
Continuous Fluorescent Sirtuin Activity Assay Based on Fatty Acylated Lysines
 , , and
, , and
Abstract
:1. Introduction
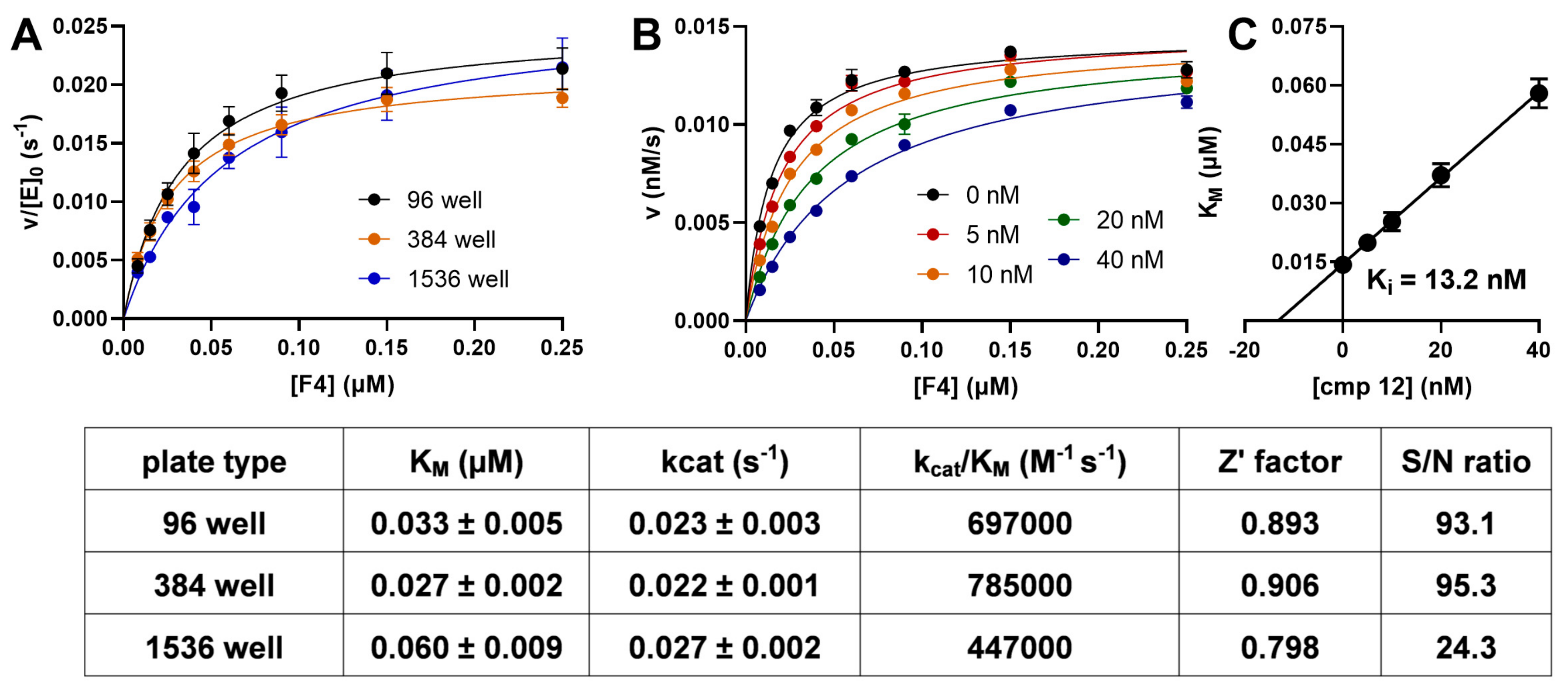
2. Results
3. Discussion
4. Materials and Methods
4.1. Enzymes and Chemicals
4.2. Synthesis
4.3. Mcm1–Mcm3
4.4. F1–F6
4.5. Recording of Fluorescence and Absorbance Spectra
4.6. Quenching Efficiency Determination
4.7. HPLC Based Deacylation Analysis
4.8. Steady-State Kinetics
4.9. Determination of IC50 Values
4.10. Determination of the Ki Values
4.11. Binding Experiments
4.12. Determination of Z′ Value and S/N Ratio
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, B.C.; Hallows, W.C.; Denu, J.M. Mechanisms and molecular probes of sirtuins. Chem. Biol. 2008, 15, 1002–1013. [Google Scholar] [CrossRef] [PubMed]
- Schutkowski, M.; Fischer, F.; Roessler, C.; Steegborn, C. New assays and approaches for discovery and design of Sirtuin modulators. Expert Opin. Drug Discov. 2014, 9, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Zessin, M.; Meleshin, M.; Simic, Z.; Kalbas, D.; Arbach, M.; Gebhardt, P.; Melesina, J.; Liebscher, S.; Bordusa, F.; Sippl, W.; et al. Continuous Sirtuin/HDAC (histone deacetylase) activity assay using thioamides as PET (Photoinduced Electron Transfer)-based fluorescence quencher. Bioorg. Chem. 2021, 117, 105425. [Google Scholar] [CrossRef]
- Riester, D.; Hildmann, C.; Grünewald, S.; Beckers, T.; Schwienhorst, A. Factors affecting the substrate specificity of histone deacetylases. Biochem. Biophys. Res. Commun. 2007, 357, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Wegener, D.; Hildmann, C.; Riester, D.; Schober, A.; Meyer-Almes, F.-J.; Deubzer, H.E.; Oehme, I.; Witt, O.; Lang, S.; Jaensch, M.; et al. Identification of novel small-molecule histone deacetylase inhibitors by medium-throughput screening using a fluorigenic assay. Biochem. J. 2008, 413, 143–150. [Google Scholar] [CrossRef]
- Ciossek, T.; Julius, H.; Wieland, H.; Maier, T.; Beckers, T. A homogeneous cellular histone deacetylase assay suitable for compound profiling and robotic screening. Anal. Biochem. 2008, 372, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Wegener, D.; Wirsching, F.; Riester, D.; Schwienhorst, A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem. Biol. 2003, 10, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Wegener, D.; Hildmann, C.; Riester, D.; Schwienhorst, A. Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem. 2003, 321, 202–208. [Google Scholar] [CrossRef]
- Bradner, J.E.; West, N.; Grachan, M.L.; Greenberg, E.F.; Haggarty, S.J.; Warnow, T.; Mazitschek, R. Chemical phylogenetics of histone deacetylases. Nat. Chem. Biol. 2010, 6, 238–243. [Google Scholar] [CrossRef]
