
Structural Changes of Sodium Warfarin in Tablets Affecting the Dissolution Profiles and Potential Safety of Generic Substitution

 ,
,
Abstract
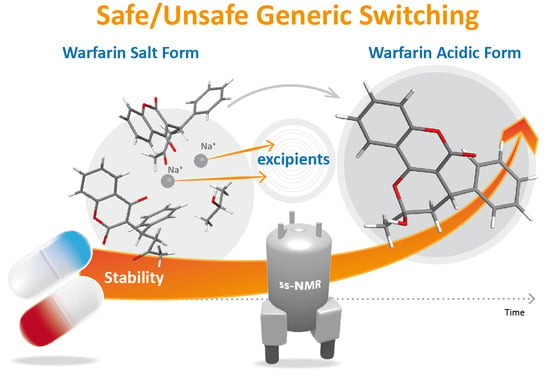
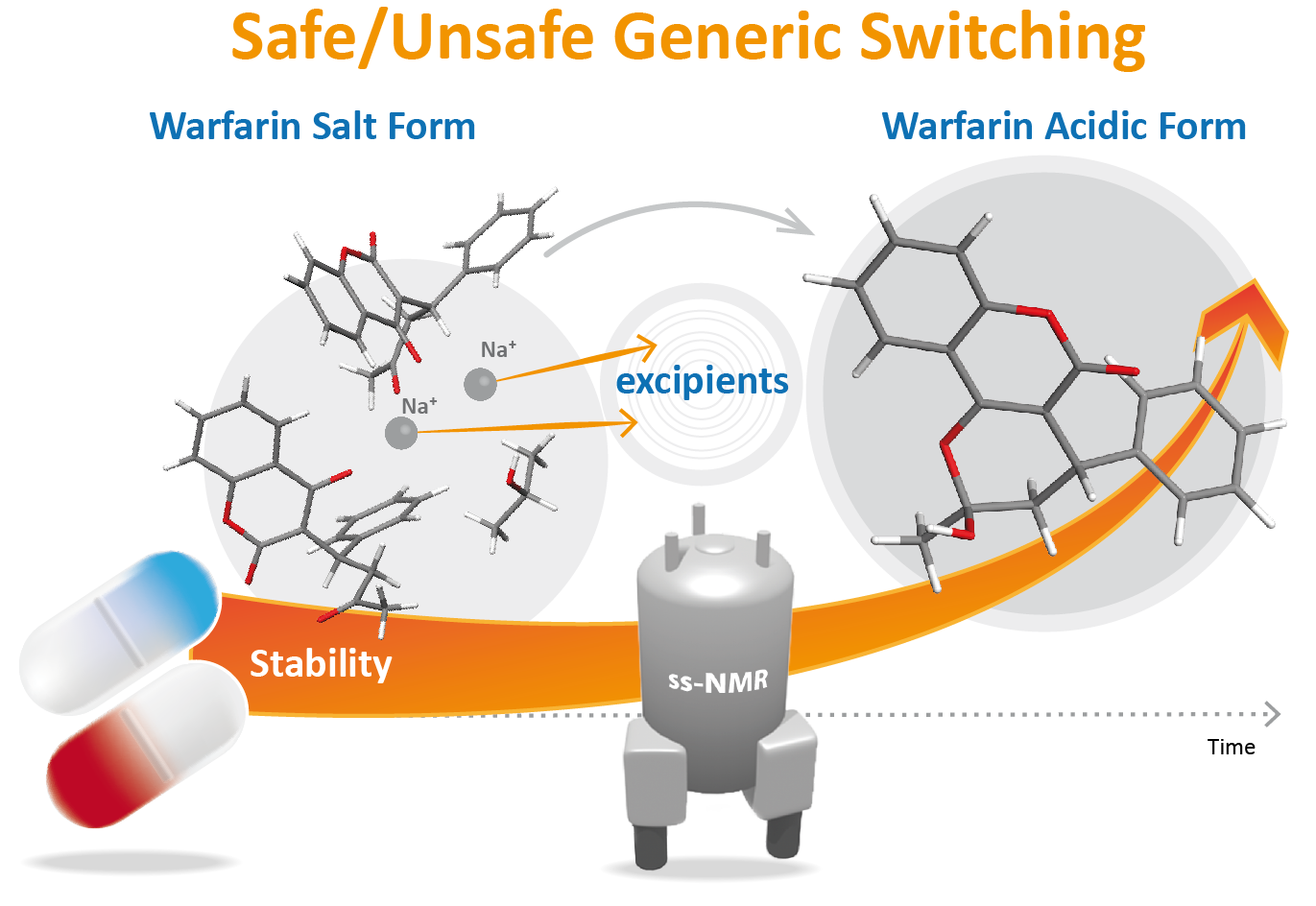
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Preparation of Tablets
2.1.2. Preparation of Binary Mixtures (Warfarin/Excipient)
2.2. Stability Studies
2.3. Physicochemical Testing of Tablets
2.4. In Vitro Drug Release
2.4.1. Dissolution Test of Tablets
2.4.2. Biphasic Dissolution Test of Tablets
2.4.3. Dissolution Test of Non-Ionized Warfarin (Acid Form) with Different Particle Sizes
2.5. Warfarin Solid State Characterization
2.5.1. Solid-State NMR Spectroscopy
2.5.2. Raman Scattering Measurements
2.5.3. Particle Size
3. Results and Discussion
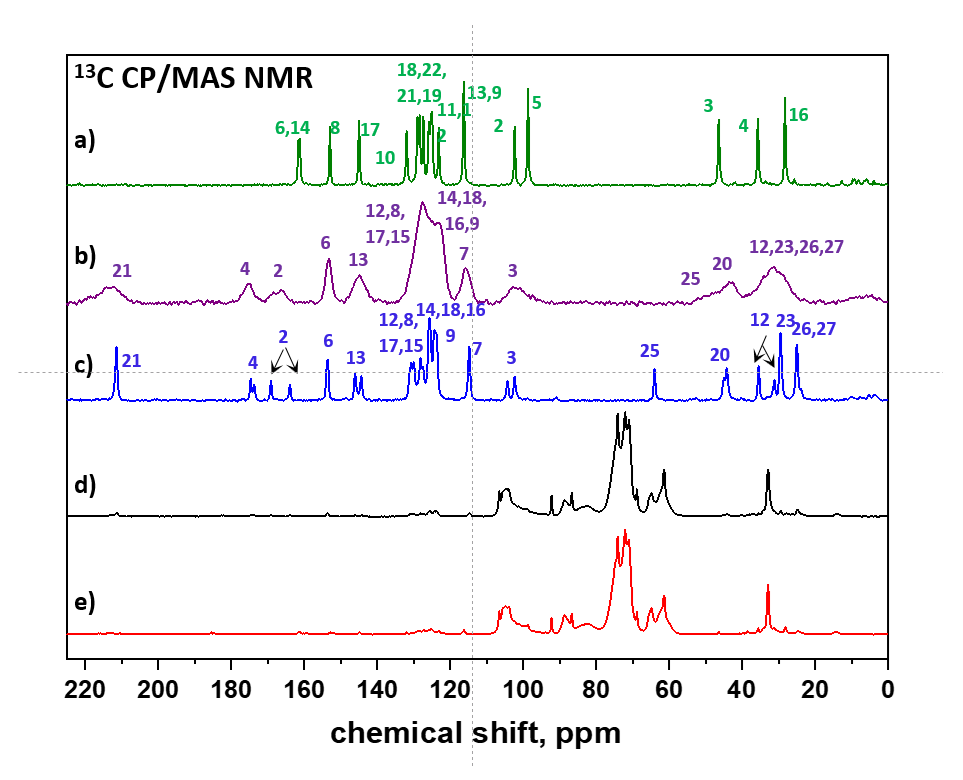
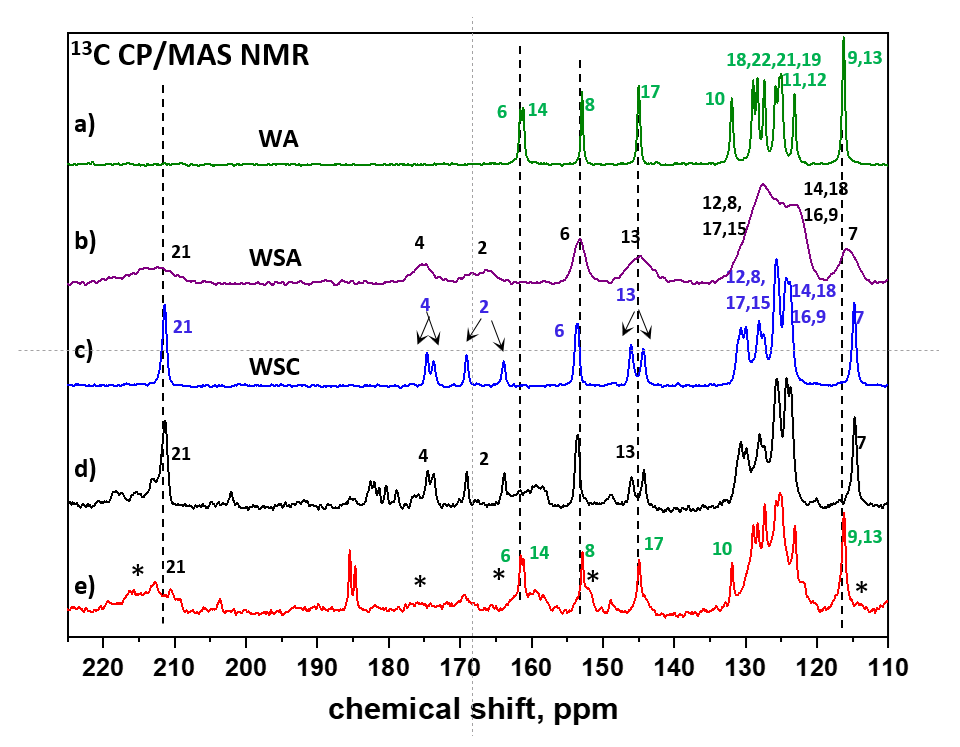
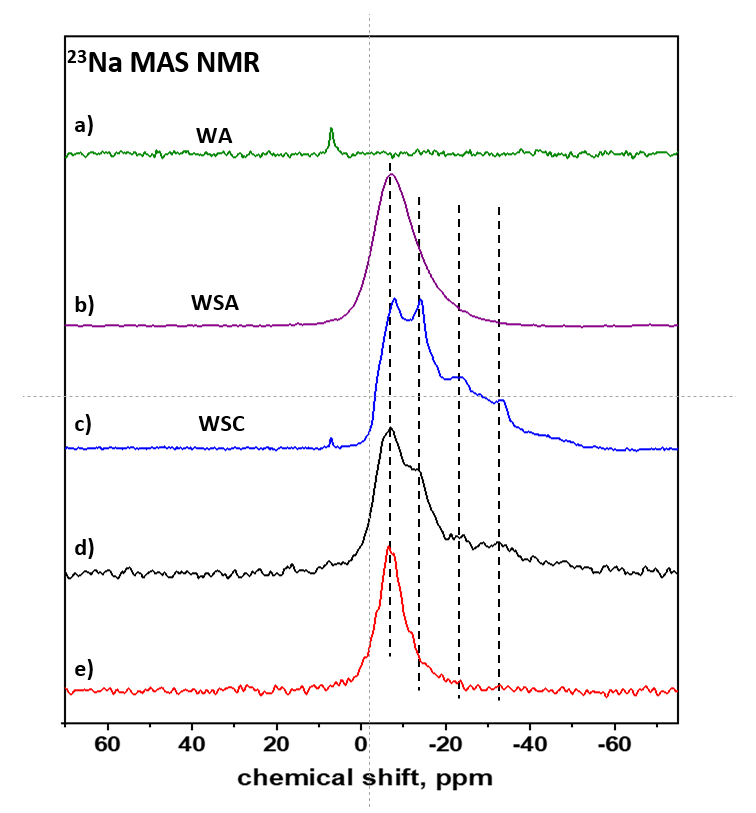
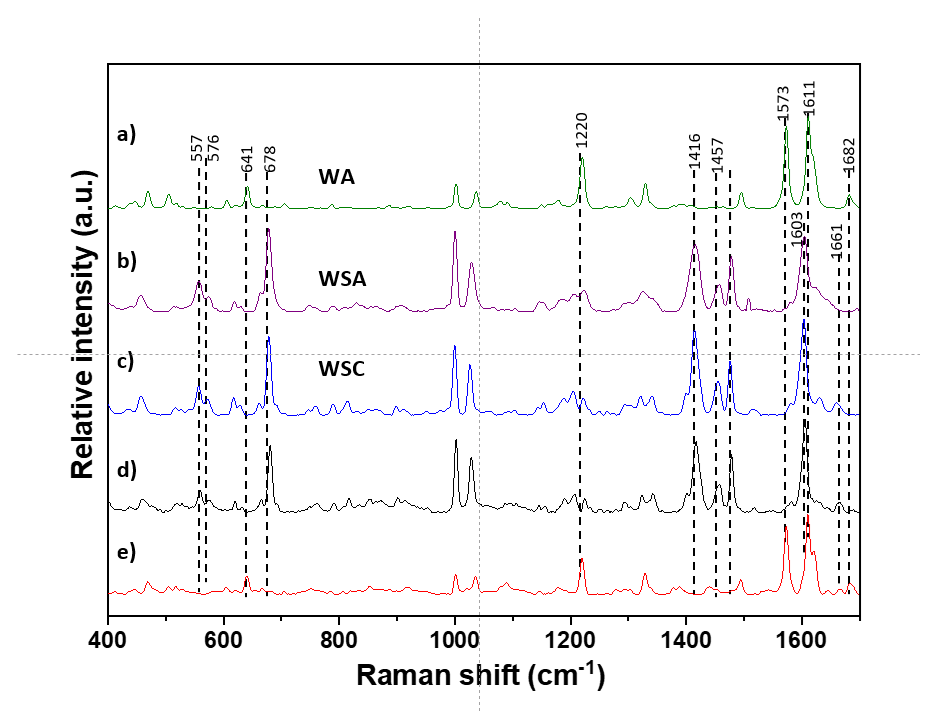
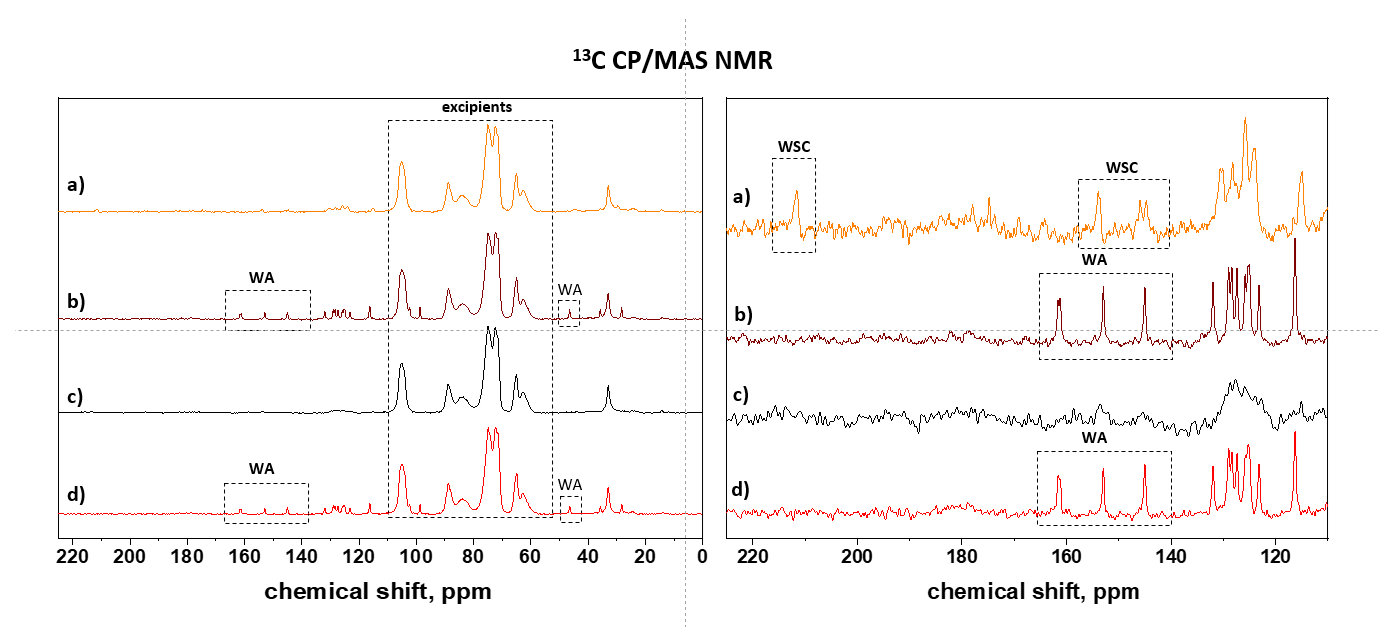
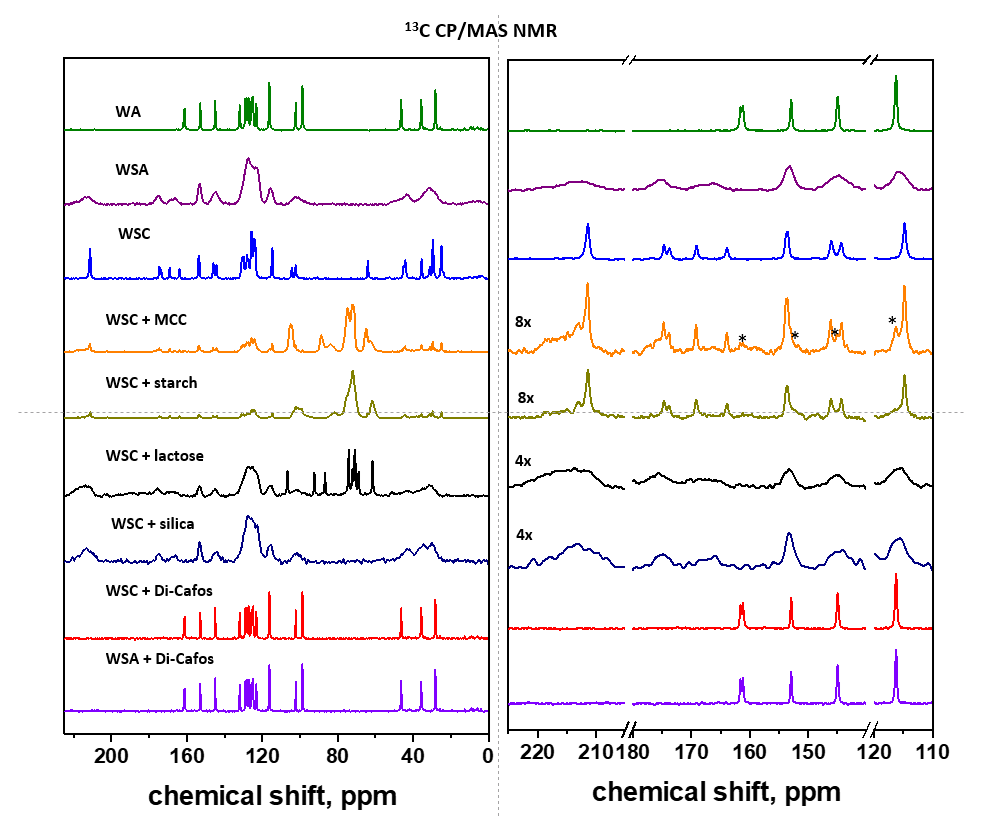
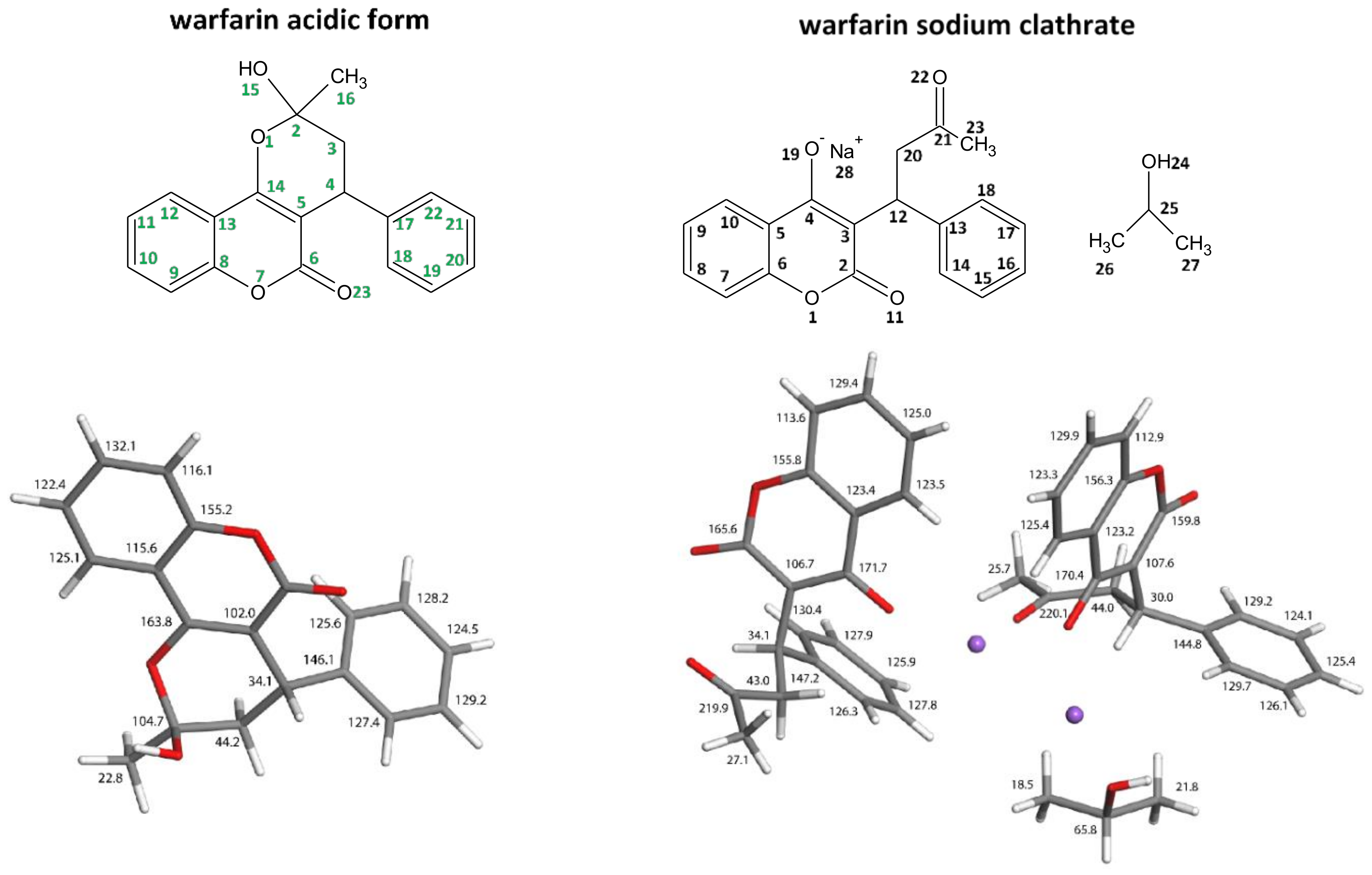
3.1. API Solid-State Change Characterization in Tablets
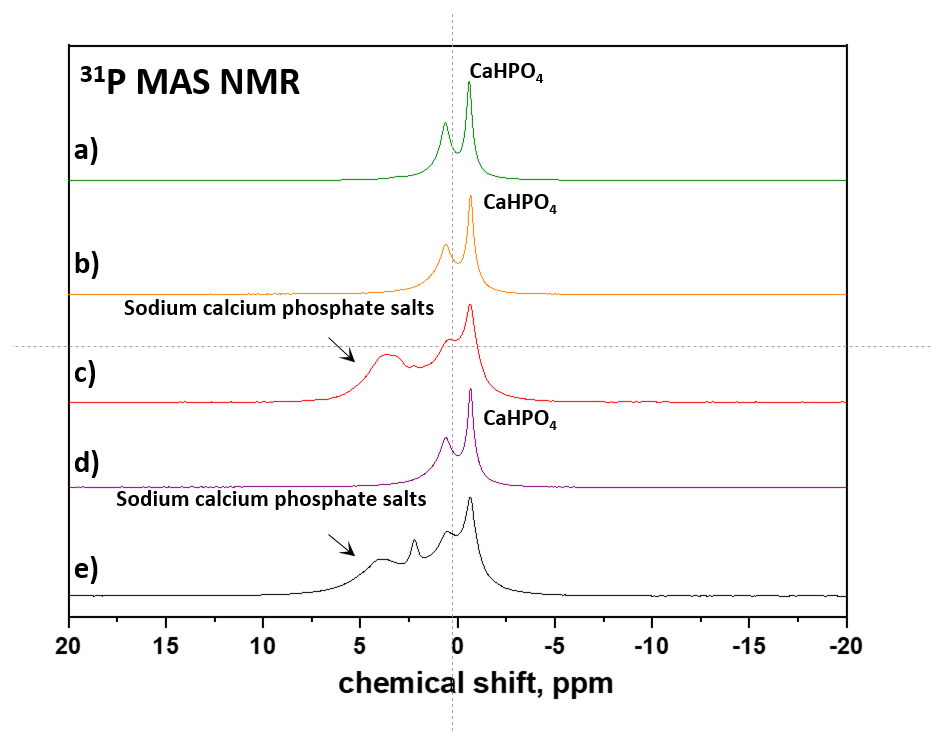
3.1.1. Commercial Tablets
3.1.2. Model Warfarin Tablets
3.2. Physicochemical Characterization of API and Tablets
3.3. Dissolution Tests
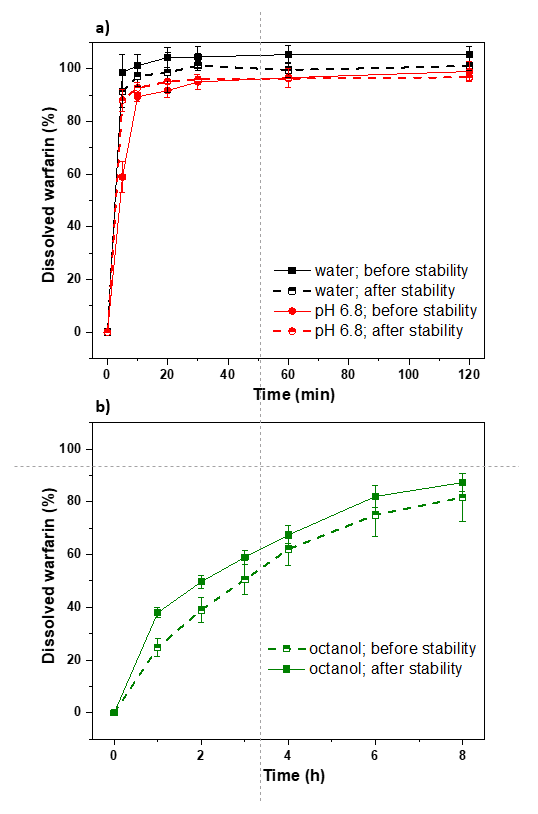
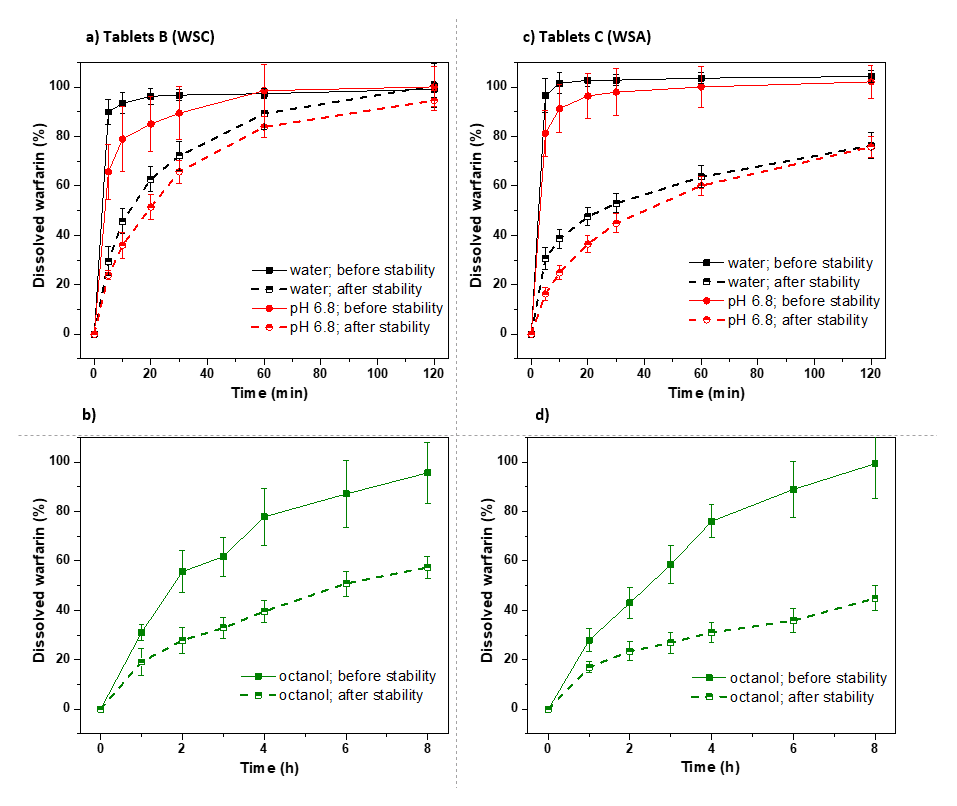
3.3.1. Commercial Tablets
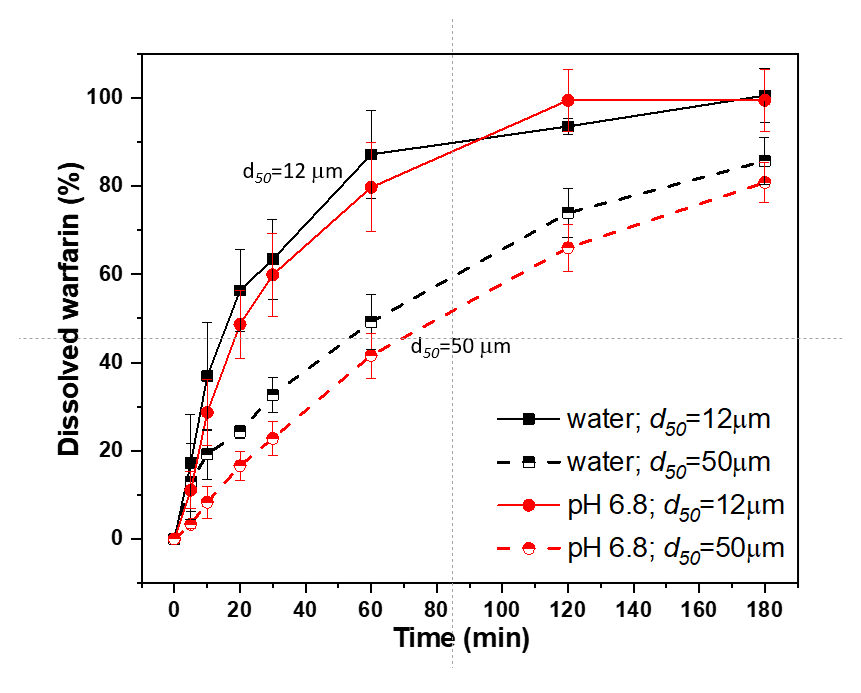
3.3.2. Model Warfarin Tablets
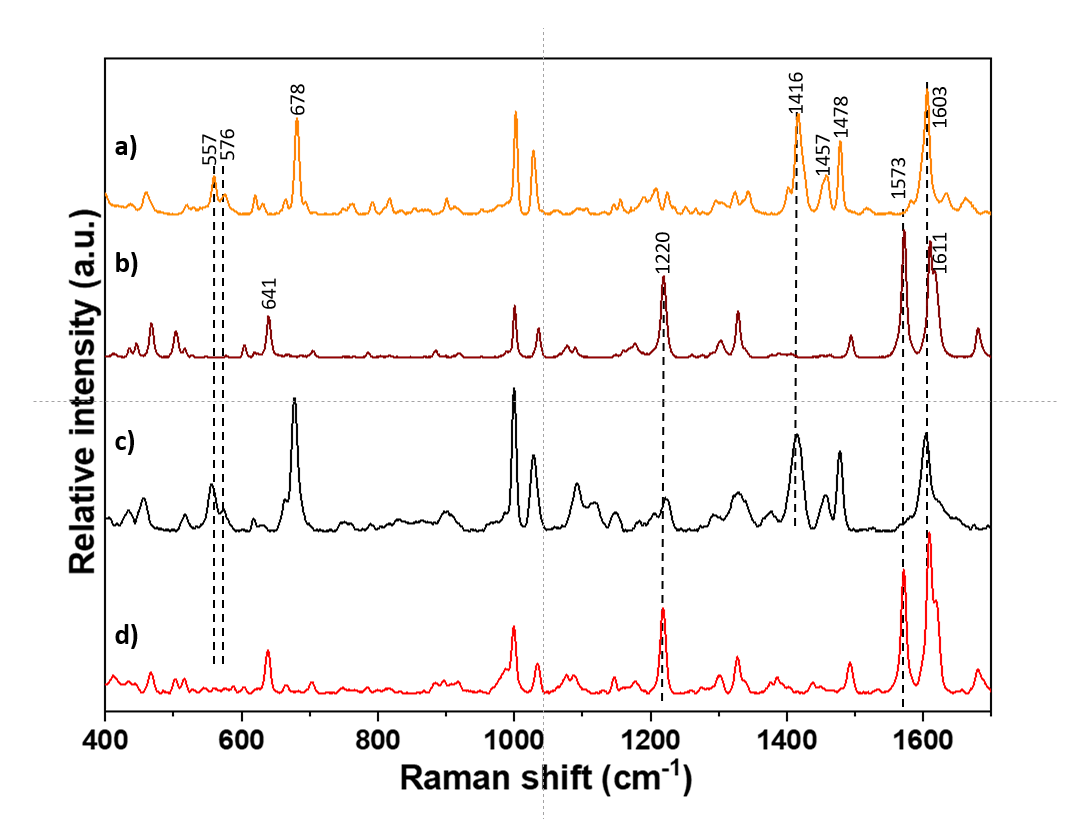
3.4. Warfarin-Excipient Interactions in Binary Mixtures
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Erener, S. Diabetes, infection risk and COVID-19. Mol. Metab. 2020, 39, 101044. [Google Scholar] [CrossRef] [PubMed]
- Bultas, J.; Karetová, D. New oral anticoagulants—Aspects surrounded by silence. Remedia 2015, 25, 127–134. [Google Scholar]
- Harper, P.; Young, L.; Merriman, E. Bleeding risk with dabigatran in the frail elderly. N. Engl. J. Med. 2012, 366, 864–866. [Google Scholar] [CrossRef]
