
Applying the Alkali-Activation Method to Encapsulate Silicon Nitride Particles in a Bioactive Matrix for Augmented Strength and Bioactivity
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
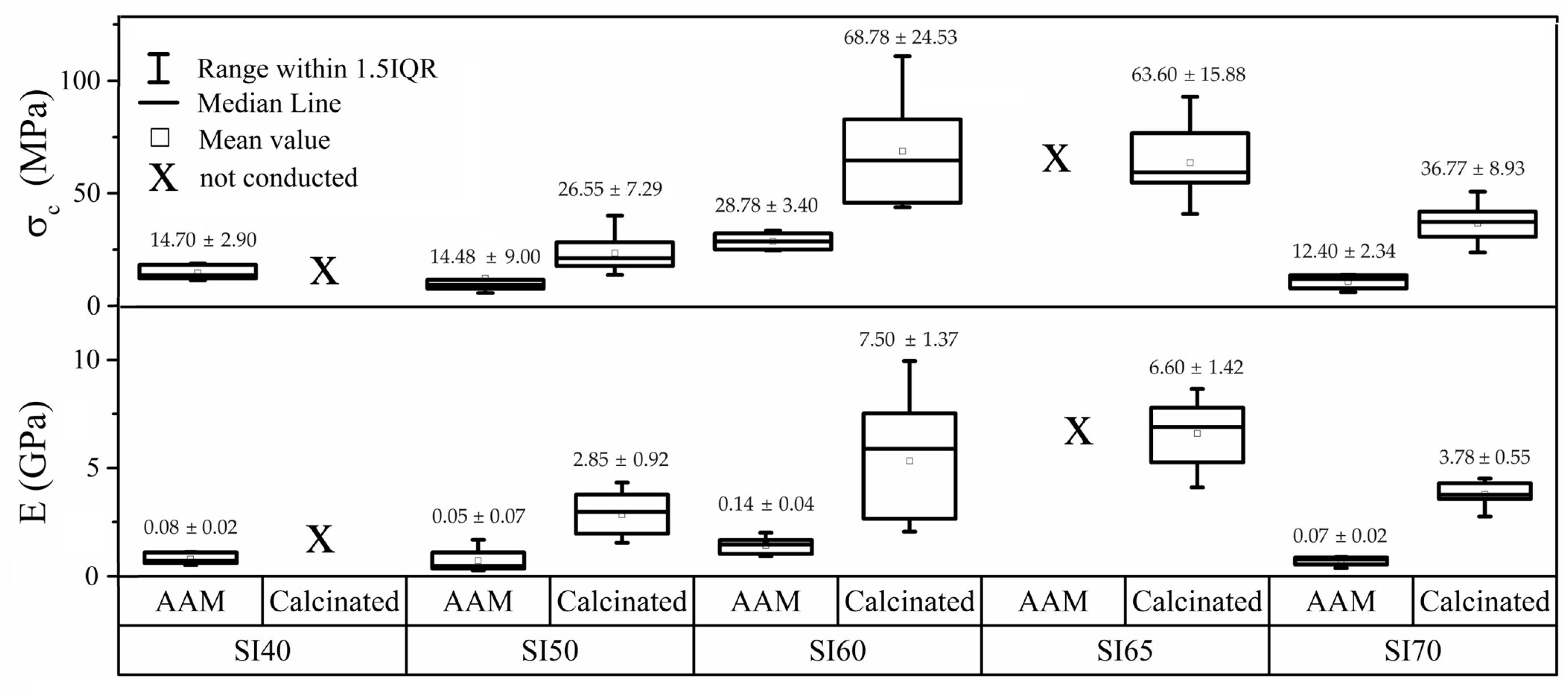
3. Results
3.1. Materials Characterization
3.2. Mechanical Tests
3.3. Bioactivity
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bergmann, C.; Stumpf, A. Topics in Mining, Metallurgy and Materials Engineering. In Dental Ceramics; Springer: Berlin/Heidelberg, Germany, 2013; Volume 49, ISBN 978-3-642-38223-9. [Google Scholar]
- Demontiero, O.; Vidal, C.; Duque, G. Aging and Bone Loss: New Insights for the Clinician. Ther. Adv. Musculoskelet. Dis. 2012, 4, 61–76. [Google Scholar] [CrossRef] [PubMed]
- Sadigov, R. Rapid Growth of the World Population and Its Socioeconomic Results. Sci. World J. 2022, 2022, 8110229. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Yamashita, K. Fabrication Processes for Bioceramics. In Bioceramics and Their Clinical Applications; Woodhead Publishing Limited: Cambridge, UK, 2008; pp. 28–52. ISBN 9781845692049. [Google Scholar]
- Jones, J.R. Review of Bioactive Glass: From Hench to Hybrids. Acta Biomater. 2013, 9, 4457–4486. [Google Scholar] [CrossRef]
- Jones, J.R. Bioactive Glasses; Woodhead Publishing Limited: Cambridge, UK, 2008. [Google Scholar]
- Roy, D.M. Alkali Activated Cements, Opportunities and Challenges. Cem. Concr. Res. 1999, 29, 249–254. [Google Scholar] [CrossRef]
- Provis, J.L. Alkali-Activated Materials. Cem. Concr. Res. 2016, 114, 40–48. [Google Scholar] [CrossRef]
- Taveri, G.; Tousek, J.; Bernardo, E.; Toniolo, N.; Boccaccini, A.R.R.; Dlouhy, I. Proving the Role of Boron in the Structure of Fly-Ash / Borosilicate Glass Based Geopolymers. Mater. Lett. 2017, 200, 105–108. [Google Scholar] [CrossRef]
- Toniolo, N.; Rincón, A.; Roether, J.A.; Ercole, P.; Bernardo, E.; Boccaccini, A.R. Extensive Reuse of Soda-Lime Waste Glass in Fly Ash-Based Geopolymers. Constr. Build. Mater. 2018, 188, 1077–1084. [Google Scholar] [CrossRef]
- Xu, H.H.K.; Wang, P.; Wang, L.; Bao, C.; Chen, Q.; Weir, M.D.; Chow, L.C.; Zhao, L.; Zhou, X.; Reynolds, M.A. Calcium Phosphate Cements for Bone Engineering and Their Biological Properties. Bone Res. 2017, 5, 17056. [Google Scholar] [CrossRef]