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef]
- Smith, B.C.; Hallows, W.C.; Denu, J.M. A continuous microplate assay for sirtuins and nicotinamide-producing enzymes. Anal. Biochem. 2009, 394, 101–109. [Google Scholar] [CrossRef]
- Wei, W.; Zhang, J.; Xu, Z.; Liu, Z.; Huang, C.; Cheng, K.; Meng, L.; Matsuda, Y.; Hao, Q.; Zhang, H.; et al. Universal Strategy to Develop Fluorogenic Probes for Lysine Deacylase/Demethylase Activity and Application in Discriminating Demethylation States. ACS Sens. 2023, 8, 28–39. [Google Scholar] [CrossRef]
- Tan, S.; Li, X. Small-Molecule Fluorescent Probes for Detecting HDAC Activity. Chem. Asian J. 2022, 17, e202200835. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Kikuchi, K. Chemical Tools with Fluorescence Switches for Verifying Epigenetic Modifications. Acc. Chem. Res. 2019, 52, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Scriba, G.K.E. Electrophoretically mediated microanalysis assay for sirtuin enzymes. Electrophoresis 2010, 31, 3874–3880. [Google Scholar] [CrossRef] [PubMed]
- Ohla, S.; Beyreiss, R.; Scriba, G.K.E.; Fan, Y.; Belder, D. An integrated on-chip sirtuin assay. Electrophoresis 2010, 31, 3263–3267. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gerber, R.; Wu, J.; Tsuruda, T.; McCarter, J.D. High-throughput assays for sirtuin enzymes: A microfluidic mobility shift assay and a bioluminescence assay. Anal. Biochem. 2008, 378, 53–59. [Google Scholar] [CrossRef]
- Blackwell, L.; Norris, J.; Suto, C.M.; Janzen, W.P. The use of diversity profiling to characterize chemical modulators of the histone deacetylases. Life Sci. 2008, 82, 1050–1058. [Google Scholar] [CrossRef]
- Khan, A.N.; Lewis, P.N. Unstructured conformations are a substrate requirement for the Sir2 family of NAD-dependent protein deacetylases. J. Biol. Chem. 2005, 280, 36073–36078. [Google Scholar] [CrossRef]
- Jackson, M.D.; Denu, J.M. Structural identification of 2′- and 3′-O-acetyl-ADP-ribose as novel metabolites derived from the Sir2 family of beta -NAD+-dependent histone/protein deacetylases. J. Biol. Chem. 2002, 277, 18535–18544. [Google Scholar] [CrossRef]
- Tanner, K.G.; Landry, J.; Sternglanz, R.; Denu, J.M. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADP-ribose. Proc. Natl. Acad. Sci. USA 2000, 97, 14178–14182. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, P.A.; Richardson, P.L.; Guo, J.; Barrett, L.W.; Xu, N.; Gunasekera, A.; Glaser, K.B. Fluorescence assay of SIRT protein deacetylases using an acetylated peptide substrate and a secondary trypsin reaction. Anal. Biochem. 2004, 332, 90–99. [Google Scholar] [CrossRef]