- Hernandez, I.; Baik, S.H.; Piñera, A.; Zhang, Y. Risk of bleeding with dabigatran in atrial fibrillation. JAMA Intern. Med. 2015, 175, 18–24. [Google Scholar] [CrossRef] [Green Version]
- Ringleb, P.A. Thrombolytics, anticoagulants, and antiplatelet agents. Stroke 2006, 37, 312–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godman, B.; Malmström, R.E.; Diogene, E.; Jayathissa, S.; McTaggart, S.; Cars, T.; Alvarez-Madrazo, S.; Baumgärtel, C.; Brzezinska, A.; Bucsics, A.; et al. Dabigatran-a continuing exemplar case history demonstrating the need for comprehensive models to optimize the utilization of new drugs. Front. Pharmacol. 2014, 5, 109. [Google Scholar] [CrossRef] [PubMed]
- Kow, C.S.; Sunter, W.; Bain, A.; Zaidi, S.T.R.; Hasan, S.S. Management of outpatient warfarin therapy amid COVID-19 pandemic: A practical guide. Am. J. Cardiovasc. Drugs 2020, 20, 301–309. [Google Scholar] [CrossRef]
- Hohnloser, S.H.; Oldgren, J.; Yang, S.; Wallentin, L.; Ezekowitz, M.; Reilly, P.; Eikelboom, J.; Brueckmann, M.; Yusuf, S.; Connolly, S.J. Myocardial ischemic events in patients with atrial fibrillation treated with dabigatran or warfarin in the RE-LY (Randomized evaluation of long-term anticoagulation therapy) trial. Circulation 2012, 125, 669–676. [Google Scholar] [CrossRef]
- Douxfils, J.; Buckinx, F.; Mullier, F.; Minet, V.; Rabenda, V.; Reginster, J.Y.; Hainaut, P.; Bruyère, O.; Dogné, J.M. Dabigatran etexilate and risk of myocardial infarction, other cardiovascular events, major bleeding, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. J. Am. Heart Assoc. 2014, 3, e000515. [Google Scholar] [CrossRef] [Green Version]
- Zeeshan, M.; Jehan, F.; O’Keeffe, T.; Khan, M.; Zakaria, E.R.; Hamidi, M.; Gries, L.; Kulvatunyou, N.; Joseph, B. The novel oral anticoagulants (NOACs) have worse outcomes compared with warfarin in patients with intracranial hemorrhage after TBI. J. Trauma Acute Care Surg. 2018, 85, 915–920. [Google Scholar] [CrossRef]