- Petzow, M.H. High Performance Non-Oxide Ceramics II; Jansen, M., Ed.; Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2002; Volume 102, ISBN 978-3-540-43132-9. [Google Scholar]
- Collins, J.F.; Gerby, R.W. New Refractory Uses For Silicon Nitride Reported. JOM 1955, 7, 612–615. [Google Scholar] [CrossRef]
- Quinn, G.D.; Swab, J.J.; Motyka, M.J. Fracture Toughness of a Toughened Silicon Nitride by ASTM C 1421. J. Am. Ceram. Soc. 2003, 86, 1043–1045. [Google Scholar] [CrossRef]
- Henstock, J.R.; Canham, L.T.; Anderson, S.I. Silicon: The Evolution of Its Use in Biomaterials. Acta Biomater. 2015, 11, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Frajkorová, F.; Bodišová, K.; Boháč, M.; Bartoníčková, E.; Sedláček, J. Preparation and Characterisation of Porous Composite Biomaterials Based on Silicon Nitride and Bioglass. Ceram. Int. 2015, 41, 9770–9778. [Google Scholar] [CrossRef]
- Das, M.; Bhimani, K.; Balla, V.K. In Vitro Tribological and Biocompatibility Evaluation of Sintered Silicon Nitride. Mater. Lett. 2018, 212, 130–133. [Google Scholar] [CrossRef]
- Cecen, B.; Topates, G.; Kara, A.; Onat, S.; Havitcioglu, H.; Didem, L. Biocompatibility of Silicon Nitride Produced via Partial Sintering & Tape Casting. Ceram. Int. 2021, 47, 3938–3945. [Google Scholar] [CrossRef]
- Gorth, D.J.; Puckett, S.; Ercan, B.; Webster, T.J.; Rahaman, M.; Sonny Bal, B. Decreased Bacteria Activity on Si3N4 Surfaces Compared with PEEK or Titanium. Int. J. Nanomedicine 2012, 7, 4829–4840. [Google Scholar] [CrossRef] [PubMed]
- Pezzotti, G.; Marin, E.; Adachi, T.; Lerussi, F.; Rondinella, A.; Boschetto, F.; Zhu, W.; Kitajima, T.; Inada, K.; McEntire, B.J.; et al. Incorporating Si 3 N 4 into PEEK to Produce Antibacterial, Osteocondutive, and Radiolucent Spinal Implants. Macromol. Biosci. 2018, 18, 1800033. [Google Scholar] [CrossRef] [PubMed]
- Bal, B.S.; Rahaman, M.N. Orthopedic Applications of Silicon Nitride Ceramics. Acta Biomater. 2012, 8, 2889–2898. [Google Scholar] [CrossRef]
- Raza, S.M.; Khurshid, Z.; Zafar, M.S.; Najeeb, S.; Azeem, S.; Yaqin, U. 15—Silicon Nitride (SiN): An Emerging Material for Dental Implant Applications; Elsevier: Amsterdam, The Netherlands, 2020; ISBN 9780128195864. [Google Scholar]