- Du, J.; Zhou, Y.; Su, X.; Yu, J.J.; Khan, S.; Jiang, H.; Kim, J.; Woo, J.; Kim, J.H.; Choi, B.H.; et al. Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.N.; Lewis, P.N. Use of substrate analogs and mutagenesis to study substrate binding and catalysis in the Sir2 family of NAD-dependent protein deacetylases. J. Biol. Chem. 2006, 281, 11702–11711. [Google Scholar] [CrossRef]
- Borra, M.T.; Denu, J.M. Quantitative assays for characterization of the Sir2 family of NAD(+)-dependent deacetylases. Methods Enzymol. 2004, 376, 171–187. [Google Scholar] [CrossRef] [PubMed]
- McDonagh, T.; Hixon, J.; DiStefano, P.S.; Curtis, R.; Napper, A.D. Microplate filtration assay for nicotinamide release from NAD using a boronic acid resin. Methods 2005, 36, 346–350. [Google Scholar] [CrossRef]
- Hoffmann, K.; Heltweg, B.; Jung, M. Improvement and Validation of the Fluorescence-Based Histone Deacetylase Assay Using an Internal Standard. Arch. Pharm. Pharm. Med. Chem. 2001, 334, 248–252. [Google Scholar] [CrossRef]
- Shao, D.; Yao, C.; Kim, M.H.; Fry, J.; Cohen, R.A.; Costello, C.E.; Matsui, R.; Seta, F.; McComb, M.E.; Bachschmid, M.M. Improved mass spectrometry-based activity assay reveals oxidative and metabolic stress as sirtuin-1 regulators. Redox Biol. 2019, 22, 101150. [Google Scholar] [CrossRef]
- Holzhauser, S.; Freiwald, A.; Weise, C.; Multhaup, G.; Han, C.-T.; Sauer, S. Discovery and characterization of protein-modifying natural products by MALDI mass spectrometry reveal potent SIRT1 and p300 inhibitors. Angew. Chem. Int. Ed. Engl. 2013, 52, 5171–5174. [Google Scholar] [CrossRef]
- Rye, P.T.; Frick, L.E.; Ozbal, C.C.; Lamarr, W.A. Advances in label-free screening approaches for studying sirtuin-mediated deacetylation. J. Biomol. Screen. 2011, 16, 1217–1226. [Google Scholar] [CrossRef]
- Fischer, F.; Gertz, M.; Suenkel, B.; Lakshminarasimhan, M.; Schutkowski, M.; Steegborn, C. Sirt5 deacylation activities show differential sensitivities to nicotinamide inhibition. PLoS ONE 2012, 7, e45098. [Google Scholar] [CrossRef]
- Machleidt, T.; Robers, M.B.; Hermanson, S.B.; Dudek, J.M.; Bi, K. TR-FRET cellular assays for interrogating posttranslational modifications of histone H3. J. Biomol. Screen. 2011, 16, 1236–1246. [Google Scholar] [CrossRef]
- Degorce, F.; Card, A.; Soh, S.; Trinquet, E.; Knapik, G.P.; Xie, B. HTRF: A technology tailored for drug discovery—A review of theoretical aspects and recent applications. Curr. Chem. Genomics 2009, 3, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Dudek, J.M.; Horton, R.A. TR-FRET biochemical assays for detecting posttranslational modifications of p53. J. Biomol. Screen. 2010, 15, 569–575. [Google Scholar] [CrossRef]