- Chokesuwattanaskul, R.; Thongprayoon, C.; Tanawuttiwat, T.; Kaewput, W.; Pachariyanon, P.; Cheungpasitporn, W. Safety and efficacy of apixaban versus warfarin in patients with end-stage renal disease: Meta-analysis. Pacing Clin. Electrophysiol. 2018, 41, 627–634. [Google Scholar] [CrossRef]
- Russo-Alvarez, G.; Martinez, K.A.; Valente, M.; Bena, J.; Hu, B.; Luxenburg, J.; Chaitoff, A.; Ituarte, C.; Brateanu, A.; Rothberg, M.B. Thromboembolic and major bleeding events with rivaroxaban versus warfarin use in a real-world setting. Ann. Pharmacother. 2018, 52, 19–25. [Google Scholar] [CrossRef]
- You, J.H. Novel oral anticoagulants versus warfarin therapy at various levels of anticoagulation control in atrial fibrillation—A cost-effectiveness analysis. J. Gen. Intern. Med. 2014, 29, 438–446. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Alexander, G.C.; Nazarian, S.; Segal, J.B.; Wu, A.W. Trends and variation in oral anticoagulant choice in patients with atrial fibrillation, 2010–2017. Pharmacotherapy 2018, 38, 907–920. [Google Scholar] [CrossRef] [PubMed]
- Siguret, V.; Pautas, E.; Gouin-Thibault, I. Warfarin therapy: Influence of pharmacogenetic and environmental factors on the anticoagulant response to warfarin. Vitam. Horm. 2008, 78, 247–264. [Google Scholar] [CrossRef]
- Ghate, S.R.; Biskupiak, J.E.; Ye, X.; Hagan, M.; Kwong, W.J.; Fox, E.S.; Brixner, D.I. Hemorrhagic and thrombotic events associated with generic substitution of warfarin in patients with atrial fibrillation: A retrospective analysis. Ann. Pharmacother. 2011, 45, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Bird, S.T.; Flowers, N.; Zhao, Y.; McKean, S.; Izem, R.; Wernecke, M.; Kozlowski, S.; MaCurdy, T.E.; Kelman, J.A.; Graham, D.J. Healthy user bias in comparative safety studies for brand-name vs. generic products: The example of warfarin. Clin. Pharmacol. Ther. 2019, 106, 1037–1045. [Google Scholar] [CrossRef]