- Amaral, M.; Lopes, M.A.; Silva, R.F.; Santos, J.D. Densification Route and Mechanical Properties of Si3N4–Bioglass Biocomposites. Biomaterials 2002, 23, 857–862. [Google Scholar] [CrossRef]
- Liu, W.; Tong, W.; He, R.; Wu, H.; Wu, S. Effect of the Y2O3 Additive Concentration on the Properties of a Silicon Nitride Ceramic Substrate. Ceram. Int. 2016, 42, 18641–18647. [Google Scholar] [CrossRef]
- Ziegler, G.; Heinrich, J.; Wötting, G. Relationships between Processing, Microstructure and Properties of Dense and Reaction-Bonded Silicon Nitride. J. Mater. Sci. 1987, 22, 3041–3086. [Google Scholar] [CrossRef]
- Lukianova, O.A.; Parkhomenko, A.A.; Krasilnikov, V.V.; Khmara, A.N.; Kuzmenko, A.P. New Method of Free Silicon Determination in Pressureless Sintered Silicon Nitride by Raman Spectroscopy and XRD. Ceram. Int. 2019, 45, 14338–14346. [Google Scholar] [CrossRef]
- McEntire, B.J.; Lakshminarayanan, R. Processing and Characterization of Silicon Nitride Bioceramics. Bioceram. Dev. Appl. 2016, 6, 1–9. [Google Scholar] [CrossRef]
- Belmonte, M.; González-Julián, J.; Miranzo, P.; Osendi, M.I. Spark Plasma Sintering: A Powerful Tool to Develop New Silicon Nitride-Based Materials. J. Eur. Ceram. Soc. 2010, 30, 2937–2946. [Google Scholar] [CrossRef]
- Lukianova, O.A.; Ivanov, O.N. The Effect of Al2O3-MgO Additives on the Microstructure of Spark Plasma Sintered Silicon Nitride. Ceram. Int. 2018, 44, 390–393. [Google Scholar] [CrossRef]
- Tatli, Z.; Çalişkan, F.; Butler, J.; Crowley, C.; Hampshire, S. SPS Sintering of Silicon Nitride with Fluoride Additive. Ceram. Int. 2014, 40, 1399–1404. [Google Scholar] [CrossRef]
- Qiu, L.; Guzonas, D.A.; Qian, J. Corrosion of Silicon Nitride in High Temperature Alkaline Solutions. J. Nucl. Mater. 2016, 476, 293–301. [Google Scholar] [CrossRef]
- Diba, M.; Goudouri, O.; Tapia, F.; Boccaccini, A.R. Magnesium-Containing Bioactive Polycrystalline Silicate-Based Ceramics and Glass-Ceramics for Biomedical Applications. Curr. Opin. Solid State Mater. Sci. 2014, 18, 147–167. [Google Scholar] [CrossRef]
- International Organization for Standardization of DIN EN ISO 7500-1 Standard. Available online: https://www.zwickroell.com/accessories/load-cells/ (accessed on 3 January 2024).