- Robers, M.B.; Loh, C.; Carlson, C.B.; Yang, H.; Frey, E.A.; Hermanson, S.B.; Bi, K. Measurement of the cellular deacetylase activity of SIRT1 on p53 via LanthaScreen® technology. Mol. Biosyst. 2011, 7, 59–66. [Google Scholar] [CrossRef]
- Rauh, D.; Fischer, F.; Gertz, M.; Lakshminarasimhan, M.; Bergbrede, T.; Aladini, F.; Kambach, C.; Becker, C.F.W.; Zerweck, J.; Schutkowski, M.; et al. An acetylome peptide microarray reveals specificities and deacetylation substrates for all human sirtuin isoforms. Nat. Commun. 2013, 4, 2327. [Google Scholar] [CrossRef] [PubMed]
- Kutil, Z.; Skultetyova, L.; Rauh, D.; Meleshin, M.; Snajdr, I.; Novakova, Z.; Mikesova, J.; Pavlicek, J.; Hadzima, M.; Baranova, P.; et al. The unraveling of substrate specificity of histone deacetylase 6 domains using acetylome peptide microarrays and peptide libraries. FASEB J. 2019, 33, 4035–4045. [Google Scholar] [CrossRef]
- Heltweg, B.; Dequiedt, F.; Verdin, E.; Jung, M. Nonisotopic substrate for assaying both human zinc and NAD+-dependent histone deacetylases. Anal. Biochem. 2003, 319, 42–48. [Google Scholar] [CrossRef]
- Toro, T.B.; Watt, T.J. KDAC8 substrate specificity quantified by a biologically relevant, label-free deacetylation assay. Protein Sci. 2015, 24, 2020–2032. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ge, J.; Lei, H.; Peng, B.; Zhang, H.; Wang, D.; Pan, S.; Chen, G.; Chen, L.; Wang, Y.; et al. Fluorescent Probes for Single-Step Detection and Proteomic Profiling of Histone Deacetylases. J. Am. Chem. Soc. 2016, 138, 15596–15604. [Google Scholar] [CrossRef]
- Baba, R.; Hori, Y.; Kikuchi, K. Intramolecular long-distance nucleophilic reactions as a rapid fluorogenic switch applicable to the detection of enzymatic activity. Chemistry 2015, 21, 4695–4702. [Google Scholar] [CrossRef]
- Baba, R.; Hori, Y.; Mizukami, S.; Kikuchi, K. Development of a fluorogenic probe with a transesterification switch for detection of histone deacetylase activity. J. Am. Chem. Soc. 2012, 134, 14310–14313. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Du, W.; Zhang, T.; Liang, G. A Bioluminescent Probe for Simultaneously Imaging Esterase and Histone Deacetylase Activity in a Tumor. Anal. Chem. 2020, 92, 15275–15279. [Google Scholar] [CrossRef] [PubMed]
- Rooker, D.R.; Klyubka, Y.; Gautam, R.; Tomat, E.; Buccella, D. Peptide-Based Fluorescent Probes for Deacetylase and Decrotonylase Activity: Toward a General Platform for Real-Time Detection of Lysine Deacylation. Chembiochem 2018, 19, 496–504. [Google Scholar] [CrossRef]