- Hellfritzsch, M.; Rathe, J.; Stage, T.B.; Thirstrup, S.; Grove, E.L.; Damkier, P.; Pottegård, A. Generic switching of warfarin and risk of excessive anticoagulation: A Danish nationwide cohort study. Pharmacoepidemiol. Drug Saf. 2016, 25, 336–343. [Google Scholar] [CrossRef]
- Hope, K.A.; Havrda, D.E. Subtherapeutic INR values associated with a switch to generic warfarin. Ann. Pharmacother. 2001, 35, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Bongiorno, R.A.; Nutescu, E.A. Generic warfarin: Implications for clinical practice and perceptions of anticoagulation providers. Semin. Thromb. Hemost. 2004, 30, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Nguyenpho, A.; Ciavarella, A.B.; Siddiqui, A.; Rahman, Z.; Akhtar, S.; Hunt, R.; Korang-Yeboah, M.; Khan, M.A. Evaluation of in-use stability of anticoagulant drug products: Warfarin sodium. J. Pharm. Sci. 2015, 104, 4232–4240. [Google Scholar] [CrossRef]
- Rahman, Z.; Korang-Yeboah, M.; Siddiqui, A.; Mohammad, A.; Khan, M.A. Understanding effect of formulation and manufacturing variables on the critical quality attributes of warfarin sodium product. Int. J. Pharm. 2015, 495, 19–30. [Google Scholar] [CrossRef]
- Kasim, N.A.; Whitehouse, M.; Ramachandran, C.; Bermejo, M.; Lennernas, H.; Hussain, A.S.; Junginger, H.E.; Stavchansky, S.A.; Midha, K.K.; Shah, V.P.; et al. Molecular properties of WHO essential drugs and provisional biopharmaceutical classification. Mol. Pharm. 2004, 1, 85–96. [Google Scholar] [CrossRef]
- Gao, D.; Maurin, M.B. Physical chemical stability of warfarin sodium. AAPS PharmSci 2001, 3, E3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haines, S.T. Substituting warfarin products: What’s the source of the problem? Ann. Pharmacother. 2011, 45, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Fan, J.; Vince, B.; Li, T.; Gao, W.; Kinjo, M.; Brown, J.; Sun, W.; Jia-ng, W.; et al. Integrating In vitro, modeling, and In vivo approaches to investigate warfarin bioequivalence. CPT Pharmacomet. Syst. Pharmacol. 