- Anstis, G.R.; Chantikul, P.; Lawn, B.R.; Marshall, D.B. A Critical Evaluation of Indentation Techniques for Measuring Fracture Toughness: I, Direct Crack Measurements. J. Am. Ceram. Soc. 1981, 64, 533–538. [Google Scholar] [CrossRef]
- Kokubo, T.; Takadama, H. How Useful Is SBF in Predicting in Vivo Bone Bioactivity? Biomaterials 2006, 27, 2907–2915. [Google Scholar] [CrossRef]
- Baino, F.; Fiume, E. Elastic Mechanical Properties of 45S5-Based Bioactive Glass-Ceramic Scaffolds. Materials 2019, 12, 3244. [Google Scholar] [CrossRef]
- Finocchiaro, C.; Barone, G.; Mazzoleni, P.; Leonelli, C.; Gharzouni, A.; Rossignol, S. FT-IR Study of Early Stages of Alkali Activated Materials Based on Pyroclastic Deposits (Mt. Etna, Sicily, Italy) Using Two Different Alkaline Solutions. Constr. Build. Mater. 2020, 262, 120095. [Google Scholar] [CrossRef]
- Gharzouni, A.; Joussein, E.; Samet, B.; Baklouti, S.; Rossignol, S. Effect of the Reactivity of Alkaline Solution and Metakaolin on Geopolymer Formation. J. Non. Cryst. Solids 2015, 410, 127–134. [Google Scholar] [CrossRef]
- Duxson, P.; Lukey, G.C.; Van Deventer, J.S.J. Physical Evolution of Na-Geopolymer Derived from Metakaolin up to 1000 °C. J. Mater. Sci. 2007, 42, 3044–3054. [Google Scholar] [CrossRef]
- Baranowska, A.; Lesniak, M.; Kochanowicz, M.; Zmojda, J.; Dorosz, D. Crystallization Kinetics and Structural Properties of the 45S5 Bioactive Glass and Glass-Ceramic Fiber Doped with Eu3+. Mater. Artic. 2020, 13, 1281. [Google Scholar] [CrossRef] [PubMed]
- Zandi Karimi, A.; Rezabeigi, E.; Drew, R.A.L. Crystallization Behavior of Combeite in 45S5 Bioglass® via Controlled Heat Treatment. J. Non. Cryst. Solids 2018, 502, 176–183. [Google Scholar] [CrossRef]
- Lee, Y.K.; Peng, Y.L.; Tomozawa, M. IR Reflection Spectroscopy of a Soda-Lime Glass Surface during Ion-Exchange. J. Non. Cryst. Solids 1997, 222, 125–130. [Google Scholar] [CrossRef]
- Shi, C.; Zhu, Y.; Qian, H.; Lu, L. Fabrication of Silicon Nitride Fiber-PMMA Composite through Free Radical Polymerization in Batch. Mater. Res. Bull. 2014, 51, 161–166. [Google Scholar] [CrossRef]
- Ricciotti, L.; Apicella, A.; Perrotta, V.; Aversa, R. Geopolymer Materials for Bone Tissue Applications: Recent Advances and Future Perspectives. Polymers 2023, 15, 1087. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Kansal, I.; Lancellotti, I.; Leonelli, C.; Azom, C. Mechanical and Biological Characterization of Geopolymers for Potential Application as Biomaterials. AZojomo 2012, 1–15. [Google Scholar]
- Qin, Y.; Qu, C.; Ma, C.; Zhou, L. One-Part Alkali-Activated Materials: State of the Art and Perspectives. Polymers 2022, 14, 5046. [Google Scholar] [CrossRef]
