risk Management; Springer: Cham, Switzerland, 2018; pp. 149–158. [Google Scholar]
- Cremer, T.; Cremer, M.; Hübner, B.; Silahtaroglu, A.; Hendzel, M.; Lanctôt, C.; Strickfaden, H.; Cremer, C. The Interchromatin Compartment Participates in the Structural and Functional Organization of the Cell Nucleus. BioEssays 2020, 42, 1900132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendandi, A.; Dante, S.; Zia, S.R.; Diaspro, A.; Rocchia, W. Chromatin Compaction Multiscale Modeling: A Complex Synergy Between Theory, Simulation, and Experiment. Front. Mol. Biosci. 2020, 7, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Steensel, B.; Furlong, E.E.M. The role of transcription in shaping the spatial organization of the genome. Nat. Rev. Mol. Cell Biol. 2019, 20, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Vergara, Z.; Gutierrez, C. Emerging roles of chromatin in the maintenance of genome organization and function in plants. Genome Biol. 2017, 18, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauer, M.H.; Gasser, S.M. Chromatin and nucleosome dynamics in DNA damage and repair. Genes Dev. 2017, 31, 2204–2221. [Google Scholar] [CrossRef] [Green Version]
- Zafar, F.; Okita, A.K.; Onaka, A.T.; Su, J.; Katahira, Y.; Nakayama, J.; Takahashi, T.S.; Masukata, H.; Nakagawa, T. Regulation of mitotic recombination between DNA repeats in centromeres. Nucleic Acids Res. 2017, 45, 11222–11235. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.; Colmenares, S.U.; Karpen, G.H. Heterochromatin: Guardian of the Genome. Annu. Rev. Cell Dev. Biol. 2018, 34, 265–288. [Google Scholar] [CrossRef] [Green Version]
- Allshire, R.C.; Madhani, H.D. Ten principles of heterochromatin formation and function. Nat. Rev. Mol. Cell Biol. 2018, 19, 229–244. [Google Scholar] [CrossRef]
- Jasin, M.; Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 2013, 5, a012740. [Google Scholar] [CrossRef]
- Pagáčová, E.; Falk, M.; Falková, I.; Lukášová, E.; Michalová, K.; Oltová, A.; Raška, I.; Kozubek, S. Frequent chromatin rearrangements in myelodysplastic syndromes–what stands behind? Folia Biol. 2014, 60 (Suppl. 1), 1–7. [Google Scholar]
- Koltsova, A.S.; Pendina, A.A.; Efimova, O.A.; Chiryaeva, O.G.; Kuznetzova, T.V.; Baranov, V.S. On the Complexity of Mechanisms and Consequences of Chromothripsis: An Update. Front. Genet. 2019, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.L.; Sperle, K.; Sternberg, N. Model for homologous recombination during transfer of DNA into mouse L cells: Role for DNA ends in the recombination process. Mol. Cell. Biol. 1984, 4, 1020–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornforth, M.N.; Durante, M. Radiation quality and intra-chromosomal aberrations: Size matters. Mutat. Res. Genet. Toxicol. Environ. Mutagenesis 2018, 836, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Brenner, D.J.; Okladnikova, N.; Hande, P.; Burak, L.; Geard, C.R.; Azizova, T. Biomarkers Specific to Densely-ionising (High LET) Radiations. Radiat. Prot. Dosim. 2001, 97, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Costes, S.V.; Chiolo, I.; Pluth, J.M.; Barcellos-Hoff, M.H.; Jakob, B. Spatiotemporal characterization of ionizing radiation induced DNA damage foci and their relation to chromatin organization. Mutat. Res. Rev. Mutat. Res. 2010, 704, 78–87. [Google Scholar] [CrossRef] [Green Version]
- Banno, K.; Kisu, I.; Yanokura, M.; Tsuji, K.; Masuda, K.; Ueki, A.; Kobayashi, Y.; Yamagami, W.; Nomura, H.; Tominaga, E.; et al. Epimutation and cancer: A new carcinogenic mechanism of Lynch syndrome. Int. J. Oncol. 2012, 41, 793–797. [Google Scholar] [CrossRef] [Green Version]
- Machnik, M.; Oleksiewicz, U. Dynamic Signatures of the Epigenome: Friend or Foe? Cells 2020, 9, 653. [Google Scholar] [CrossRef] [Green Version]
- Lønning, P.E.; Eikesdal, H.P.; Løes, I.M.; Knappskog, S. Constitutional Mosaic Epimutations–a hidden cause of cancer? Cell Stress 2019, 3, 118–135. [Google Scholar] [CrossRef] [Green Version]
- Misteli, T. Higher-order genome organization in human disease. Cold Spring Harb. Perspect. Biol. 2010, 2, a000794. [Google Scholar] [CrossRef] [Green Version]
- Engreitz, J.M.; Agarwala, V.; Mirny, L.A. Three-Dimensional Genome Architecture Influences Partner Selection for Chromosomal Translocations in Human Disease. PLoS ONE 2012, 7, e44196. [Google Scholar] [CrossRef] [Green Version]