2017, 6, 523–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franc, A.; Muselík, J.; Zeman, J.; Lukášová, I.; Kurhajec, S.; Bartoníčková, E.; Galvánková, L.; Mika, F.; Dominik, M.; Vetchý, D. The effect of amorphous and crystal sodium warfarin and its content uniformity on bioequivalence of tablets. Eur. J. Pharm. Sci. 2018, 125, 120–129. [Google Scholar] [CrossRef]
- Muselík, J.; Franc, A.; Doležel, P.; Goněc, R.; Krondlová, A.; Lukášová, I. Influence of process parameters on content uniformity of a low dose active pharmaceutical ingredient in a tablet formulation according to GMP. Acta Pharm. 2014, 64, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X. Bioequivalence and characterization of generic drugs: Substitutability of generic drugs: Perceptions and reality. In CERSI Workshop FDA.; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2016. [Google Scholar]
- Brus, J. Heating of samples induced by fast magic-angle spinning. Solid State Nucl. Magn. Reson. 2000, 16, 151–160. [Google Scholar] [CrossRef]
- Urbanova, M.; Gajdosova, M.; Steinhart, M.; Vetchy, D.; Brus, J. Molecular-level control of ciclopirox olamine release from poly(ethylene oxide)-based mucoadhesive buccal films: Exploration of structure-property relationships with solid-state NMR. Mol. Pharm. 2016, 13, 1551–1563. [Google Scholar] [CrossRef]
- Brus, J.; Urbanova, M.; Sedenkova, I.; Brusova, H. New perspectives of 19F MAS NMR in the characterization of amorphous forms of atorvastatin in dosage formulations. Int. J. Pharm. 2011, 409, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Hušák, M.; Jegorov, A.; Czernek, J.; Rohlíček, J.; Žižková, S.; Vraspír, P.; Kolesa, P.; Fitch, A.; Brus, J. Successful strategy for high degree of freedom crystal structure determination from powder X-ray diffraction data: A case study for selexipag form I with 38 DOF. Cryst. Growth Des. 2019, 19, 4625–4631. [Google Scholar] [CrossRef]