- Baino, F.; Fiume, E. Mechanical Characterization of 45S5 Bioactive Glass-Derived Scaffolds. Mater. Lett. 2019, 245, 14–17. [Google Scholar] [CrossRef]
- Rezwan, K.; Chen, Q.Z.; Blaker, J.J.; Boccaccini, A.R. Biodegradable and Bioactive Porous Polymer/Inorganic Composite Scaffolds for Bone Tissue Engineering. Biomaterials 2006, 27, 3413–3431. [Google Scholar] [CrossRef] [PubMed]
- Zandi Karimi, A.; Rezabeigi, E.; Drew, R.A.L. Aluminum-Free Glass Ionomer Cements Containing 45S5 Bioglass® and Its Bioglass-Ceramic. J. Mater. Sci. Mater. Med. 2021, 32, 76. [Google Scholar] [CrossRef] [PubMed]
- Yli-Urpo, H.; Lassila, L.V.J.; Närhi, T.; Vallittu, P.K. Compressive Strength and Surface Characterization of Glass Ionomer Cements Modified by Particles of Bioactive Glass. Dent. Mater. 2005, 21, 201–209. [Google Scholar] [CrossRef]
- Abo-Mosallam, H.A.; Salama, S.N.; Salman, S.M. Formulation and Characterization of Glass-Ceramics Based on Na 2Ca2Si3O9-Ca5(PO4)3F-Mg2SiO4-System in Relation to Their Biological Activity. J. Mater. Sci. Mater. Med. 2009, 20, 2385–2394. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Si3N4 (wt%) | Composition of the Matrix (wt%) | Molarity of the Alkaline Activator (M) | |||
|---|---|---|---|---|---|---|
| SiO2 | Ca(OH)2 | MgO | NaOH | |||
| SI40 | 40.00 | 23.25 | 16.75 | 3.10 | 16.90 | 30.72 |
| SI50 | 50.00 | 19.38 | 13.95 | 2.58 | 14.09 | 25.60 |
| SI60 | 60.00 | 15.50 | 11.16 | 2.07 | 11.26 | 20.48 |
| SI65 | 65.00 | 13.56 | 9.77 | 1.82 | 9.86 | 17.92 |
| SI70 | 70.00 | 11.63 | 8.37 | 1.55 | 8.45 | 15.36 |
| SI80 | 80.00 | 7.75 | 5.58 | 1.03 | 5.63 | 10.24 |
| Composition | SI50 | SI60 | SI70 |
|---|---|---|---|
| Relative density (%) | 75.67 ± 0.21 | 71.74 ± 0.61 | 68.31 ± 0.07 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de la Torre, G.M.O.; Tatarková, M.; Netriová, Z.; Barlog, M.; Bertolla, L.; Hnatko, M.; Taveri, G. Applying the Alkali-Activation Method to Encapsulate Silicon Nitride Particles in a Bioactive Matrix for Augmented Strength and Bioactivity. Materials 2024, 17, 328. https://doi.org/10.3390/ma17020328
de la Torre GMO, Tatarková M, Netriová Z, Barlog M, Bertolla L, Hnatko M, Taveri G. Applying the Alkali-Activation Method to Encapsulate Silicon Nitride Particles in a Bioactive Matrix for Augmented Strength and Bioactivity. Materials. 2024; 17(2):328. https://doi.org/10.3390/ma17020328
Chicago/Turabian Stylede la Torre, Guido Manuel Olvera, Monika Tatarková, Zuzana Netriová, Martin Barlog, Luca Bertolla, Miroslav Hnatko, and Gianmarco Taveri. 2024. "Applying the Alkali-Activation Method to Encapsulate Silicon Nitride Particles in a Bioactive Matrix for Augmented Strength and Bioactivity" Materials 17, no. 2: 328. https://doi.org/10.3390/ma17020328