- Rooker, D.R.; Buccella, D. Real-time detection of histone deacetylase activity with a small molecule fluorescent and spectrophotometric probe. Chem. Sci. 2015, 6, 6456–6461. [Google Scholar] [CrossRef]
- Feldman, J.L.; Baeza, J.; Denu, J.M. Activation of the protein deacetylase SIRT6 by long-chain fatty acids and widespread deacylation by mammalian sirtuins. J. Biol. Chem. 2013, 288, 31350–31356. [Google Scholar] [CrossRef]
- Kutil, Z.; Novakova, Z.; Meleshin, M.; Mikesova, J.; Schutkowski, M.; Barinka, C. Histone Deacetylase 11 Is a Fatty-Acid Deacylase. ACS Chem. Biol. 2018, 13, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Yruela, C.; Galleano, I.; Madsen, A.S.; Olsen, C.A. Histone Deacetylase 11 Is an ε-N-Myristoyllysine Hydrolase. Cell Chem. Biol. 2018, 25, 849–856. [Google Scholar] [CrossRef]
- Cao, J.; Sun, L.; Aramsangtienchai, P.; Spiegelman, N.A.; Zhang, X.; Huang, W.; Seto, E.; Lin, H. HDAC11 regulates type I interferon signaling through defatty-acylation of SHMT2. Proc. Natl. Acad. Sci. USA 2019, 116, 5487–5492. [Google Scholar] [CrossRef]
- Schuster, S.; Roessler, C.; Meleshin, M.; Zimmermann, P.; Simic, Z.; Kambach, C.; Schiene-Fischer, C.; Steegborn, C.; Hottiger, M.O.; Schutkowski, M. A continuous sirtuin activity assay without any coupling to enzymatic or chemical reactions. Sci. Rep. 2016, 6, 22643. [Google Scholar] [CrossRef]
- Kutil, Z.; Mikešová, J.; Zessin, M.; Meleshin, M.; Nováková, Z.; Alquicer, G.; Kozikowski, A.; Sippl, W.; Bařinka, C.; Schutkowski, M. Continuous Activity Assay for HDAC11 Enabling Reevaluation of HDAC Inhibitors. ACS Omega 2019, 4, 19895–19904. [Google Scholar] [CrossRef]
- Petersson, E.J.; Goldberg, J.M.; Wissner, R.F. On the use of thioamides as fluorescence quenching probes for tracking protein folding and stability. Phys. Chem. Chem. Phys. 2014, 16, 6827–6837. [Google Scholar] [CrossRef]
- Zessin, M.; Kutil, Z.; Meleshin, M.; Nováková, Z.; Ghazy, E.; Kalbas, D.; Marek, M.; Romier, C.; Sippl, W.; Bařinka, C.; et al. One-Atom Substitution Enables Direct and Continuous Monitoring of Histone Deacylase Activity. Biochemistry 2019, 58, 4777–4789. [Google Scholar] [CrossRef]
- Smith, B.C.; Denu, J.M. Mechanism-based inhibition of Sir2 deacetylases by thioacetyl-lysine peptide. Biochemistry 2007, 46, 14478–14486. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Hu, J.; Zhang, X.; Lin, H. Thiomyristoyl peptides as cell-permeable Sirt6 inhibitors. Org. Biomol. Chem. 2014, 12, 7498–7502. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Hu, J.; He, B.; Negrón Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.