- Hušák, M.; Jegorov, A.; Rohlíček, J.; Fitch, A.; Czernek, J.; Kobera, L.; Brus, J. Determining the crystal structures of peptide analogs of boronic acid in the absence of single crystals: Intricate motifs of ixazomib citrate revealed by XRPD guided by ss-NMR. Cryst. Growth Des. 2018, 18, 3616–3625. [Google Scholar] [CrossRef]
- Urbanova, M.; Brus, J.; Sedenkova, I.; Policianova, O.; Kobera, L. Characterization of solid polymer dispersions of active pharmaceutical ingredients by 19F MAS NMR and factor analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2013, 100, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Giannini, D.D.; Chan, K.K.; Roberts, J.D. Carbon-13 nuclear magnetic resonance spectroscopy. Structure of the anticoagulant warfarin and related compounds in solution. Proc. Natl. Acad. Sci. USA 1974, 71, 4221–4223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valente, E.J.; Trager, W.F.; Jensen, L.H. The crystal and molecular structure and absolute configuration of (−)-(S)-warfarin. Acta Crystallogr. 1975, 31, 954–960. [Google Scholar] [CrossRef]
- Brus, J.; Czernek, J.; Kobera, L.; Urbanova, M.; Abbrent, S.; Husak, M. Predicting the crystal structure of decitabine by powder NMR crystallography: Influence of long-range molecular packing symmetry on NMR parameters. Cryst. Growth Des. 2016, 16, 7102–7111. [Google Scholar] [CrossRef]
- Deshpande, M.D.; Scheicher, R.H.; Ahuja, R.; Pandey, R. Binding strength of sodium ions in cellulose for different water contents. J. Phys. Chem. B 2008, 112, 8985–8989. [Google Scholar] [CrossRef]
- Franc, A.; Kurhajec, S.; Pavloková, S.; Sabadková, D.; Muselík, J. Influence of concentration and type of microcrystalline cellulose on the physical properties of tablets containing Cornelian cherry fruits. Acta Pharm. 2017, 67, 187–202. [Google Scholar] [CrossRef] [Green Version]