- Iarovaia, O.V.; Rubtsov, M.; Ioudinkova, E.; Tsfasman, T.; Razin, S.V.; Vassetzky, Y.S. Dynamics of double strand breaks and chromosomal translocations. Mol. Cancer 2014, 13, 249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chagin, V.O.; Reinhart, B.; Becker, A.; Mortusewicz, O.; Jost, K.L.; Rapp, A.; Leonhardt, H.; Cardoso, M.C. Processive DNA synthesis is associated with localized decompaction of constitutive heterochromatin at the sites of DNA replication and repair. Nucleus 2019, 10, 231–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bach, M.; Savini, C.; Krufczik, M.; Cremer, C.; Rösl, F.; Hausmann, M. Super-Resolution Localization Microscopy of γ-H2AX and Heterochromatin after Folate Deficiency. Int. J. Mol. Sci. 2017, 18, 1726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cann, K.L.; Dellaire, G. Heterochromatin and the DNA damage response: The need to relax. Biochem. Cell Biol. 2011, 89, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Nickoloff, J.A.; De Haro, L.P.; Wray, J.; Hromas, R. Mechanisms of leukemia translocations. Curr. Opin. Hematol. 2008, 15, 338–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozubek, S.; Bártová, E.; Kozubek, M.; Lukásová, E.; Cafourková, A.; Koutná, I.; Skalníková, M. Spatial distribution of selected genetic loci in nuclei of human leukemia cells after irradiation. Radiat. Res. 2001, 155, 311–319. [Google Scholar] [PubMed]
- Kozubek, S.; Lukásová, E.; Marecková, A.; Skalníková, M.; Kozubek, M.; Bártová, E.; Kroha, V.; Krahulcová, E.; Slotová, J. The topological organization of chromosomes 9 and 22 in cell nuclei has a determinative role in the induction of t(9,22) translocations and in the pathogenesis of t(9,22) leukemias. Chromosoma 1999, 108, 426–435. [Google Scholar] [CrossRef]
- Lukášová, E.; Kozubek, S.; Kozubek, M.; Kroha, V.; Marečková, A.; Skalníková, M.; Bártová, E.; Šlotová, J. Chromosomes participating in translocations typical of malignant hemoblastoses are also involved in exchange aberrations induced by fast neutrons. Radiat. Res. 1999, 151, 375–384. [Google Scholar] [CrossRef]
- Nathanailidou, P.; Taraviras, S.; Lygerou, Z. Chromatin and Nuclear Architecture: Shaping DNA Replication in 3D. Trends Genet. 2020, 36, 967–980. [Google Scholar] [CrossRef]
- Meaburn, K.J.; Misteli, T.; Soutoglou, E. Spatial genome organization in the formation of chromosomal translocations. Semin. Cancer Biol. 2007, 17, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Rowley, M.J.; Corces, V.G. Organizational principles of 3D genome architecture. Nat. Rev. Genet. 2018, 19, 789–800. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.D.; Hu, M.; Ren, B. Genome-wide mapping and analysis of chromosome architecture. Nat. Rev. Mol. Cell Biol. 2016, 17, 743–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takata, H.; Hanafusa, T.; Mori, T.; Shimura, M.; Iida, Y.; Ishikawa, K.; Yoshikawa, K.; Yoshikawa, Y.; Maeshima, K. Chromatin Compaction Protects Genomic DNA from Radiation Damage. PLoS ONE 2013, 8, e75622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krigerts, J.; Salmina, K.; Freivalds, T.; Rumnieks, F.; Inashkina, I.; Zayakin, P.; Hausmann, M.; Erenpreisa, J. Early Critical Phase Transitions of Pericentromere-Associated Domains in MCF-7 Breast Cancer Cells Committed to Differentiation by Heregulin. Preprints 2020, 2020050248. [Google Scholar] [CrossRef]
- Williamson, A.K.; Zhu, Z.; Yuan, Z.-M. Epigenetic mechanisms behind cellular sensitivity to DNA damage. CST 2018, 2, 176–180. [Google Scholar] [CrossRef]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R.; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR Substrate Analysis Reveals Extensive Protein Networks Responsive to DNA Damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Hildenbrand, G.; Metzler, P.; Pilarczyk, G.; Bobu, V.; Kriz, W.; Hosser, H.; Fleckenstein, J.; Krufczik, M.; Bestvater, F.; Wenz, F.; et al. Dose enhancement effects of gold nanoparticles specifically targeting RNA in breast cancer cells. PLoS ONE 2018, 13, e0190183. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Repair Pathway | Chromatin Domain Type (Architecture and Function) | |||
|---|---|---|---|---|
| Euchromatin | Heterochromatin | |||
| cNHEJ |
| x | x | x |
| ATM-NHEJ | x | x |
| x |
| HR/RNA-HR | highly precise | highly precise | x | x |
| ATM-HR | x | x |
|
|
| Alternative pathways (A-Ej) |
|
|
|
|
| Low-LET | High-LET | Low-LET | High-LET | |
| Radiation quality | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falk, M.; Hausmann, M. A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers 2021, 13, 18. https://doi.org/10.3390/cancers13010018
Falk M, Hausmann M. A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation? Cancers. 2021; 13(1):18. https://doi.org/10.3390/cancers13010018
Chicago/Turabian StyleFalk, Martin, and Michael Hausmann. 2021. "A Paradigm Revolution or Just Better Resolution—Will Newly Emerging Superresolution Techniques Identify Chromatin Architecture as a Key Factor in Radiation-Induced DNA Damage and Repair Regulation?" Cancers 13, no. 1: 18. https://doi.org/10.3390/cancers13010018