-L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Hong, J.Y.; Hu, J.; Jing, H.; Wang, M.; Price, I.R.; Cao, J.; Yang, M.; Zhang, X.; Lin, H. A Small-Molecule SIRT2 Inhibitor That Promotes K-Ras4a Lysine Fatty-Acylation. ChemMedChem 2019, 14, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, M.; Ikegawa, S.; Ieda, N.; Nakagawa, H. A Fluorescent Probe for Imaging Sirtuin Activity in Living Cells, Based on One-Step Cleavage of the Dabcyl Quencher. Chembiochem 2016, 17, 1961–1967. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, Y.; Kawaguchi, M.; Ieda, N.; Nakagawa, H. A Set of Highly Sensitive Sirtuin Fluorescence Probes for Screening Small-Molecular Sirtuin Defatty-Acylase Inhibitors. ACS Med. Chem. Lett. 2021, 12, 617–624. [Google Scholar] [CrossRef]
- Kannan, S.; Melesina, J.; Hauser, A.-T.; Chakrabarti, A.; Heimburg, T.; Schmidtkunz, K.; Walter, A.; Marek, M.; Pierce, R.J.; Romier, C.; et al. Discovery of inhibitors of Schistosoma mansoni HDAC8 by combining homology modeling, virtual screening, and in vitro validation. J. Chem. Inf. Model. 2014, 54, 3005–3019. [Google Scholar] [CrossRef]
- Wu, J.; Zheng, Y.G. Fluorescent reporters of the histone acetyltransferase. Anal. Biochem. 2008, 380, 106–110. [Google Scholar] [CrossRef]
- Peng, C.; Lu, Z.; Xie, Z.; Cheng, Z.; Chen, Y.; Tan, M.; Luo, H.; Zhang, Y.; He, W.; Yang, K.; et al. The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteom. 2011, 10, M111.012658. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Peng, C.; Anderson, K.A.; Chhoy, P.; Xie, Z.; Dai, L.; Park, J.; Chen, Y.; Huang, H.; Zhang, Y.; et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014, 19, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A. Fatty acid binding to plasma albumin. J. Lipid Res. 1975, 16, 165–179. [Google Scholar] [CrossRef]
- Moniot, S.; Schutkowski, M.; Steegborn, C. Crystal structure analysis of human Sirt2 and its ADP-ribose complex. J. Struct. Biol. 2013, 182, 136–143. [Google Scholar] [CrossRef]
- Vogelmann, A.; Schiedel, M.; Wössner, N.; Merz, A.; Herp, D.; Hammelmann, S.; Colcerasa, A.; Komaniecki, G.; Hong, J.Y.; Sum, M.; et al. Development of a NanoBRET assay to validate inhibitors of Sirt2-mediated lysine deacetylation and defatty-acylation that block prostate cancer cell migration. RSC Chem. Biol. 2022, 3, 468–485. [Google Scholar] [CrossRef]
- Rumpf, T.; Schiedel, M.; Karaman, B.; Roessler, C.; North, B.J.; Lehotzky, A.; Oláh, J.; Ladwein, K.I.; Schmidtkunz, K.; Gajer, M.; et al. Selective Sirt2 inhibition by ligand-induced rearrangement of the active site. Nat. Commun. 2015, 6, 6263. [Google Scholar] [CrossRef]
- Spiegelman, N.A.; Price, I.R.; Jing, H.; Wang, M.; Yang, M.; Cao, J.; Hong, J.Y.; Zhang, X.; Aramsangtienchai, P.; Sadhukhan, S.; et al. Direct Comparison of SIRT2 Inhibitors: Potency, Specificity, Activity-Dependent Inhibition, and On-Target Anticancer Activities. ChemMedChem 2018, 13, 1890–1894. [Google Scholar] [CrossRef] [PubMed]