- Franc, A.; Muselłk, J.; Goněc, R.; Vetchý, D. Biphasic dissolution method for quality control and assurance of drugs containing active substances in the form of weak acid salts. Acta Pharm. 2016, 66, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Investigation of Bioequivalence; European Medicines Agency: London, UK, 2010. [Google Scholar]
- Food and Drug Administration. Dissolution testing of immediate release solid oral dosage forms. In Guidance for Industry; Food and Drug Administration: Rockville, MD, USA, 1997. [Google Scholar]
- Vercaigne, L.M.; Zhanel, G.G. Clinical significance of bioequivalence and interchangeability of narrow therapeutic range drugs: Focus on warfarins. J. Pharm. Pharm. Sci. 1998, 1, 92–94. [Google Scholar] [PubMed]
- Urbanova, M.; Pavelkova, M.; Czernek, J.; Kubova, K.; Vyslouzil, J.; Pechova, A.; Molinkova, D.; Vyslouzil, J.; Vetchy, D.; Brus, J. Interaction pathways and structure-chemical transformations of alginate gels in physiological environments. Biomacromolecules 2019, 20, 4158–4170. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Guo, H.; Pujari-Palmer, M.; Stevensson, B.; Grins, J.; Engqvist, H.; Edén, M. Advanced solid-state 1H/31P NMR characterization of pyrophosphate-doped calcium phosphate cements for biomedical applications: The structural role of pyrophosphate. Ceram. Int. 2019, 45, 20642–20655. [Google Scholar] [CrossRef]
- Awa, K.; Shinzawa, H.; Ozaki, Y. The effect of microcrystalline cellulose crystallinity on the hydrophilic property of tablets and the hydrolysis of acetylsalicylic acid as active pharmaceutical ingredient inside tablets. AAPS PharmSciTech 2015, 16, 865–870. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tablets A | Tablets B | Tablets C | |
|---|---|---|---|
| Before stability study | |||
| API content (%) | 100.6 ± 2.0 | 101.4 ± 1.6 | 98.6 ± 1.6 |
| Mass uniformity (mg) | 181.0 ± 3.8 | 271.5 ± 1.9 | 269.1 ± 1.4 |
| Friability (%) | 0.12 | 0.60 | 0.43 |