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.-C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519. [Google Scholar] [CrossRef]
- Kalbas, D.; Meleshin, M.; Liebscher, S.; Zessin, M.; Melesina, J.; Schiene-Fischer, C.; Bülbül, E.F.; Bordusa, F.; Sippl, W.; Schutkowski, M. Small Changes Make the Difference for SIRT2: Two Different Binding Modes for 3-Arylmercapto-Acylated Lysine Derivatives. Biochemistry 2022, 61, 1705–1722. [Google Scholar] [CrossRef]
- Yamagata, K.; Goto, Y.; Nishimasu, H.; Morimoto, J.; Ishitani, R.; Dohmae, N.; Takeda, N.; Nagai, R.; Komuro, I.; Suga, H.; et al. Structural basis for potent inhibition of SIRT2 deacetylase by a macrocyclic peptide inducing dynamic structural change. Structure 2014, 22, 345–352. [Google Scholar] [CrossRef]
- Suenkel, B.; Fischer, F.; Steegborn, C. Inhibition of the human deacylase Sirtuin 5 by the indole GW5074. Bioorg. Med. Chem. Lett. 2013, 23, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Dose, A.; Jost, J.O.; Spieß, A.C.; Henklein, P.; Beyermann, M.; Schwarzer, D. Facile synthesis of colorimetric histone deacetylase substrates. Chem. Commun. 2012, 48, 9525–9527. [Google Scholar] [CrossRef] [PubMed]
- Halley, F.; Reinshagen, J.; Ellinger, B.; Wolf, M.; Niles, A.L.; Evans, N.J.; Kirkland, T.A.; Wagner, J.M.; Jung, M.; Gribbon, P.; et al. A bioluminogenic HDAC activity assay: Validation and screening. J. Biomol. Screen. 2011, 16, 1227–1235. [Google Scholar] [CrossRef]
- Milne, J.C.; Lambert, P.D.; Schenk, S.; Carney, D.P.; Smith, J.J.; Gagne, D.J.; Jin, L.; Boss, O.; Perni, R.B.; Vu, C.B.; et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007, 450, 712–716. [Google Scholar] [CrossRef]
- Roessler, C.; Tüting, C.; Meleshin, M.; Steegborn, C.; Schutkowski, M. A Novel Continuous Assay for the Deacylase Sirtuin 5 and Other Deacetylases. J. Med. Chem. 2015, 58, 7217–7223. [Google Scholar] [CrossRef]
- Galleano, I.; Schiedel, M.; Jung, M.; Madsen, A.S.; Olsen, C.A. A Continuous, Fluorogenic Sirtuin 2 Deacylase Assay: Substrate Screening and Inhibitor Evaluation. J. Med. Chem. 2016, 59, 1021–1031. [Google Scholar] [CrossRef]
- Xuan, W.; Yao, A.; Schultz, P.G. Genetically Encoded Fluorescent Probe for Detecting Sirtuins in Living Cells. J. Am. Chem. Soc. 2017, 139, 12350–12353. [Google Scholar] [CrossRef]
- Spinck, M.; Ecke, M.; Sievers, S.; Neumann, H. Highly Sensitive Lysine Deacetylase Assay Based on Acetylated Firefly Luciferase. Biochemistry 2018, 57, 3552–3555. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Substrate | KM ± SD (nM) | kcat ± SD × 103 (s−1) | kcat/KM (M−1s−1) |