| Disintegration (min) | less than 7 | less than 1 | less than 1 |
| Hardness (N) | 55.1 ± 10.9 | 46.0 ± 6.8 | 40.3 ± 8.1 |
| Thickness (mm) | 3.35 ± 0.03 | 2.29 ± 0.07 | 2.26 ± 0.01 |
| Diameter (mm) | 8.04 ± 0.01 | 9.97 ± 0.03 | 10.01 ± 0.01 |
| After stability study | |||
| API content (%) | 103.6 ± 2.8 | 97.8 ± 2.4 | 98.6 ± 3.4 |
| Mass uniformity (mg) | 180.3 ± 2.2 | 267.4 ± 5.1 | 270.0 ± 1.9 |
| Friability (%) | 0.26 | 0.64 | 0.77 |
| Disintegration (min) | less than 5 | less than 2 | less than 2 |
| Hardness (N) | 36.6 ± 3.8 | 47.6 ± 1.4 | 36.8 ± 3.3 |
| Thickness (mm) | 3.46 ± 0.03 | 2.40 ± 0.03 | 2.29 ± 0.01 |
| Diameter (mm) | 8.164 ± 0.02 | 10.08 ± 0.01 | 10.10 ± 0.01 |
| API Form | Particle Size [µm] 1 | ||
|---|---|---|---|
| d10 | d50 | d90 | |
| WSC | 2.3 | 12.9 | 230.3 |
| WSA | 2.1 | 60.7 | 279.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muselík, J.; Urbanova, M.; Bartoníčková, E.; Palovčík, J.; Vetchý, D.; Czernek, J.; Janisova, L.; Velychkivska, N.; Franc, A.; Brus, J. Structural Changes of Sodium Warfarin in Tablets Affecting the Dissolution Profiles and Potential Safety of Generic Substitution. Pharmaceutics 2021, 13, 1364. https://doi.org/10.3390/pharmaceutics13091364
Muselík J, Urbanova M, Bartoníčková E, Palovčík J, Vetchý D, Czernek J, Janisova L, Velychkivska N, Franc A, Brus J. Structural Changes of Sodium Warfarin in Tablets Affecting the Dissolution Profiles and Potential Safety of Generic Substitution. Pharmaceutics. 2021; 13(9):1364. https://doi.org/10.3390/pharmaceutics13091364
Chicago/Turabian StyleMuselík, Jan, Martina Urbanova, Eva Bartoníčková, Jakub Palovčík, David Vetchý, Jiří Czernek, Larisa Janisova, Nadiia Velychkivska, Aleš Franc, and Jiří Brus. 2021. "Structural Changes of Sodium Warfarin in Tablets Affecting the Dissolution Profiles and Potential Safety of Generic Substitution" Pharmaceutics 13, no. 9: 1364. https://doi.org/10.3390/pharmaceutics13091364