|---|---|---|---|---|
| SIRT2 | F1 | 16 ± 3 | 8.4 ± 1.9 | 536,000 |
| F2 | 39 ± 5 | 6.7 ± 1.0 | 175,000 | |
| F4 | 33 ± 5 | 23 ± 3 | 697,000 | |
| F5 | 43 ± 5 | 15 ± 2 | 335,400 | |
| Mcm1 | 17 ± 2 | 6.8 ± 1.5 | 400,000 | |
| SIRT3 | F1 | >20 µM | >17 | n.d. |
| F2 | >20 µM | >6 | n.d. | |
| F4 | 530 ± 70 | 23 ± 6 | 44,000 | |
| F5 | 990 ± 70 | 27 ± 6 | 27,000 |
| cmp | IC50 Value ± SD in µM or Inhibition in % at a Given Concentration | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| [S] | K (µM) | [E] (nM) | NAM | S2iL5 | cmp12 | SMyr | KK-22 | SirReal2 | AGK2 |
| F4 (1 µM) | 0.033 | 10 | 100 ± 5 | 0.34 ± 0.02 | 0.32 ± 0.04 | 0.015 ± 0.001 | 16 ± 1.2 | 0% @20 µM | 0% @20 µM |
| F4 (40 nM) | 0.033 | 1 | 211 ± 7 | 0.034 ± 0.004 | 0.015 ± 0.002 | 0.00078 ± 0.00005 | 1.5 ± 1 | 25 ± 2% @20 µM | 42 ± 4% @20 µM |
| S2 (1 µM) | 0.0053 | 10 | 340 ± 7 | 1.3 ± 0.3 | 0.93 ± 0.2 | 0.028 ± 0.005 | 30 ± 2 | 18 ± 2% @20 µM | 0% @20 µM |
| S2 (10 nM) | 0.0053 | 0.5 | 600 ± 70 | 0.028 ± 0.015 | 0.019 ± 0.006 | 0.00058 ± 0.00013 | 2.1 ± 0.1 | 1.1 ± 0.1 | 32 ± 2% @20 µM |
| Mcm1 (1 µM) | 0.017 | 10 | 43 ± 6 | 2.4 ± 0.5 | 1.7 ± 0.2 | 0.093 ± 0.002 | 7 ± 3% @20 µM | 0% @20 µM | 0% @20 µM |
| Mcm1_T (1 µM) | 0.017 | 10 | 45 ± 3 | 1.9 ± 0.1 | 14 ± 3 | 0.058 ± 0.025 | 0% @20 µM | 0% @20 µM | 0 % @20 µM |
| S1 (1 µM) | 0.15 | 10 | 120 ± 10 | 0.56 ± 0.02 | 0.41 ± 0.04 | 0.0099 ± 0.003 | 12 ± 0.5 | 5.5 ± 0.8 | 33 ± 5 % @20 µM |
| C1 (20 µM) | - | 100 | 12 ± 1 | 0.21 ± 0.01 | 0.048 ± 0.005 | 0.056 ± 0.011 | 0.41 ± 0.03 | 0.089 ± 0.009 | 12 ± 1 |
| C2 (1 µM) | - | 100 | 42 ± 6 | 0.42 ± 0.02 | 0.24 ± 0.03 | 0.33 ± 0.01 | 24 ± 1% | 22 ± 3% @20 µM | 6 ± 6% @ 6 µM |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zessin, M.; Meleshin, M.; Hilscher, S.; Schiene-Fischer, C.; Barinka, C.; Jung, M.; Schutkowski, M. Continuous Fluorescent Sirtuin Activity Assay Based on Fatty Acylated Lysines. Int. J. Mol. Sci. 2023, 24, 7416. https://doi.org/10.3390/ijms24087416
Zessin M, Meleshin M, Hilscher S, Schiene-Fischer C, Barinka C, Jung M, Schutkowski M. Continuous Fluorescent Sirtuin Activity Assay Based on Fatty Acylated Lysines. International Journal of Molecular Sciences. 2023; 24(8):7416. https://doi.org/10.3390/ijms24087416
Chicago/Turabian StyleZessin, Matthes, Marat Meleshin, Sebastian Hilscher, Cordelia Schiene-Fischer, Cyril Barinka, Manfred Jung, and Mike Schutkowski. 2023. "Continuous Fluorescent Sirtuin Activity Assay Based on Fatty Acylated Lysines" International Journal of Molecular Sciences 24, no. 8: 7416. https://doi.org/10.3390/ijms24087416